

In Silico Screening and Optimization of Cell-Penetrating Peptides Using Deep Learning Methods

Abstract
:1. Introduction
2. Materials and Methods
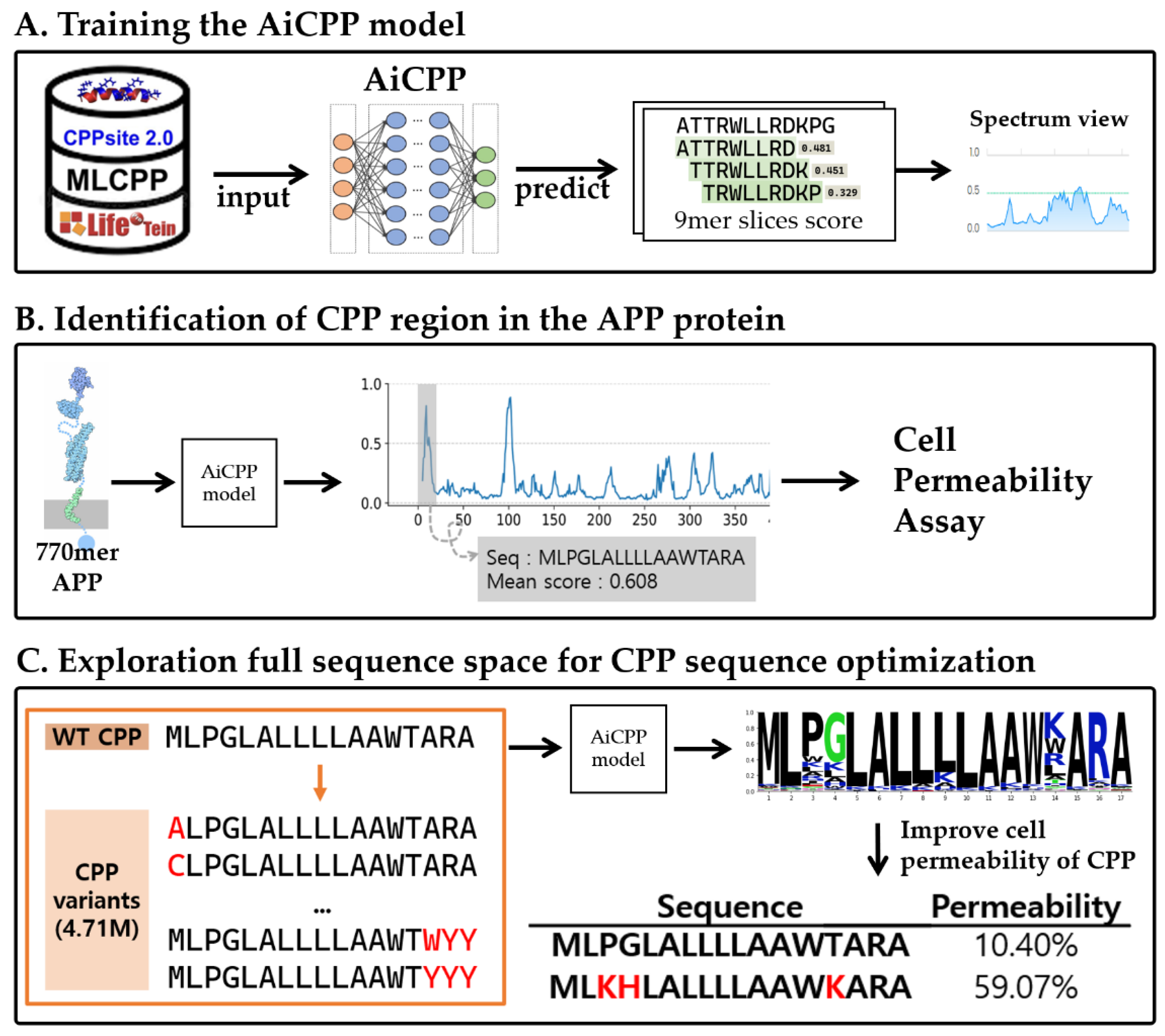
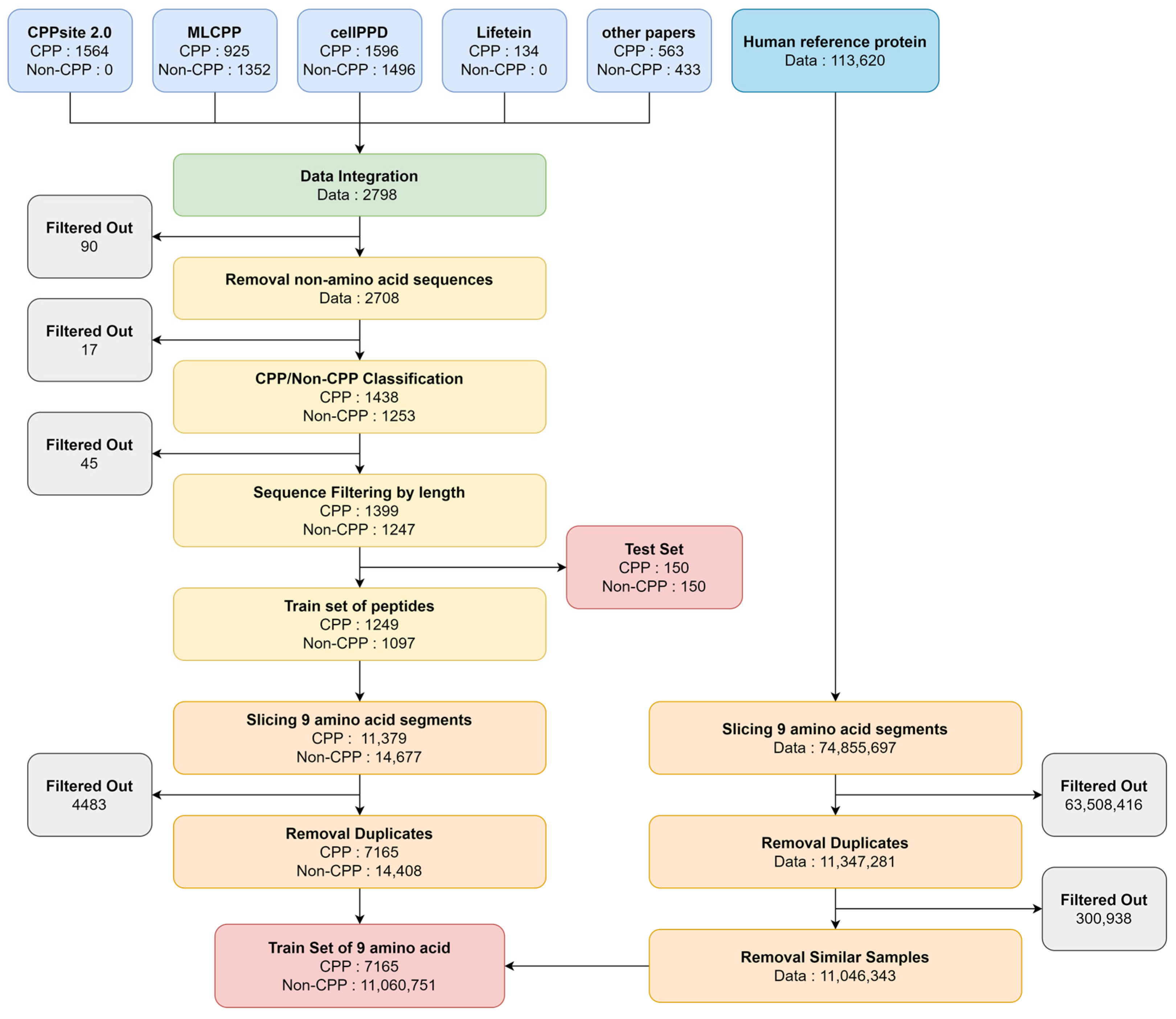
2.1. CPP Dataset Preparation
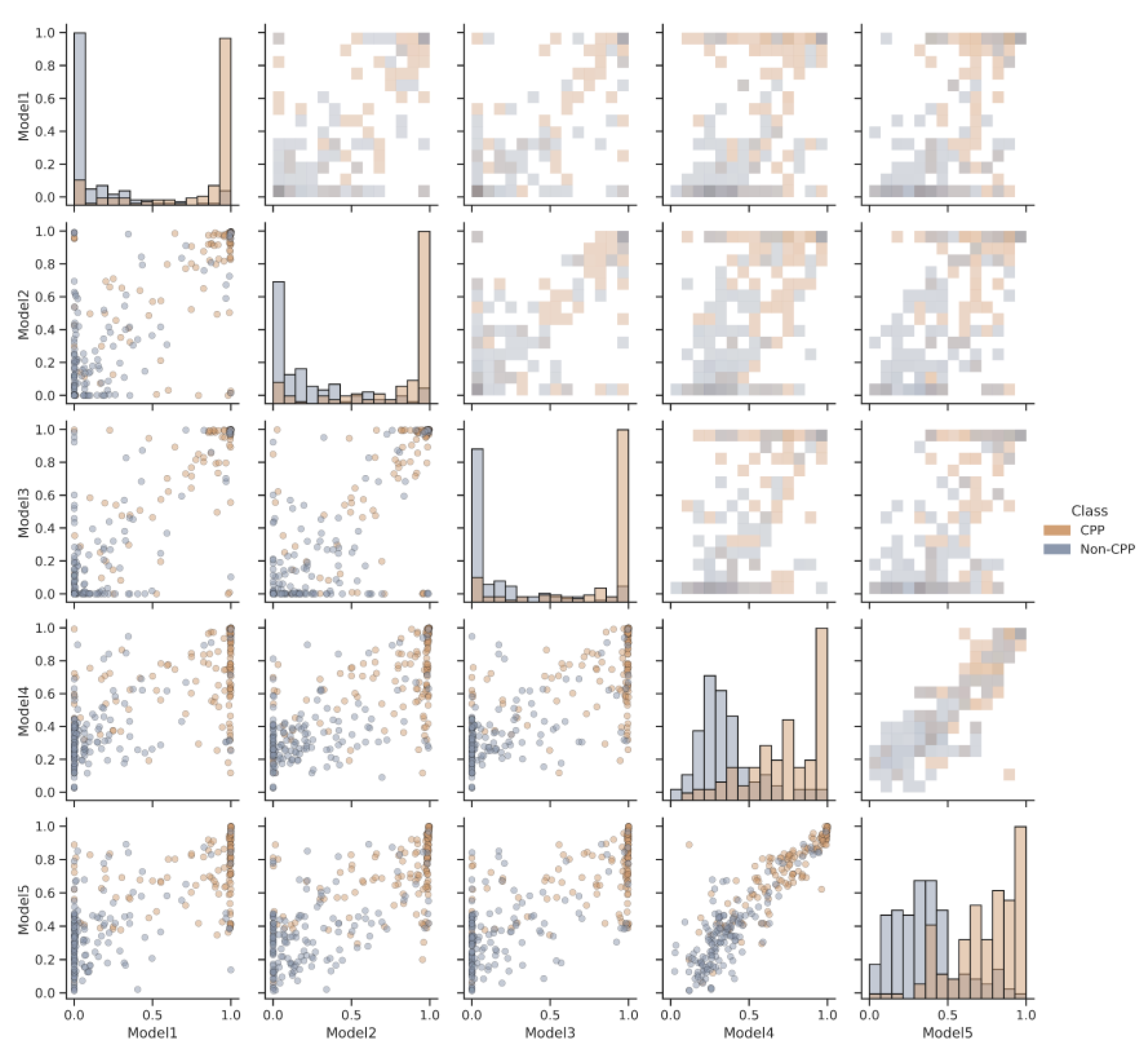
2.2. Model Algorithms and Assessment
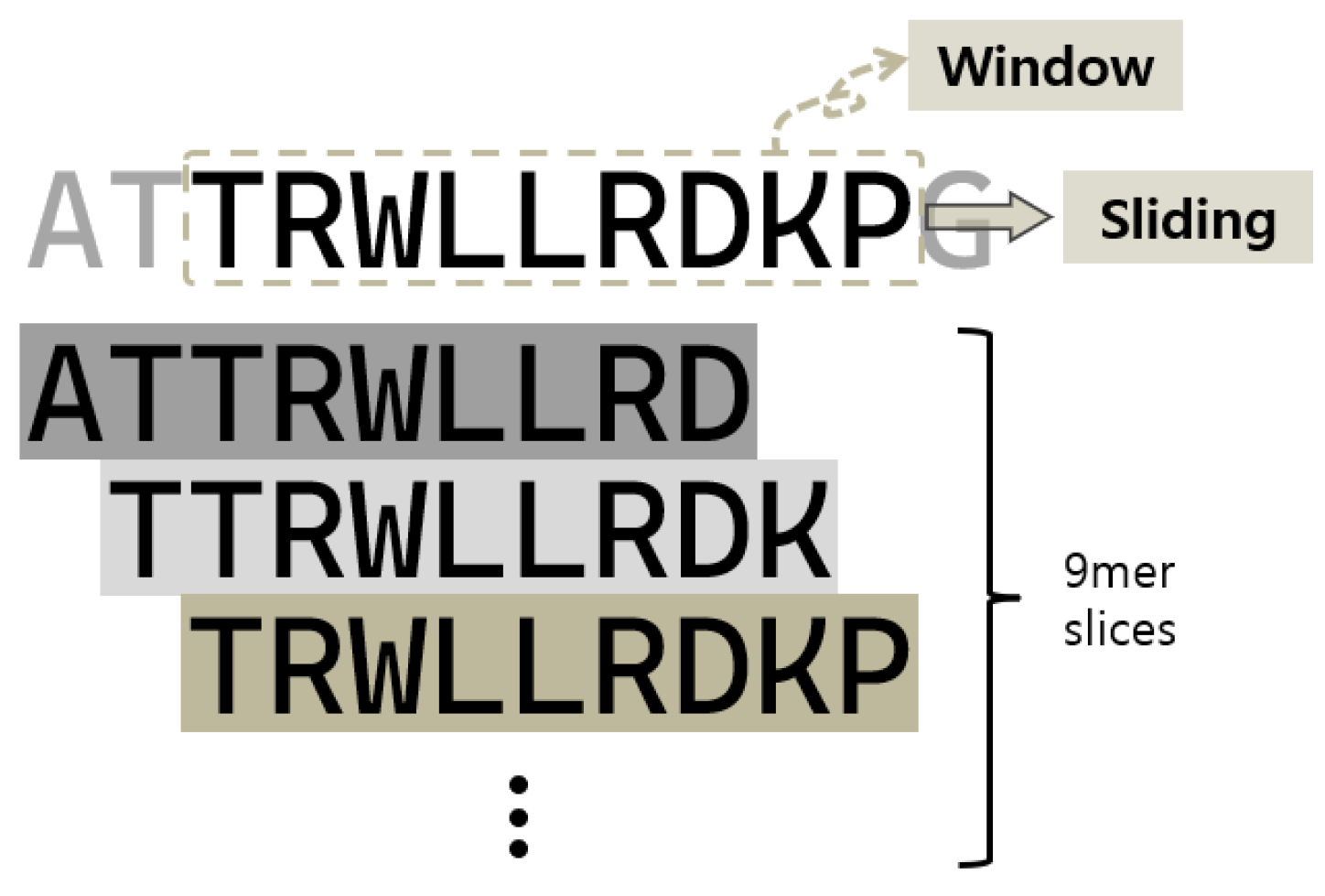
2.3. Calculation of Cell-Penetrating Propensity for Each Residue in the Peptide Sequence
2.4. Discovery and Optimization of CPP
2.5. Peptide Synthesis
2.6. Cell Culture and Peptide Treatment
2.7. Cell Permeability Assay
3. Results
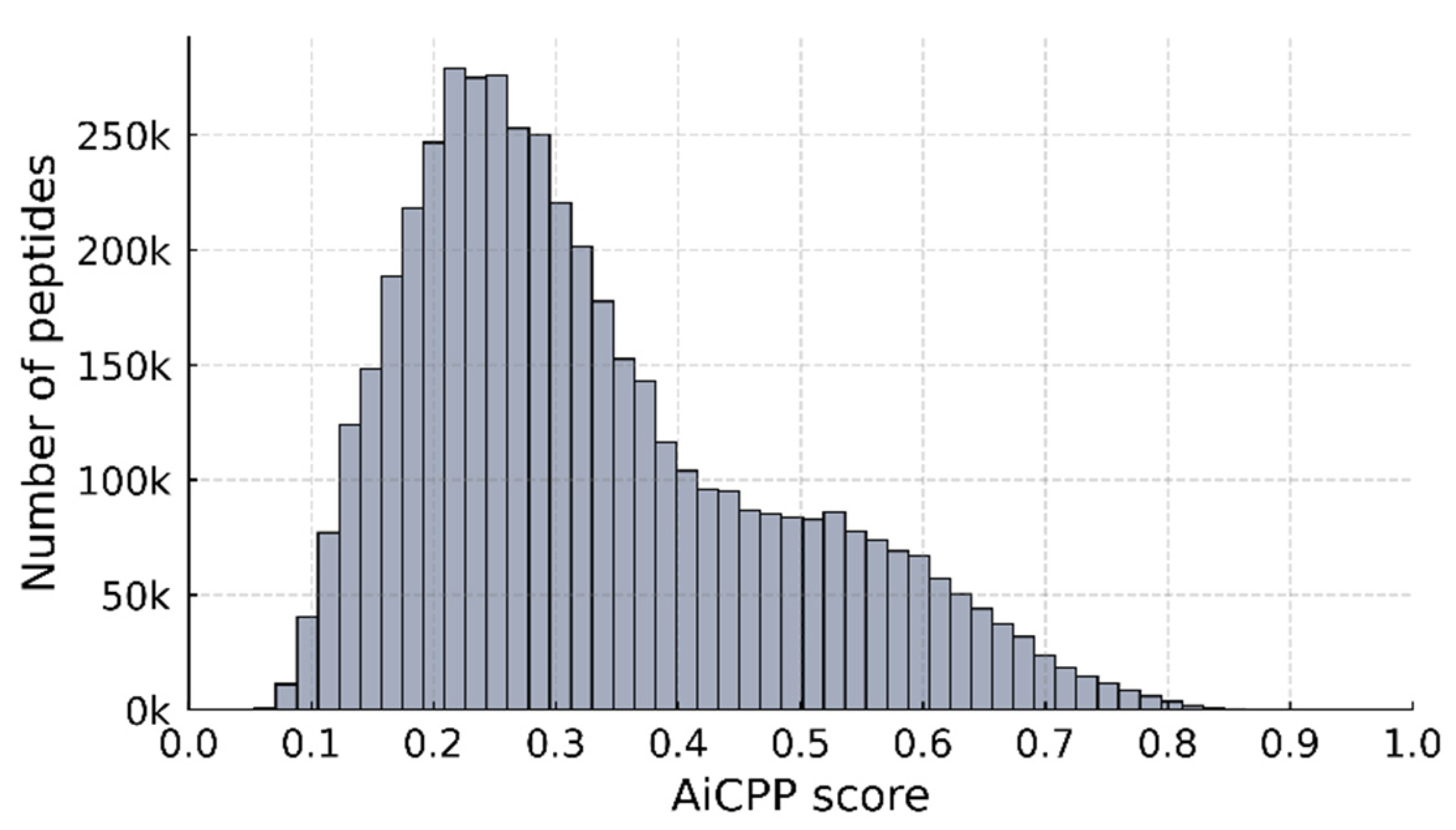
3.1. Performance of AiCPP in CPP Prediction
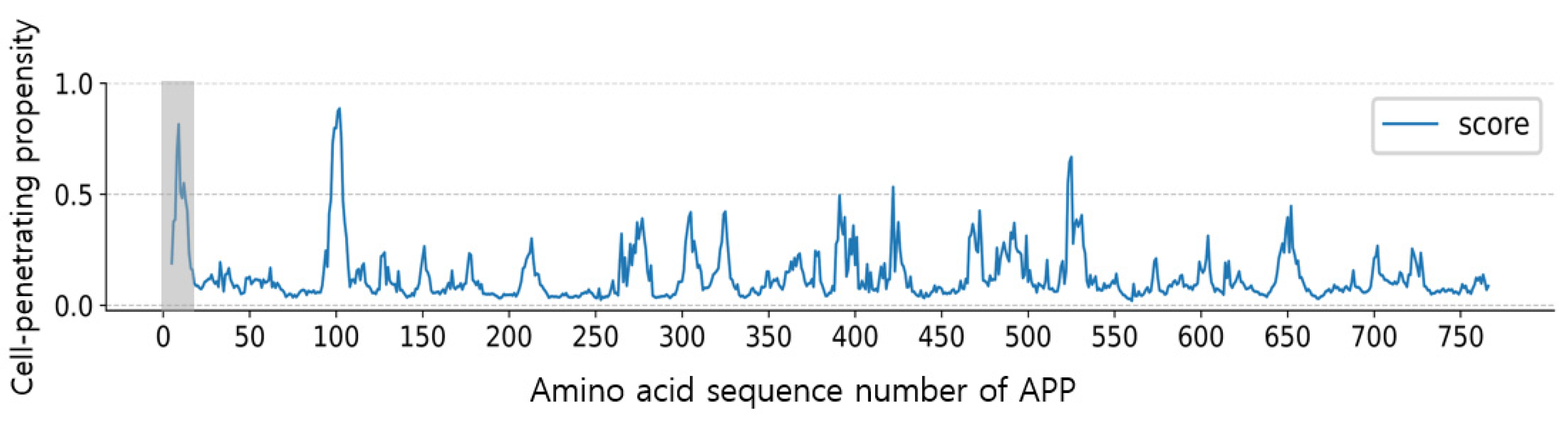
3.2. CPP Screening and Optimization Using AiCPP for Human APP Protein
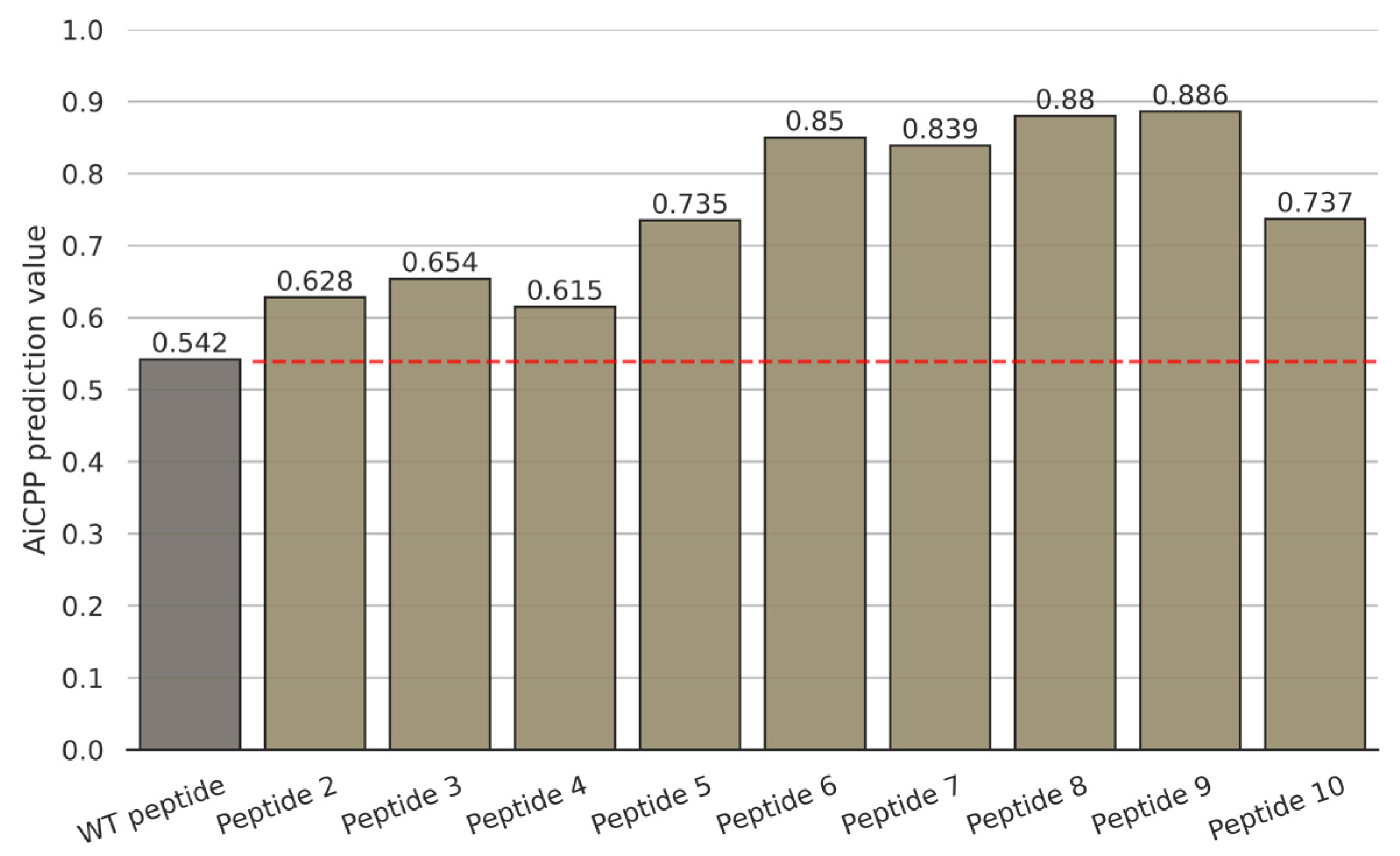
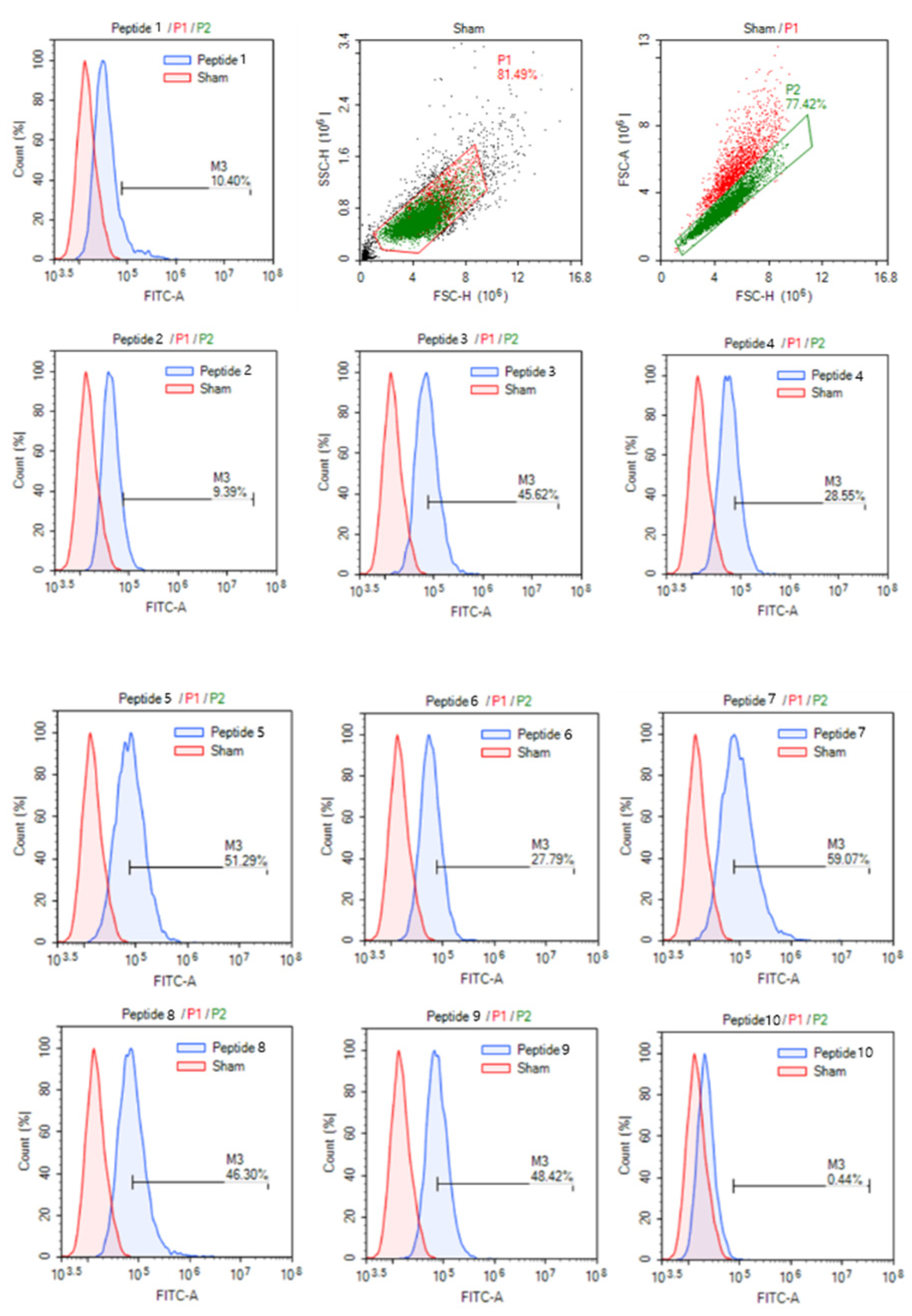
3.3. Enhanced Cell-Permeability of Modifided Peptides in MCF-7 Cells by AiCPP Optimization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nasrollahi, S.A.; Fouladdel, S.; Taghibiglou, C.; Azizi, E.; Farboud, E.S. A peptide carrier for the delivery of elastin into fibroblast cells. J. Dermatol. 2012, 51, 923–929. [Google Scholar] [CrossRef]
