
G Protein-Dependent Activation of the PKA-Erk1/2 Pathway by the Striatal Dopamine D1/D3 Receptor Heteromer Involves Beta-Arrestin and the Tyrosine Phosphatase Shp-2
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. Cell Cultures, Transfection and Treatments
2.4. Primary Mouse Striatal Neuron Cultures and Treatments
2.5. Beta-Arrestin 1 and Beta-Arrestin 2 Gene Silencing in Primary Striatal Neurons Using RNA Interference
2.6. Protein Preparation, Electrophoresis and Western Blot (WB)
2.7. Co-Immunoprecipitation and WB
2.8. Proximity Ligation Assay (PLA)
2.9. Statistical Analysis
3. Results
3.1. In Hek293 Cells Expressing the D1R/D3R Heteromer, the Simultaneous Stimulation of D1R and D3R Selectively Activates the PKA/Erk1/2 Pathway
3.2. In Hek293 Cells Expressing the D1R/D3R Heteromer, the Individual Stimulation of D1R and D3R Specifically Activates the PKA/Erk1/2 Pathway
3.3. In Hek-D1R/D3R Cells, D3R Interaction with D1R Increases the Ability of DA and D1R Agonists in Activating the ERK1/2 Pathway
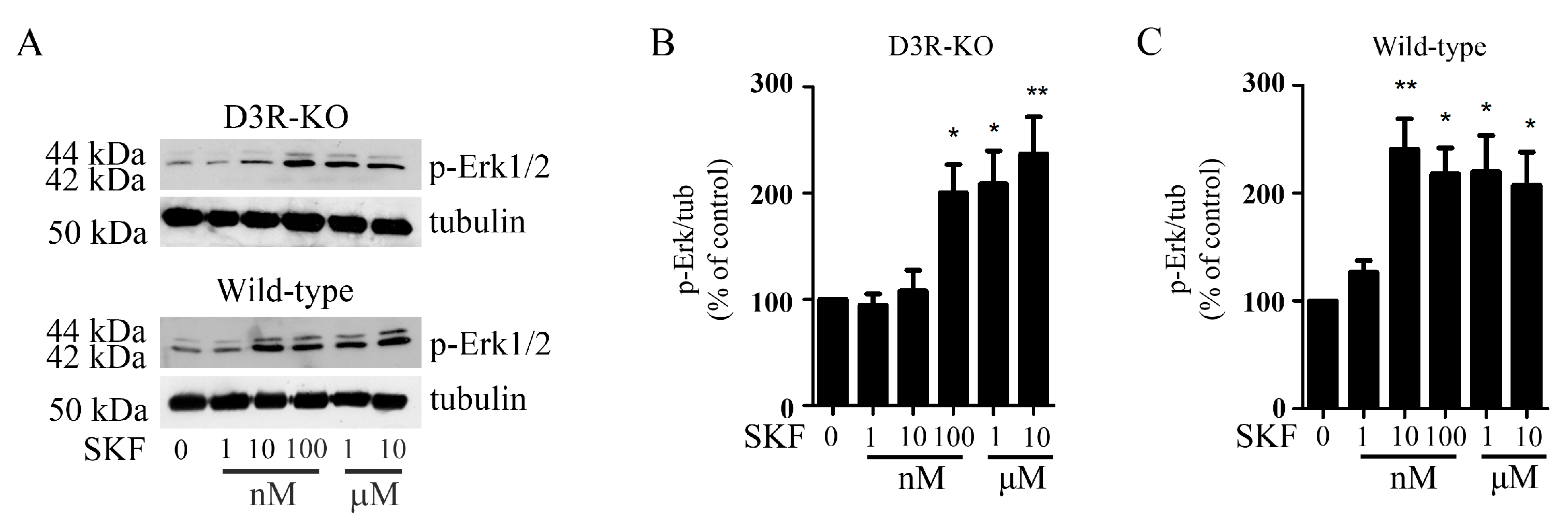
3.4. In Hek-D1R/D3R Cells, Activation of Erk1/2 Induced with D1R and D3R Co-Stimulation Requires the Tyrosine Phosphatase Shp-2
3.5. In Primary Cultures of Striatal Neurons, Activation of the Erk1/2 Pathway with Either D1R or D3R Requires PKA
3.6. In Primary Striatal Neurons, D3R Dimerization with D1R Increases the Ability of D1R Agonists in Activating the ERK1/2 Pathway
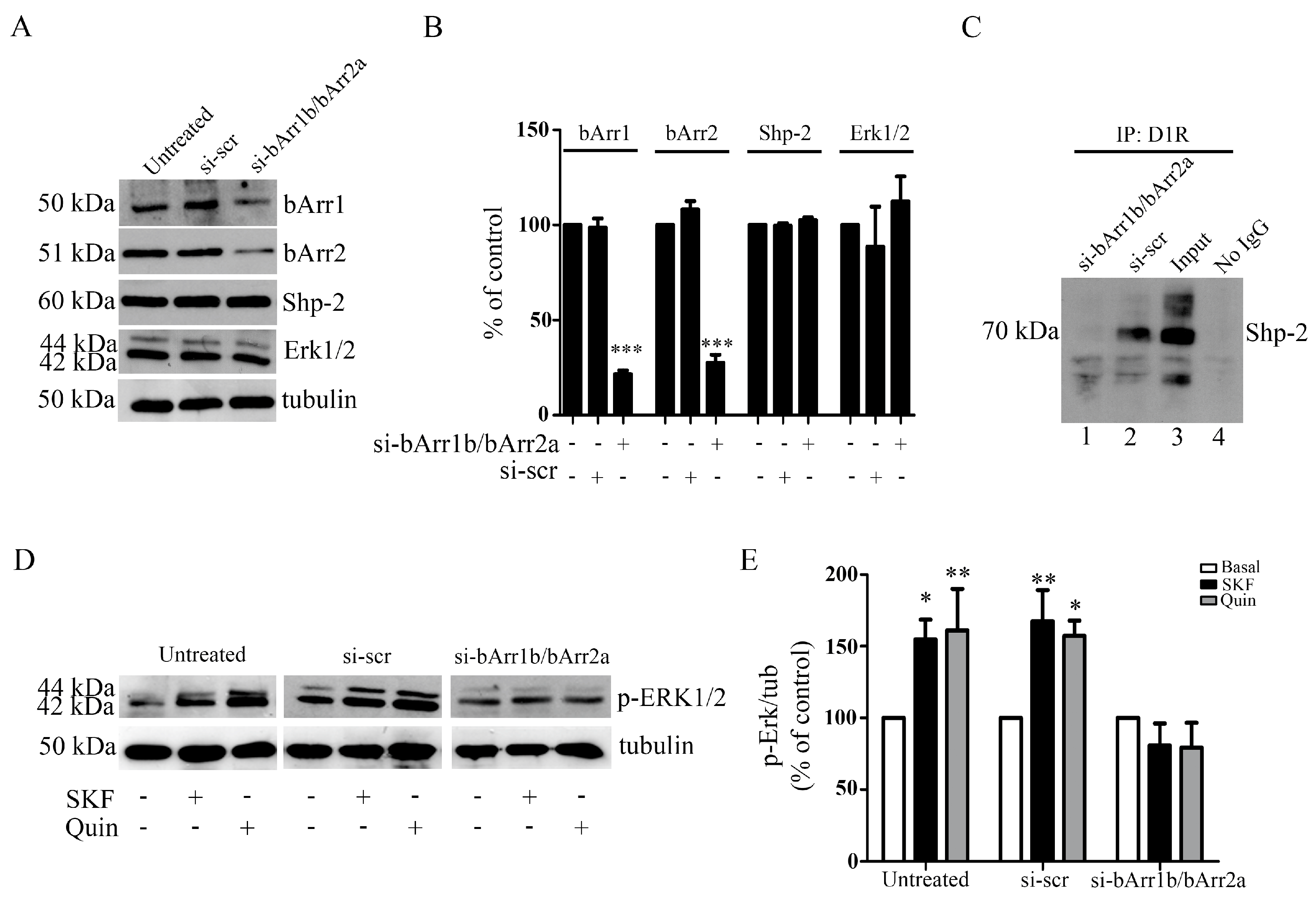
3.7. In Primary Striatal Neurons, Beta-Arrestin1 and Beta-Arrestin2 and Shp2 Are Required for D1R-D3R Heteromer-Induced Activation of PKA-Erk1/2 Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenningsson, P.; Nishi, A.; Fisone, G.; Girault, J.A.; Nairn, A.C.; Greengard, P. DARPP-32: An integrator of neurotransmission. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 269–296. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Mattanza, C.; Collo, G.; Savoia, P.; Spano, P.; Missale, C. The tyrosine phosphatase Shp-2 interacts with the dopamine D1 receptor and triggers D1-mediated Erk signaling in striatal neurons. J. Neurochem. 2011, 117, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Barbon, A.; Missale, C. Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Neurobiol. Dis. 2013, 54, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.M. Drugs for psychosis and mood: Unique actions at D3, D2, and D1 dopamine receptor subtypes. CNS. Spectr. 2017, 22, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Nicola, S.M.; Surmeier, J.; Malenka, R.C. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu. Rev. Neurosci. 2000, 23, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Flores, G.; Barbeau, D.; Quirion, R.; Srivastava, L.K. Decreased binding of dopamine D3 receptors in limbic subregions after neonatal bilateral lesion of rat hippocampus. J. Neurosci. 1996, 16, 2020–2026, PMCID: PMC6578499. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Koeltzow, T.E.; Santiago, G.T.; Moratalla, R.; Cooper, D.C.; Hu, X.T.; White, N.M.; Graybiel, A.M.; White, F.J.; Tonegawa, S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron 1997, 19, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cussac, D.; Newman-Tancredi, A.; Pasteau, V.; Millan, M.J. Human dopamine D3 receptors mediate mitogen-activated protein kinase activation via a phosphatidylinositol 3-kinase and an atypical protein kinase C-dependent mechanism. Mol. Pharmacol. 