1. Introduction
Enantiomers are molecules with opposite stereochemistry at every chiral center. They have identical physical properties, but have non-superimposable mirror image structures and rotate polarized light in opposite directions. Enantiomers of hydrophobic ligands have equal solubility in lipids and are equally effective at disrupting lipid bilayers [
1,
2,
3]. Enantioselectivity has thus been used as a criterion for demonstrating that a hydrophobic ligand modulates membrane protein activity by interacting with specific chiral protein binding sites rather than by perturbing membrane properties [
2,
4].
The demonstration that the anesthetic neurosteroid allopregnanolone (3α-hydroxy-5α-pregnan-20-one ALLO) is an enantioselective modulator of GABA
A receptor currents and an enantiospecific anesthetic provided the initial evidence that neurosteroids bind to specific sites on GABA
A receptors to enhance GABA currents and produce anesthesia [
5,
6]. Pregnanolone (3α-hydroxy-5β-pregnan-20-one; PREG), the 5β-epimer of ALLO, is also a positive allosteric modulator of GABA
A currents and an anesthetic, but has minimal enantioselectivity either as a modulator of GABA
A receptors or as an anesthetic [
7].
It has subsequently been shown using site-directed mutagenesis [
8], neurosteroid analogue photoaffinity labeling [
9,
10,
11] and x-ray crystallography [
12,
13,
14] that PAM neurosteroids bind in an intersubunit site between the third transmembrane helix (TM3) of a GABA
A β-subunit and the first transmembrane helix (TM1) of an adjacent α-subunit. Occupancy of this site is the major contributor to the PAM effect of neurosteroids and mutations to the Q242 and W246 residues in this site largely ablate PAM activity [
8,
15,
16,
17]. Photoaffinity labeling studies have shown that PAM neurosteroid binding to intrasubunit sites in the α-subunit (between TM1 and TM4) and the β-subunit (between TM3 and TM4) also contributes to modulation of GABA
A receptors [
9,
15,
16,
18].
To determine the molecular mechanism underlying the differential enantioselective effects of ALLO and PREG, we examined the binding of ALLO, PREG and their enantiomers (ent-ALLO and ent-PREG) to the three neurosteroid binding sites on α1β3 GABAA receptors. Site-specific binding was determined by measuring the ability of the PAM neurosteroids and their enantiomers to prevent neurosteroid analogue photolabeling of peptides in each of the binding sites. We also used KK152, the enantiomer of the ALLO-analogue photolabeling reagent KK123, to confirm enantiomer binding to the intrasubunit sites. The binding results were correlated with electrophysiological studies examining the effects of the PAM neurosteroids and their enantiomers on GABAA currents in wild type receptors and in receptors with an α1(Q242L) mutation in the intersubunit binding site. Collectively, the experimental data demonstrate that the difference in enantioselectivity between ALLO and PREG results from the differential binding of ent-ALLO and ent-PREG in the canonical β3-α1 intersubunit binding site. The structural basis for this difference was explored using rigid body molecular docking.
2. Materials and Methods
2.1. Receptor Expression in Xenopus laevis Oocytes and Electrophysiological Recordings
GABAA receptors were expressed in oocytes from the African clawed frog (X. laevis). The oocytes were purchased as quarter ovaries from Xenopus1 (Dexter, MI, USA). The ovaries were digested in a 2% w/v (mg/mL) solution of collagenase A in ND96 solution (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) containing 100 U/mL penicillin and 100 μg/mL streptomycin for 30 to 40 min at 37 °C.
The cDNAs containing the human α
1 or β
3 subunits were linearized with XbaI (NEB Labs, Ipswich, MA, USA), and the cRNAs were generated using T7 mMessage mMachine (Ambion, Austin, TX, USA). The α
1(Q242L) and α
1(V227W) mutations were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA), and the coding region was fully sequenced prior to use. The functional properties of the α
1(Q242L) and α
1(V227W) mutants have been reported in previous publications [
8,
9,
16,
17]. The oocytes were injected with a total of 12 ng cRNA. The ratio of cRNAs was 5:1 (α
1:β
3) to minimize the expression of β
3 homomeric receptors. Following injection, the oocytes were incubated in ND96 at 16 °C for 2 days prior to conducting electrophysiological recordings.