- Lehto, T.; Kurrikoff, K.; Langel, Ü. Cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2012, 9, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Padari, K.; Pooga, M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol. Ther. 2012, 20, 525–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Xu, W.; Pan, R.; Chen, P. Design and characterization of a new peptide vector for short interfering RNA delivery. J. Nanobiotechnol. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, N.Q.; Gao, W.; Xiang, B.; Qi, X.R. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int. J. Nanomed. 2012, 7, 1613–1621. [Google Scholar]
- Dekiwadia, C.D.; Lawrie, A.C.; Fecondo, J.V. Peptide-mediated cell-penetration and targeted delivery of gold nanoparticles into lysosomes. J. Pept. Sci. 2012, 18, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Mäe, M.; Andaloussi, S.E.; Lehto, T.; Langel, U. Chemically modified cell-penetrating peptides for the delivery of nucleic acids. Expert Opin. Drug Deliv. 2009, 6, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Raucher, D.; Ryu, J.S. Cell-penetrating peptides: Strategies for anticancer treatment. Trends Mol. Med. 2015, 21, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef]
- Cerrato, C.P.; Lehto, T.; Langel, Ü. Peptide-based vectors: Recent developments. Biomol. Concepts 2014, 5, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Habault, J.; Poyet, J.L. Recent Advances in Cell-penetrating Peptide-Based Anticancer Therapies. Molecules 2019, 24, 927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissmann, S. Cell-penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef] [PubMed]
- Dobchev, D.; Mager, I.; Tulp, I.; Karelson, G.; Tamm, T.; Tamm, K.; Janes, J.; Langel, U.; Karelson, M. Prediction of Cell-Penetrating Peptides Using Artificial Neural Networks. Curr. Comput.-Aided Drug Des. 2010, 6, 79–89. [Google Scholar] [CrossRef]
- Sanders, W.S.; Johnston, C.I.; Bridges, S.M.; Burgess, S.C.; Willeford, K.O. Prediction of cell-penetrating peptides by support vector machines. PLoS Comput. Biol. 2011, 7, e1002101. [Google Scholar] [CrossRef] [Green Version]