1999, 56, 1025–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beom, S.; Cheong, D.; Torres, G.; Caron, M.G.; Kim, K.M. Comparative studies of molecular mechanisms of dopamine D2 and D3 receptors for the activation of extracellular signal-regulated kinase. J. Biol. Chem. 2004, 279, 28304–28314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collo, G.; Zanetti, S.; Missale, C.; Spano, P. Dopamine D3 receptor-preferring agonists increase dendrite arborization of mesencephalic dopaminergic neurons via extracellular signal-regulated kinase phosphorylation. Eur. J. Neurosci. 2008, 28, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Collo, G.; Cavalleri, L.; Spano, P. Structural plasticity in mesencephalic dopaminergic neurons produced by drugs of abuse: Critical role of BDNF and dopamine. Front. Pharmacol. 2014, 5, 259, PMCID: PMC4243500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutti, V.; Bono, F.; Tomasoni, Z.; Bontempi, L.; Guglielmi, A.; Bolognin, S.; Schwamborn, J.C.; Missale, C.; Fiorentini, C. Structural Plasticity of Dopaminergic Neurons Requires the Activation of the D3R-nAChR Heteromer and the PI3K-ERK1/2/Akt-Induced Expression of c-Fos and p70S6K Signaling Pathway. Mol. Neurobiol. 2022, 59, 2129–2149, PMCID: PMC9016044. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Busi, C.; Gorruso, E.; Gotti, C.; Spano, P.; Missale, C. Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol. Pharmacol. 2008, 74, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcellino, D.; Ferré, S.; Casadó, V.; Cortés, A.; Le Foll, B.; Mazzola, C.; Drago, F.; Saur, O.; Stark, H.; Soriano, A.; et al. Identification of dopamine D1–D3 receptor heteromers. Indications for a role of synergistic D1–D3 receptor interactions in the striatum. J. Biol. Chem. 2008, 283, 26016–26025, PMCID: PMC2533781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guitart, X.; Navarro, G.; Moreno, E.; Yano, H.; Cai, N.S.; Sánchez-Soto, M.; Kumar-Barodia, S.; Naidu, Y.T.; Mallol, J.; Cortés, A.; et al. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: The dopamine D1–D3 receptor heterotetramer. Mol. Pharmacol. 2014, 86, 417–429, PMCID: PMC4164978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, J.C.; Diaz, J.; Bordet, R.; Griffon, N.; Perachon, S.; Pilon, C.; Ridray, S.; Sokoloff, P. Functional implications of multiple dopamine receptor subtypes: The D1/D3 receptor coexistence. Brain. Res. Brain. Res. Rev. 1998, 26, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Ridray, S.; Schwartz, J.C.; Sokoloff, P. Involvement of the direct striatonigral pathway in levodopa-induced sensitization in 6-hydroxydopamine-lesioned rats. Eur. J. Neurosci. 2000, 12, 2117–2123. [Google Scholar] [CrossRef] [PubMed]
