
AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way

Abstract
:1. Introduction
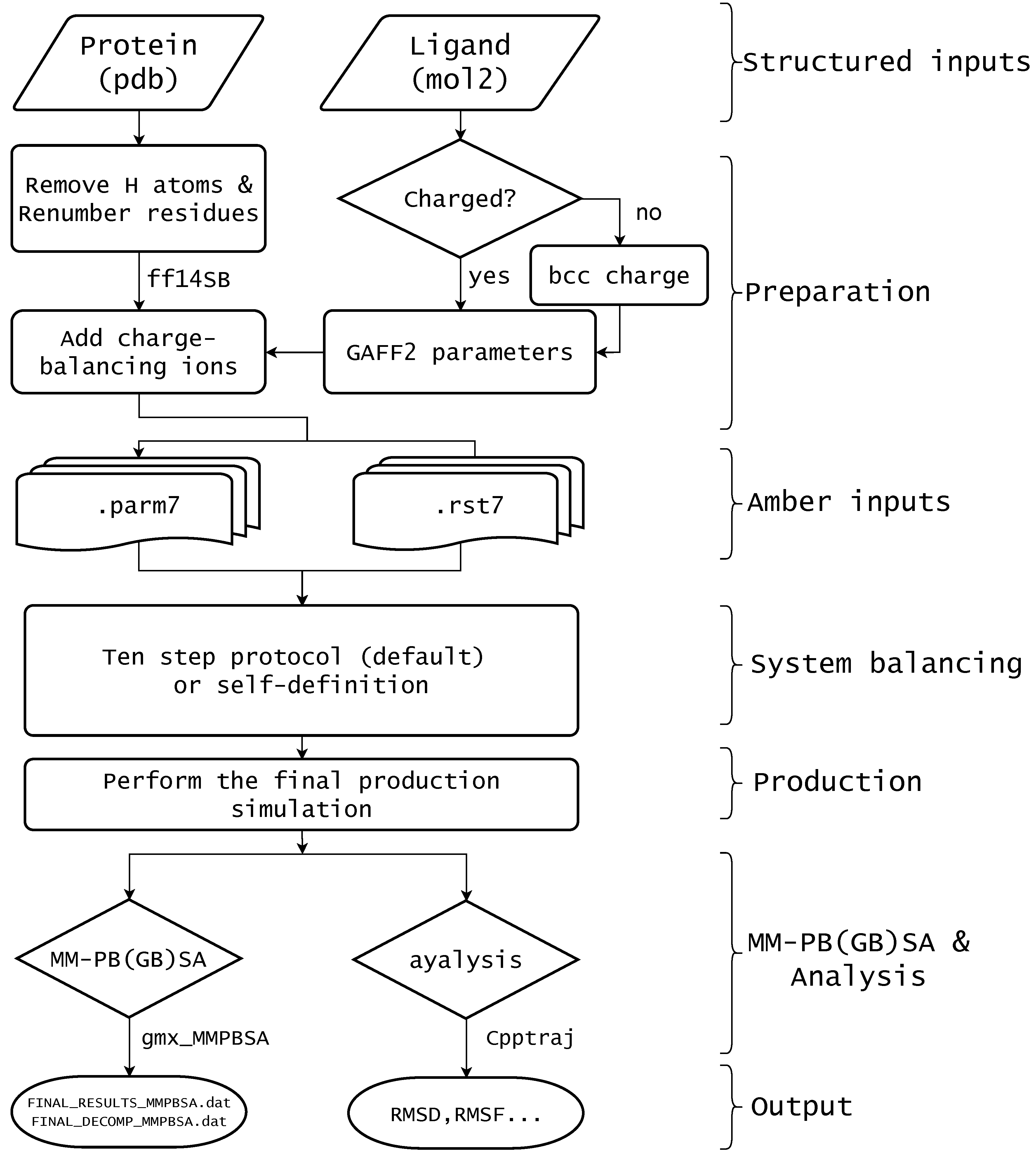
2. Materials and Methods
2.1. Thermostat Methods
2.2. Barostat Methods
2.3. Ten Step Simulation Preparation Protocol
2.4. AM1-BCC Method
2.5. MM-PB(GB)SA Calculations
3. Results
3.1. Install
3.2. Core Classes in the AmberMDrun
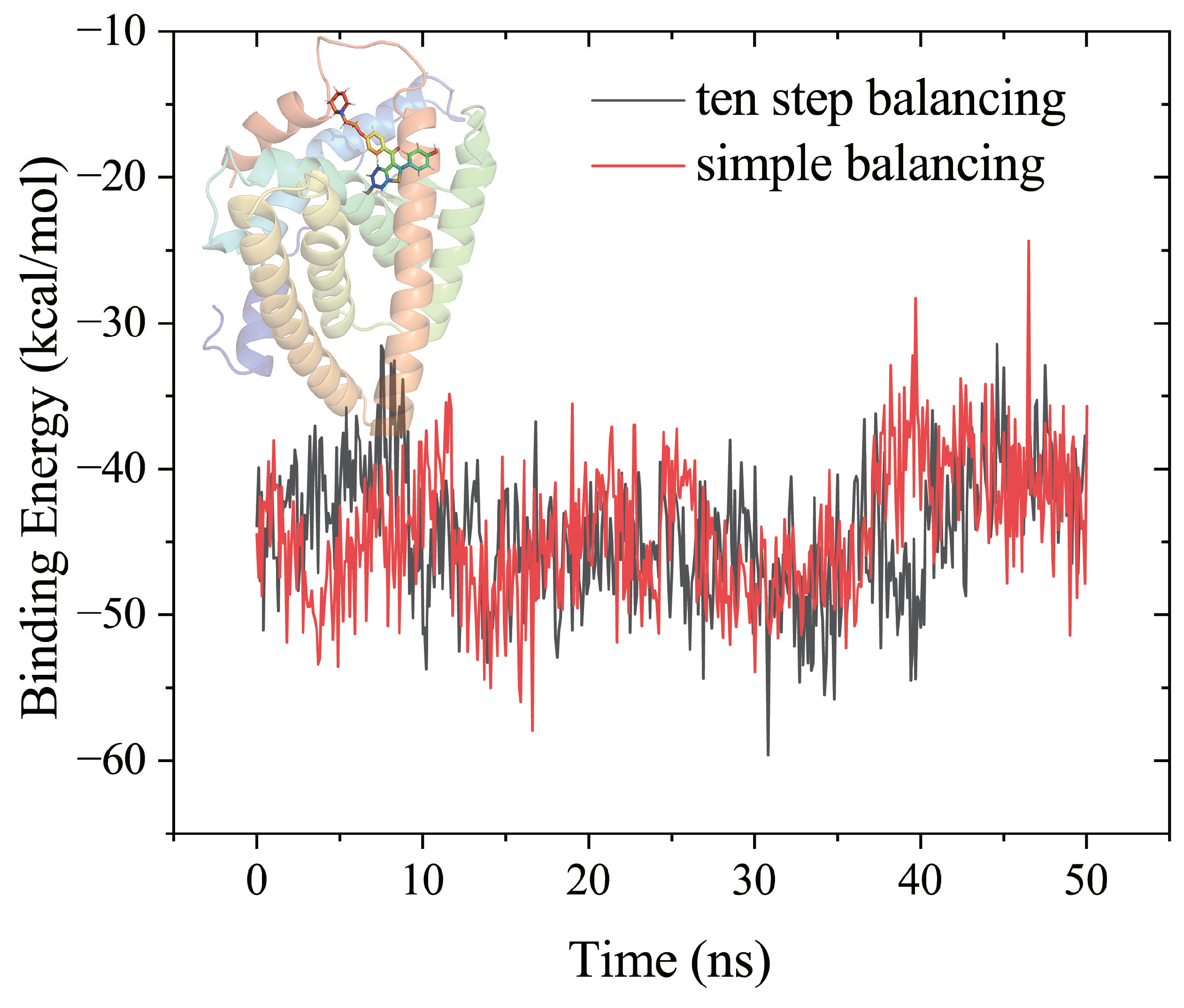
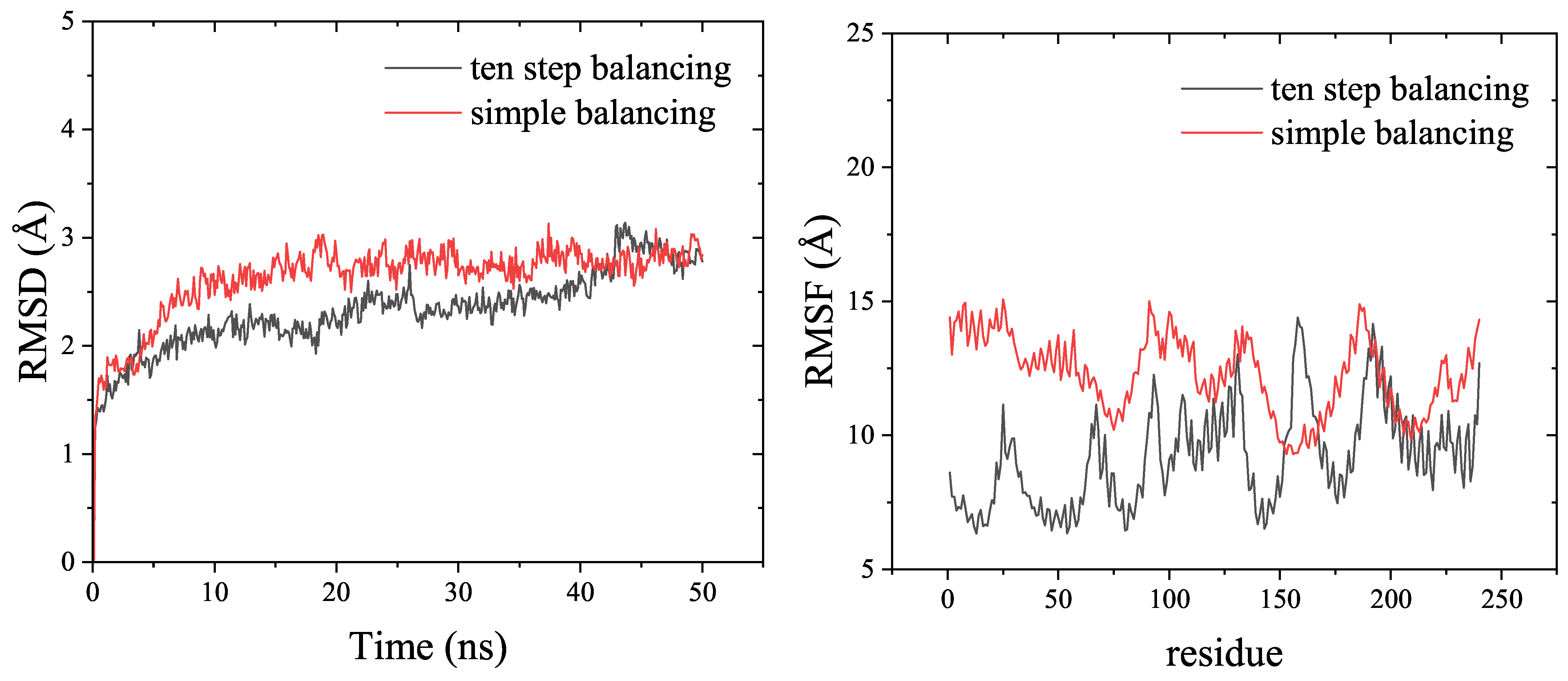
3.3. Example
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ESP | electrostatic potential |
| AM1 | Austin Model 1 |
| BCC | Bond charge correction |
| van der Waals energy | |
| Electrostatic energy | |
| Polar solvation energy | |
| Non-polar solvation energy | |
| Total gas phase free energy | |
| Total solvation free energy | |
| Total energy |
References
- Maginn, E.J. From discovery to data: What must happen for molecular simulation to become a mainstream chemical engineering tool. AIChE J. 2009, 55, 1304–1310. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.a.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Aktulga, H.; Belfon, K.; Ben-Shalom, I.; Berryman, J.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cisneros, G.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Roe, D.R.; Brooks, B.R. A protocol for preparing explicitly solvated systems for stable molecular dynamics simulations. J. Chem. Phys. 2020, 153, 054123. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Uberuaga, B.P.; Anghel, M.; Voter, A.F. Synchronization of trajectories in canonical molecular-dynamics simulations: Observation, explanation, and exploitation. J. Chem. Phys. 2004, 120, 6363–6374. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Kim, S.; Voter, A.F.; Roitberg, A.E. Bad Seeds Sprout Perilous Dynamics: Stochastic Thermostat Induced Trajectory Synchronization in Biomolecules. J. Chem. Theory Comput. 2009, 5, 1624–1631. [Google Scholar] [CrossRef]
- Kleinerman, D.S.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Implementations of Nosé–Hoover and Nosé–Poincaré thermostats in mesoscopic dynamic simulations with the united-residue model of a polypeptide chain. J. Chem. Phys. 2008, 128, 245103. [Google Scholar] [CrossRef] [PubMed]
- Posch, H.A.; Hoover, W.G.; Vesely, F.J. Canonical dynamics of the Nosé oscillator: Stability, order, and chaos. Phys. Rev. A 1986, 33, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Lingenheil, M.; Denschlag, R.; Reichold, R.; Tavan, P. The “Hot-Solvent/Cold-Solute” Problem Revisited. J. Chem. Theory Comput. 2008, 4, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Basconi, J.E.; Shirts, M.R. Effects of Temperature Control Algorithms on Transport Properties and Kinetics in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2013, 9, 2887–2899. [Google Scholar] [CrossRef] [PubMed]
- Omelyan, I.; Kovalenko, A. Multiple time step molecular dynamics in the optimized isokinetic ensemble steered with the molecular theory of solvation: Accelerating with advanced extrapolation of effective solvation forces. J. Chem. Phys. 2013, 139, 244106. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Tuckerman, M.E. Molecular dynamics based enhanced sampling of collective variables with very large time steps. J. Chem. Phys. 2018, 148, 024106. [Google Scholar] [CrossRef]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [Green Version]