- Autam, A.; Chaudhary, K.; Kumar, R.; Sharma, A.; Kapoor, P.; Tyagi, A.; Raghava, G.P. In silico approaches for designing highly effective cell-penetrating peptides. J. Transl. Med. 2013, 11, 74. [Google Scholar]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef]
- Holton, T.A.; Pollastri, G.; Shields, D.C.; Mooney, C. CPPpred: Prediction of cell-penetrating peptides. Bioinformatics 2013, 29, 3094–3096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, M.; Ahmad, S.; Ali, F.; Fang, G.; Li, M.; Yu, D.J. TargetCPP: Accurate prediction of cell-penetrating peptides from optimized multiscale features using the gradient boost decision tree. J. Comput.-Aided Mol. Des. 2020, 34, 841–856. [Google Scholar] [CrossRef] [PubMed]
- Schissel, C.K.; Mohapatra, S.; Wolfe, J.M.; Fadzen, C.M.; Bellovoda, K.; Wu, C.L.; Wood, J.A.; Malmberg, A.B.; Loas, A.; Gómez-Bombarelli, R.; et al. Deep learning to design nuclear-targeting abiotic miniproteins. Nat. Chem. 2021, 13, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Porosk, L.; Gaidutšik, I.; Langel, Ü. Approaches for the discovery of new cell-penetrating peptides. Expert Opin. Drug Discov. 2021, 16, 553–565. [Google Scholar] [CrossRef]