- Heidbreder, C. Selective antagonism at dopamine D3 receptors as a target for drug addiction pharmacotherapy: A review of preclinical evidence. CNS. Neurol. Disord. Drug. Targets. 2008, 7, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci. 2001, 2, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Mutti, V.; Tomasoni, Z.; Sbrini, G.; Missale, C.; Fiorentini, C. Recent Advances in Dopamine D3 Receptor Heterodimers: Focus on Dopamine D3 and D1 Receptor-Receptor Interaction and Striatal Function. Curr. Top. Behav. Neurosci. 2023, 60, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Farré, D.; Muñoz, A.; Moreno, E.; Reyes-Resina, I.; Canet-Pons, J.; Dopeso-Reyes, I.G.; Rico, A.J.; Lluís, C.; Mallol, J.; Navarro, G.; et al. Stronger Dopamine D1 Receptor-Mediated Neurotransmission in Dyskinesia. Mol. Neurobiol. 2015, 52, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Guitart, X.; Moreno, E.; Rea, W.; Sánchez-Soto, M.; Cai, N.S.; Quiroz, C.; Kumar, V.; Bourque, L.; Cortés, A.; Canela, E.I.; et al. Biased G Protein-Independent Signaling of Dopamine D1–D3 Receptor Heteromers in the Nucleus Accumbens. Mol. Neurobiol. 2019, 56, 6756–6769, PMCID: PMC6728209. [Google Scholar] [CrossRef] [PubMed]
- Fanni, S.; Scheggi, S.; Rossi, F.; Tronci, E.; Traccis, F.; Stancampiano, R.; De Montis, M.G.; Devoto, P.; Gambarana, C.; Bortolato, M.; et al. 5alpha-reductase inhibitors dampen l-DOPA-induced dyskinesia via normalization of dopamine D1-receptor signaling pathway and D1–D3 receptor interaction. Neurobiol. Dis. 2019, 121, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Scheggi, S.; Rossi, F.; Corsi, S.; Fanni, S.; Tronci, E.; Ludovica, C.; Vargiu, R.; Gambarana, C.; Muñoz, A.; Stancampiano, R.; et al. BDNF Overexpression Increases Striatal D3 Receptor Level at Striatal Neurons and Exacerbates D1-Receptor Agonist-Induced Dyskinesia. J. Parkinsons. Dis. 2020, 10, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Jones-Tabah, J.; Mohammad, H.; Paulus, E.G.; Clarke, P.B.S.; Hébert, T.E. The Signaling and Pharmacology of the Dopamine D1 Receptor. Front. Cell. Neurosci. 2022, 15, 806618, PMCID: PMC8801442. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Savoia, P.; Savoldi, D.; Bono, F.; Busi, C.; Barbon, A.; Missale, C. Shp-2 knockdown prevents l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov. Disord. 2016, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Bordet, R.; Ridray, S.; Carboni, S.; Diaz, J.; Sokoloff, P.; Schwartz, J.C. Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc. Natl. Acad. Sci. USA 1997, 94, 3363–3367, PMCID: PMC20375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payer, D.E.; Guttman, M.; Kish, S.J.; Tong, J.; Adams, J.R.; Rusjan, P.; Houle, S.; Furukawa, Y.; Wilson, A.A.; Boileau, I. D3 dopamine receptor-preferring [11C]PHNO PET imaging in Parkinson patients with dyskinesia. Neurology 2016, 86, 224–230, PMCID: PMC4733157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Knight, W.C.; Li, H.; Guo, Y.; Perlmutter, J.S.; Benzinger, T.L.S.; Morris, J.C.; Xu, J. Dopamine D1 + D3 receptor density may correlate with Parkinson disease clinical features. Ann. Clin. Transl. Neurol. 2021, 8, 224–237, PMCID: PMC7818081. [Google Scholar] [CrossRef] [PubMed]