- Vitalis, A.; Pappu, R.V. Chapter 3 Methods for Monte Carlo Simulations of Biomacromolecules. In Annual Reports in Computational Chemistry, Chapter 3 Methods for Monte Carlo Simulations of Biomacromolecules; Wheeler, R.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 5, pp. 49–76. [Google Scholar] [CrossRef] [Green Version]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of DNA, RNA, and Phosphoramidate-DNA Helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Account. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, J.; Miller, J.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of RNA Hairpin Loops and Helices. J. Biomol. Struct. Dyn. 1998, 16, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson–Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Connolly, M.L. Analytical molecular surface calculation. J. Appl. Crystallogr. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Lee, M.S.; Olson, M.A. Calculation of Absolute Protein-Ligand Binding Affinity Using Path and Endpoint Approaches. Biophys. J. 2006, 90, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.I.; McGee, T.D.J.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- McGee, D.; Miller, B.J.S., III. Python Script MMPBSA.py. 2009. Available online: https://ambermd.org/tutorials/advanced/tutorial3/py_script/index.php/ (accessed on 9 February 2023).
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef]
- Zhiyong, C.; Zhang, Z.; Zhou, T.; Zhou, X.; Zhang, Y.; Meng, H.; Wang, W.; Liu, Y. A TastePeptides-Meta system including an umami/bitter classification model Umami_YYDS, a TastePeptidesDB database and an open-source package Auto_Taste_ML. Food Chem. 2022, 405, 134812. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| MIN | NVT | NPT | |