- López-Vidal, E.M.; Schissel, C.K.; Mohapatra, S.; Bellovoda, K.; Wu, C.L.; Wood, J.A.; Malmberg, A.B.; Loas, A.; Gómez-Bombarelli, R.; Pentelute, B.L. Deep Learning Enables Discovery of a Short Nuclear Targeting Peptide for Efficient Delivery of Antisense Oligomers. JACS Au 2021, 1, 2009–2020. [Google Scholar] [CrossRef]
- Pandey, P.; Patel, V.; George, N.V.; Mallajosyula, S.S. KELM-CPPpred: Kernel Extreme Learning Machine Based Prediction Model for Cell-Penetrating Peptides. J. Proteome Res. 2018, 17, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, E.C.L.; Santana, K.; Josino, L.; Lima ELima, A.H.; de Souza de Sales Júnior, C. Predicting cell-penetrating peptides using machine learning algorithms and navigating in their chemical space. Sci. Rep. 2021, 11, 7628. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A. Exploring novel and potent cell-penetrating peptides in the proteome of SARS-COV-2 using bioinformatics approaches. PLoS ONE 2021, 16, e0247396. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s Disease Mutations within the Amyloid Precursor Protein Alter the Aggregation and Conformation of the Amyloid-β Peptide. J. Biol. Chem. 2017, 292, 3172–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Wei, Y.; Zhong, R.; Li, L.; Pang, H.B. Transportan Peptide Stimulates the Nanomaterial Internalization into Mammalian Cells in the Bystander Manner through Macropinocytosis. Pharmaceutics 2021, 13, 552. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Fernández-Carneado, J.; López-Iglesias, C.; Kogan, M.J.; Giralt, E. Mechanistic aspects of CPP-mediated intracellular drug delivery: Relevance of CPP self-assembly. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2006, 1758, 264–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Embedding Dimension | Convolution Layer | LSTM Layer | Attention Layer | Parameters | |
|---|---|---|---|---|---|
| Model 1 | 10 | 15 | 2 | 3 | 11,386 |
| Model 2 | 10 | 15 | 0 | 2 | 5218 |
| Model 3 | 10 | 15 | 3 | 6 | 7405 |
| Model 4 | 3 | 0 | 0 | 1 | 1275 |
| Model 5 | 6 | 0 | 0 | 1 | 1054 |