- Jeanneteau, F.; Diaz, J.; Sokoloff, P.; Griffon, N. Interactions of GIPC with dopamine D2, D3 but not D4 receptors define a novel mode of regulation of G protein-coupled receptors. Mol. Biol. Cell. 2004, 15, 696–705, PMCID: PMC329290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bontempi, L.; Savoia, P.; Bono, F.; Fiorentini, C.; Missale, C. Dopamine D3 and acetylcholine nicotinic receptor heteromerization in midbrain dopamine neurons: Relevance for neuroplasticity. Eur. Neuropsychopharmacol. 2017, 27, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Accili, D.; Fishburn, C.S.; Drago, J.; Steiner, H.; Lachowicz, J.E.; Park, B.H.; Gauda, E.B.; Lee, E.J.; Cool, M.H.; Sibley, D.R.; et al. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 1945–1949, PMCID: PMC39888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Rusnak, M.; Luedtke, R.R.; Sidhu, A. D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J. Biol. Chem. 2004, 279, 39317–39330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.C.; Lao, C.L.; Chen, J.C. The D3 dopamine receptor inhibits dopamine release in PC-12/hD3 cells by autoreceptor signaling via PP-2B, CK1, and Cdk-5. J. Neurochem. 2009, 110, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Brami-Cherrier, K.; Valjent, E.; Garcia, M.; Pagès, C.; Hipskind, R.A.; Caboche, J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: A new route to cAMP response element-binding protein phosphorylation. J. Neurosci. 2002, 22, 8911–8921, PMCID: PMC6757682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.D.; Sealfon, S.C. Agonist-specific transactivation of phosphoinositide 3-kinase signaling pathway mediated by the dopamine D2 receptor. J. Biol. Chem. 2003, 278, 47053–47061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannoury la Cour, C.; Salles, M.J.; Pasteau, V.; Millan, M.J. Signaling pathways leading to phosphorylation of Akt and GSK-3β by activation of cloned human and rat cerebral D2 and D3 receptors. Mol. Pharmacol. 2011, 79, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Collo, G.; Bono, F.; Cavalleri, L.; Plebani, L.; Merlo Pich, E.; Millan, M.J.; Spano, P.F.; Missale, C. Pre-synaptic dopamine D3 receptor mediates cocaine-induced structural plasticity in mesencephalic dopaminergic neurons via ERK and Akt pathways. J. Neurochem. 2012, 120, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Ayoub, M.A.; Fujita, W.; Jaeger, W.C.; Pfleger, K.D.; Devi, L.A. G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 403–425, PMCID: PMC5147582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, M.L.; Hasbi, A.; O’Dowd, B.F.; George, S.R. Heteromeric dopamine receptor signaling complexes: Emerging neurobiology and disease relevance. Neuropsychopharmacology 2014, 39, 156–168, PMCID: PMC3857642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, T.; Yamada, M.; Ohnishi, H.; Sano, S.; Uetsuki, T.; Hatanaka, H. Shp-2 specifically regulates several tyrosine-phosphorylated proteins in brain-derived neurotrophic factor signaling in cultured cerebral cortical neurons. J. Neurochem. 