Electrophysiological recordings were conducted using standard two-electrode voltage clamp. Borosilicate capillary glass tubing (G120F-4, OD = 1.20 mm, ID = 0.69 mm; Warner Instruments, Hamden, CT, USA) was used for voltage and current electrodes. The electrodes were filled with 3 M KCl and had resistances of 0.3–1 MΩ. The oocytes were clamped at −60 mV. The chamber (RC-1Z; Warner Instruments) was perfused with ND96 at 5–8 mL min
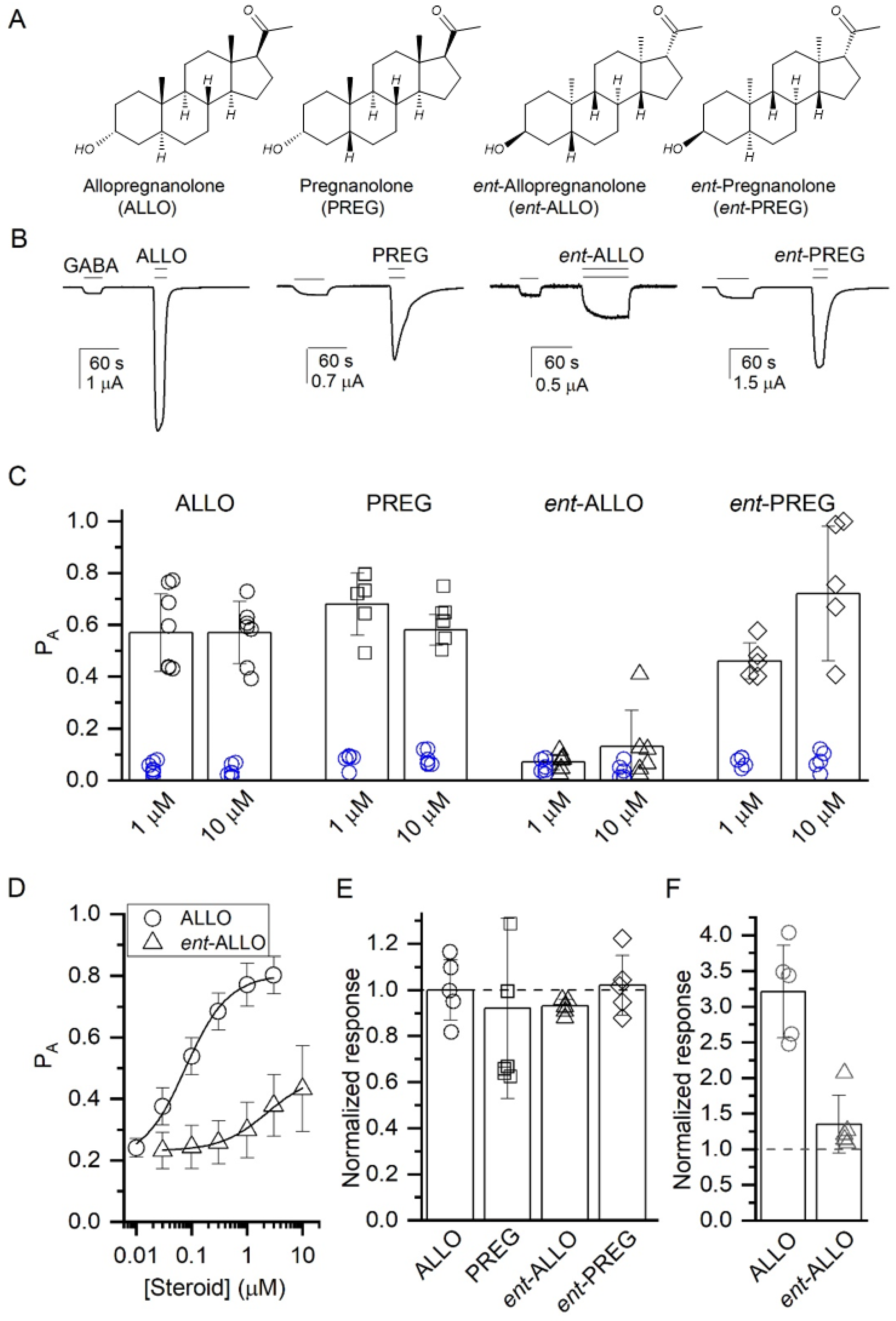
−1. Solutions were gravity-applied from 30-mL glass syringes with glass luer slips via Teflon tubing, to minimize drug absorption. The current responses were amplified with an OC-725C amplifier (Warner Instruments), digitized with a Digidata 1200 series digitizer (Molecular Devices, San Jose, CA, USA) and stored using pClamp (Molecular Devices). The peak amplitude was determined using Clampfit (Molecular Devices). The stock solution of GABA was made in ND96 bath solution at 500 mM, stored in aliquots at −20 °C, and diluted as needed on the day of experiment. Activation by steroids was tested by co-applying a steroid with 0.2–0.5 µM GABA for wild type receptors (
Figure 1B,C) and 0.5–3.0 µM GABA for the α
1(Q242L)β
3 receptors (
Figure 1E) to achieve a target probability of being in the active state (P
A) of 0.05–0.1. For steroid concentration-response curves (
Figure 1D), steroid was co-applied with 0.8–3 µM GABA to achieve a target P
A of ~0.2. The steroids were dissolved in DMSO at 10 mM and stored at room temperature.
The effects of steroids were estimated by calculating the ratio of the peak responses to GABA + steroid and GABA alone. Descriptive analysis of steroid-potentiation was carried out by fitting the steroid concentration-response data to the Hill equation. Mechanistic analysis of steroid-potentiation was conducted in the framework of a cyclic two-state (Resting-Active) concerted transition model [
19,
20]. In brief, the raw current amplitudes were converted to units of P
A by normalizing the responses to GABA or GABA + steroid to the response to 1 mM GABA + 50 µM propofol that was considered to generate a response with peak P
A of ~1 [
21]. The concentration response data were then fitted to the state function:
where L* is the P
A of the response to GABA alone in the same cell and is calculated as (1 − P
A,GABA)/P
A,GABA, [steroid] is the concentration of steroid, K