|---|---|---|---|
| Params | imin = 1 | imin = 0 | imin = 0 |
| ntb = 1 | ntb = 2 | ||
| temp = 298.15 1 | ✘ | ✔ | ✔ |
| cut = 8.0 2 | ✔ | ✔ | ✔ |
| ntpr = 50 3 | ✔ | ✔ | ✔ |
| ntwr = 500 4 | ✔ | ✔ | ✔ |
| ntwx = 500 5 | ✔ | ✔ | ✔ |
| maxcyc = 1000 6 | ✔ | ✘ | ✘ |
| ncyc = 10 7 | ✔ | ✘ | ✘ |
| ntmin = 10 8 | ✔ | ✘ | ✘ |
| nstlim = 5000 9 | ✘ | ✔ | ✔ |
| dt = 0.002 10 | ✘ | ✔ | ✔ |
| irest = False 11 | ✘ | ✔ | ✔ |
| tautp = 1.0 12 | ✘ | ✔ | ✔ |
| taup = 1.0 13 | ✘ | ✘ | ✔ |
| gamma_ln = 5.0 14 | ✘ | ✔ | ✔ |
| nscm = 0 15 | ✘ | ✔ | ✔ |
| ntc = 2 16 | ✘ | ✔ | ✔ |
| ntf = 2 17 | ✘ | ✔ | ✔ |
| thermostat 18 | ✘ | ✔ | ✔ |
| barostat 19 | ✘ | ✘ | ✔ |
| igamd = false 20 | ✘ | ✘ | ✘ |
| Result | vdW | TOTAL | |||||
|---|---|---|---|---|---|---|---|
| Tutorial 1 | −56.29 | −38.03 | 57.64 | −5.04 | −94.32 | 52.60 | −41.73 |
| Simple balancing 2 | −55.48 | −41.00 | 53.79 | −5.58 | −96.48 | 48.21 | −48.27 |
| Ten step balancing 3 | −58.62 | −36.00 | 54.00 | −5.70 | −94.63 | 48.30 | −46.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-W.; Lu, W.-C. AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way. Biomolecules 2023, 13, 635. https://doi.org/10.3390/biom13040635
Zhang Z-W, Lu W-C. AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way. Biomolecules. 2023; 13(4):635. https://doi.org/10.3390/biom13040635
Chicago/Turabian StyleZhang, Zhi-Wei, and Wen-Cai Lu. 2023. "AmberMDrun: A Scripting Tool for Running Amber MD in an Easy Way" Biomolecules 13, no. 4: 635. https://doi.org/10.3390/biom13040635