| Name | Sequence | m/z | Ion | Molecular Formula |
|---|---|---|---|---|
| Peptide 1 | N′-MLPGLALLLLAAWTARA-C′ | 2282.76 | (M + H)+ | C111H164N24O24S2 |
| Peptide 2 | N′-MLPGLALLKLAAWTARA-C′ | 2297.83 | (M + H)+ | C111H165N25O24S2 |
| Peptide 3 | N′-MLPGLALLLLAAWKARA-C′ | 2309.89 | (M + H)+ | C113H169N25O23S2 |
| Peptide 4 | N′-MLPGLALLLLAAWRARA-C′ | 2337.84 | (M + H)+ | C113H169N27O23S2 |
| Peptide 5 | N′-MLKGLALLLLAAWKARA-C′ | 2340.89 | (M + H)+ | C114H174N26O23S2 |
| Peptide 6 | N′-MLPKLALLLLAAWKARA-C′ | 2380.95 | (M + H)+ | C117H178N26O23S2 |
| Peptide 7 | N′-MLKHLALLLLAAWKARA-C′ | 2420.97 | (M + H)+ | C118H178N28O23S2 |
| Peptide 8 | N′-MLAKLALLLLAAWKARA-C′ | 2354.91 | (M + H)+ | C115H176N26O23S2 |
| Peptide 9 | N′-MLKKLALLLLAAWKARA-C′ | 2412.01 | (M + H)+ | C118H183N27O23S2 |
| Peptide 10 | N′-MKPGLALLLLAAKKARA-C′ | 2266.81 | (M + H)+ | C108H172N26O23S2 |
| AUC | MCC | ACC | SEN | SPE | PRE | |
|---|---|---|---|---|---|---|
| Model 1 | 0.893 | 0.711 | 0.853 | 0.800 | 0.907 | 0.896 |
| Model 2 | 0.878 | 0.647 | 0.823 | 0.827 | 0.820 | 0.821 |
| Model 3 | 0.878 | 0.674 | 0.837 | 0.813 | 0.860 | 0.853 |