2000, 74, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hu, D.; Fan, H.; Zhang, Y.; LeSage, G.D.; Caudle, Y.; Stuart, C.; Liu, Z.; Yin, D. β-Arrestin 2 negatively regulates Toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10. J. Biol. Chem. 2014, 289, 23075–23085, PMCID: PMC4132806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhou, R.; Zhang, Y.; Hao, D.; Wang, Y.; Huang, S.; Liu, N.; Xia, C.; Yissachar, N.; Huang, F.; et al. β-arrestin 2 quenches TLR signaling to facilitate the immune evasion of EPEC. Gut Microbes 2020, 11, 1423–1437, PMCID: PMC7524320. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.C.; Su, L.L.; Zou, L.; Liu, Y.; Wu, N.; Kong, L.; Zhuang, Z.H.; Sun, L.; Liu, H.P.; Hu, J.H.; et al. An essential function for beta-arrestin 2 in the inhibitory signaling of natural killer cells. Nat. Immunol. 2008, 9, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. 2019, 10, 53, PMCID: PMC6387912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Parmentier, M.; Springael, J.Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 2007, 282, 30062–30069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barki-Harrington, L.; Luttrell, L.M.; Rockman, H.A. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: A functional role for receptor-receptor interaction in vivo. Circulation 2003, 108, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, B.; Laszlovszky, I.; Krámos, B.; Visegrády, A.; Bobok, A.; Lévay, G.; Lendvai, B.; Román, V. Neuronal Dopamine D3 Receptors: Translational Implications for Preclinical Research and CNS Disorders. Biomolecules 2021, 11, 104, PMCID: PMC7830622. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Cardinale, A.; Picconi, B.; Gardoni, F. From cell lines to pluripotent stem cells for modelling Parkinson’s Disease. J. Neurosci. Methods 2020, 340, 108741. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Gragg, M.; Stoneman, M.R.; Biener, G.; Oliver, J.A.; Miszta, P.; Filipek, S.; Raicu, V.; Park, P.S. Quaternary structures of opsin in live cells revealed by FRET spectrometry. Biochem. J. 2016, 473, 3819–3836, PMCID: PMC5085863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleno, R.; Devost, D.; Pétrin, D.; Zhang, A.; Bourque, K.; Shinjo, Y.; Aoki, J.; Inoue, A.; Hébert, T.E. Conformational biosensors reveal allosteric interactions between heterodimeric AT1 angiotensin and prostaglandin F2α receptors. J. Biol. Chem. 2017, 292, 12139–12152, PMCID: PMC5519365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleno, R.; Hébert, T.E. The Dynamics of GPCR Oligomerization and Their Functional Consequences. Int. Rev. Cell Mol. Biol. 2018, 338, 141–171. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Dam, J.; Ayoub, M.A.; Guillaume, J.L.; Jockers, R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 2006, 7, 1094–1098, PMCID: PMC1679777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandi, C.; Cao, Y.; Martemyanov, K.A. Orphan receptor GPR179 forms macromolecular complexes with components of metabotropic signaling cascade in retina ON-bipolar neurons. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7153–7161, PMCID: PMC3813323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Furukawa, C.; Takano, A.; Ishikawa, N.; Kato, T.; Hayama, S.; Suzuki, C.; Yasui, W.; Inai, K.; Sone, S.; et al. The neuromedin U-growth hormone secretagogue receptor 1b/neurotensin receptor 1 oncogenic signaling pathway as a therapeutic target for lung cancer. Cancer Res. 2006, 66, 9408–9419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueschbell, B.; Manga, P.; Penner, E.; Schiedel, A.C. Evidence for Protein-Protein Interaction between Dopamine Receptors and the G Protein-Coupled Receptor 143. Int. J. Mol. Sci. 2021, 22, 8328, PMCID: PMC8348196. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.; Albarran-Zeckler, R.; Walsh, H.E.; Smith, R.G. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 2012, 73, 317–332, PMCID: PMC3269786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Cordomí, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 2016, 14, 26, PMCID: PMC4822319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24, PMCID: PMC6389107. [Google Scholar] [CrossRef] [PubMed]
- Dunwiddie, T.V.; Masino, S.A. The role and regulation of adenosine in the central nervous system. Annu. Rev. Neurosci. 2001, 24, 31–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297, PMCID: PMC5482185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monville, C.; Torres, E.M.; Dunnett, S.B. Validation of the l-dopa-induced dyskinesia in the 6-OHDA model and evaluation of the effects of selective dopamine receptor agonists and antagonists. Brain. Res. Bull. 2005, 68, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Bono, F.; Mutti, V.; Fiorentini, C.; Missale, C. Dopamine D3 Receptor Heteromerization: Implications for Neuroplasticity and Neuroprotection. Biomolecules 2020, 10, 1016, PMCID: PMC7407647. [Google Scholar] [CrossRef] [PubMed]
- Scarselli, M.; Novi, F.; Schallmach, E.; Lin, R.; Baragli, A.; Colzi, A.; Griffon, N.; Corsini, G.U.; Sokoloff, P.; Levenson, R.; et al. D2/D3 dopamine receptor heterodimers exhibit unique functional properties. J. Biol. Chem. 2001, 276, 30308–30314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine A2A receptor and dopamine D3 receptor interactions: Evidence of functional A2A/D3 heteromeric complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hillefors-Berglund, M.; von Euler, G. Modulation of dopamine D3 receptor binding by N-ethylmaleimide and neurotensin. Brain Res. 1994, 643, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Budzinski, J.; Maschauer, S.; Kobayashi, H.; Couvineau, P.; Vogt, H.; Gmeiner, P.; Roggenhofer, A.; Prante, O.; Bouvier, M.; Weikert, D. Bivalent ligands promote endosomal trafficking of the dopamine D3 receptor-neurotensin receptor 1 heterodimer. Commun. Biol. 2021, 4, 1062, PMCID: PMC8433439. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Resina, I.; Awad Alkozi, H.; Del Ser-Badia, A.; Sánchez-Naves, J.; Lillo, J.; Jiménez, J.; Pintor, J.; Navarro, G.; Franco, R. Expression of Melatonin and Dopamine D3 Receptor Heteromers in Eye Ciliary Body Epithelial Cells and Negative Correlation with Ocular Hypertension. Cells 2020, 9, 152, PMCID: PMC7016594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wong, A.H.; Liu, F. Interactions between NMDA and dopamine receptors: A potential therapeutic target. Brain Res. 2012, 1476, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Hasbi, A.; Fan, T.; Alijaniaram, M.; Nguyen, T.; Perreault, M.L.; O’Dowd, B.F.; George, S.R. Calcium signaling cascade links dopamine D1–D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc. Natl. Acad. Sci. USA 2009, 106, 21377–21382, PMCID: PMC2795506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Pei, L.; Fletcher, P.J.; Kapur, S.; Seeman, P.; Liu, F. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: Increased dopamine D2 receptor dimerization. Mol. Brain 2010, 3, 25, PMCID: PMC2942879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreault, M.L.; Hasbi, A.; Alijaniaram, M.; Fan, T.; Varghese, G.; Fletcher, P.J.; Seeman, P.; O’Dowd, B.F.; George, S.R. The dopamine D1–D2 receptor heteromer localizes in dynorphin/enkephalin neurons: Increased high affinity state following amphetamine and in schizophrenia. J. Biol. Chem. 2010, 285, 36625–36634, PMCID: PMC2978591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés, A.; Casadó-Anguera, V.; Moreno, E.; Casadó, V. The heterotetrameric structure of the adenosine A1-dopamine D1 receptor complex: Pharmacological implication for restless legs syndrome. Adv. Pharmacol. 2019, 84, 37–78. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Hoffmann, H.; Gonzalez-Sepúlveda, M.; Navarro, G.; Casadó, V.; Cortés, A.; Mallol, J.; Vignes, M.; McCormick, P.J.; Canela, E.I.; et al. Dopamine D1-histamine H3 receptor heteromers provide a selective link to MAPK signaling in GABAergic neurons of the direct striatal pathway. J. Biol. Chem. 2011, 286, 5846–5854, PMCID: PMC3037697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Lemus, E.; Arias-Montaño, J.A. Histamine H3 receptor activation inhibits dopamine D1 receptor-induced cAMP accumulation in rat striatal slices. Neurosci. Lett. 2004, 364, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Delgado, D.; Puigdellívol, M.; Moreno, E.; Rodríguez-Ruiz, M.; Botta, J.; Gasperini, P.; Chiarlone, A.; Howell, L.A.; Scarselli, M.; Casadó, V.; et al. Modulation of dopamine D1 receptors via histamine H3 receptors is a novel therapeutic target for Huntington’s disease. eLife 2020, 9, e51093, PMCID: PMC7282811. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, F.; Tomasoni, Z.; Mutti, V.; Sbrini, G.; Kumar, R.; Longhena, F.; Fiorentini, C.; Missale, C. G Protein-Dependent Activation of the PKA-Erk1/2 Pathway by the Striatal Dopamine D1/D3 Receptor Heteromer Involves Beta-Arrestin and the Tyrosine Phosphatase Shp-2. Biomolecules 2023, 13, 473. https://doi.org/10.3390/biom13030473
Bono F, Tomasoni Z, Mutti V, Sbrini G, Kumar R, Longhena F, Fiorentini C, Missale C. G Protein-Dependent Activation of the PKA-Erk1/2 Pathway by the Striatal Dopamine D1/D3 Receptor Heteromer Involves Beta-Arrestin and the Tyrosine Phosphatase Shp-2. Biomolecules. 2023; 13(3):473. https://doi.org/10.3390/biom13030473
Chicago/Turabian StyleBono, Federica, Zaira Tomasoni, Veronica Mutti, Giulia Sbrini, Rajesh Kumar, Francesca Longhena, Chiara Fiorentini, and Cristina Missale. 2023. "G Protein-Dependent Activation of the PKA-Erk1/2 Pathway by the Striatal Dopamine D1/D3 Receptor Heteromer Involves Beta-Arrestin and the Tyrosine Phosphatase Shp-2" Biomolecules 13, no. 3: 473. https://doi.org/10.3390/biom13030473