R,steroid is the equilibrium dissociation constant of the steroid in the resting receptor,
csteroid is the ratio of the equilibrium dissociation constant in the active receptor to K
R,steroid, and N
steroid is the number of steroid binding sites (by convention, constrained to 2). A higher value of
csteroid indicates lower efficacy. Free energy change provided by the steroid can be calculated as ∆G = NRT × ln(
csteroid). The curve-fitting was carried out using Origin 2020 (OriginLab Corp, Northampton, MA, USA). The data are presented as mean ± SD.
2.2. Cell Culture, Protein Expression and Membrane Preparation
A tetracycline-inducible cell line expressing human α
1-8xHis-FLAG and human β
3 GABA
A receptor subunits in HEK-T-Rex™-293 cells was generated and propagated as previously described [
9]. Briefly, stably transfected cells were cultured under the following conditions: cells were maintained in DMEM/F-12 50/50 medium containing 10% fetal bovine serum (tetracycline-free, Takara, Mountain View, CA, USA), penicillin (100 units/mL), streptomycin (100 g/mL), blasticidin (2 mg/mL), hygromycin (50 µg/mL) and zeocin (20 µg/mL) at 37 °C in a humidified atmosphere containing 5% CO
2. Cells were passaged twice each week, maintaining subconfluent cultures. For protein production, cells were plated into dishes. When the cells reached 50% confluence, GABA receptor expression was induced with 1 µg/mL doxycycline with the addition of 5 mM sodium butyrate and the cells were grown to confluence. 48 to 72 h after induction the cells were harvested and washed twice with a buffer containing 10 mM potassium phosphate, 100 mM potassium chloride (pH 7.5) plus protease inhibitors (Sigma-Aldrich, St. Louis, MO, USA). Cells were collected by centrifugation at 1000×
g at 4 °C for 5 min and homogenized with a glass mortar and a Teflon pestle for ten strokes on ice. Membranes were collected by centrifugation at 40,000×
g at 4 °C for 30 min and resuspended in a buffer containing 10 mM potassium phosphate, 100 mM potassium chloride (pH 7.5). Protein concentration was determined with micro-BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA). GABA
A receptor content of the membranes was determined by measuring the B
max of [
3H]muscimol binding as previously described [
9] and assuming that each mole of receptor contains two muscimol binding sites. Membranes were stored at −80 °C.