| Model 4 | 0.879 | 0.660 | 0.830 | 0.827 | 0.833 | 0.832 |
| Model 5 | 0.891 | 0.660 | 0.830 | 0.827 | 0.833 | 0.832 |
| AiCPP (ensemble) | 0.927 | 0.722 | 0.860 | 0.827 | 0.893 | 0.886 |
| MLCPP | 0.882 | 0.633 | 0.810 | 0.914 | 0.705 | 0.758 |
| CellPPD | 0.724 | 0.452 | 0.723 | 0.656 | 0.792 | 0.762 |
| CPPred | 0.845 | 0.564 | 0.780 | 0.722 | 0.839 | 0.820 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Park, J.-H.; Kim, M.S.; Cho, K.; Shin, J.-M. In Silico Screening and Optimization of Cell-Penetrating Peptides Using Deep Learning Methods. Biomolecules 2023, 13, 522. https://doi.org/10.3390/biom13030522
Park H, Park J-H, Kim MS, Cho K, Shin J-M. In Silico Screening and Optimization of Cell-Penetrating Peptides Using Deep Learning Methods. Biomolecules. 2023; 13(3):522. https://doi.org/10.3390/biom13030522
Chicago/Turabian StylePark, Hyejin, Jung-Hyun Park, Min Seok Kim, Kwangmin Cho, and Jae-Min Shin. 2023. "In Silico Screening and Optimization of Cell-Penetrating Peptides Using Deep Learning Methods" Biomolecules 13, no. 3: 522. https://doi.org/10.3390/biom13030522