2.3. Photolabeling and Purification of α1β3 GABAAR
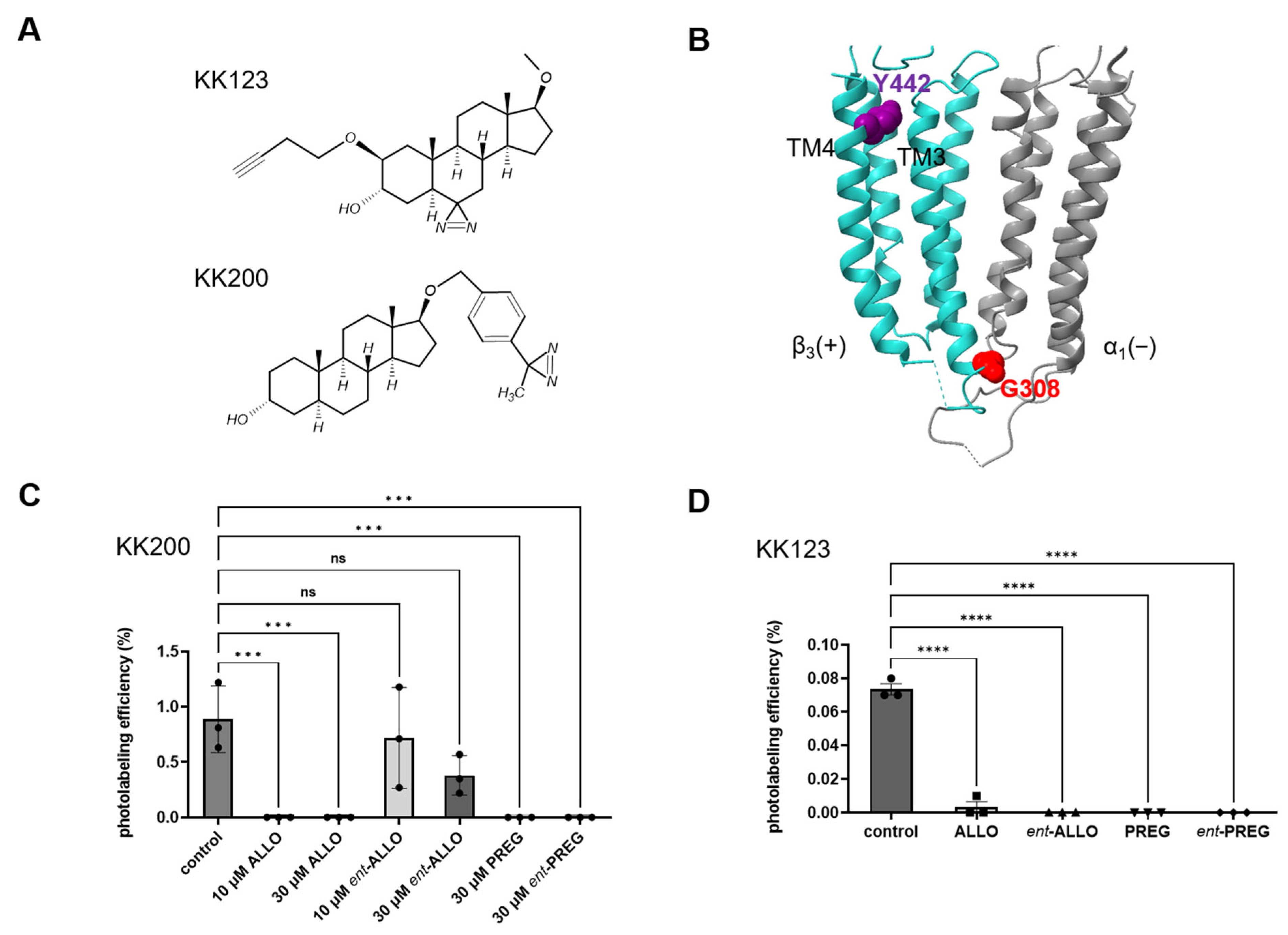
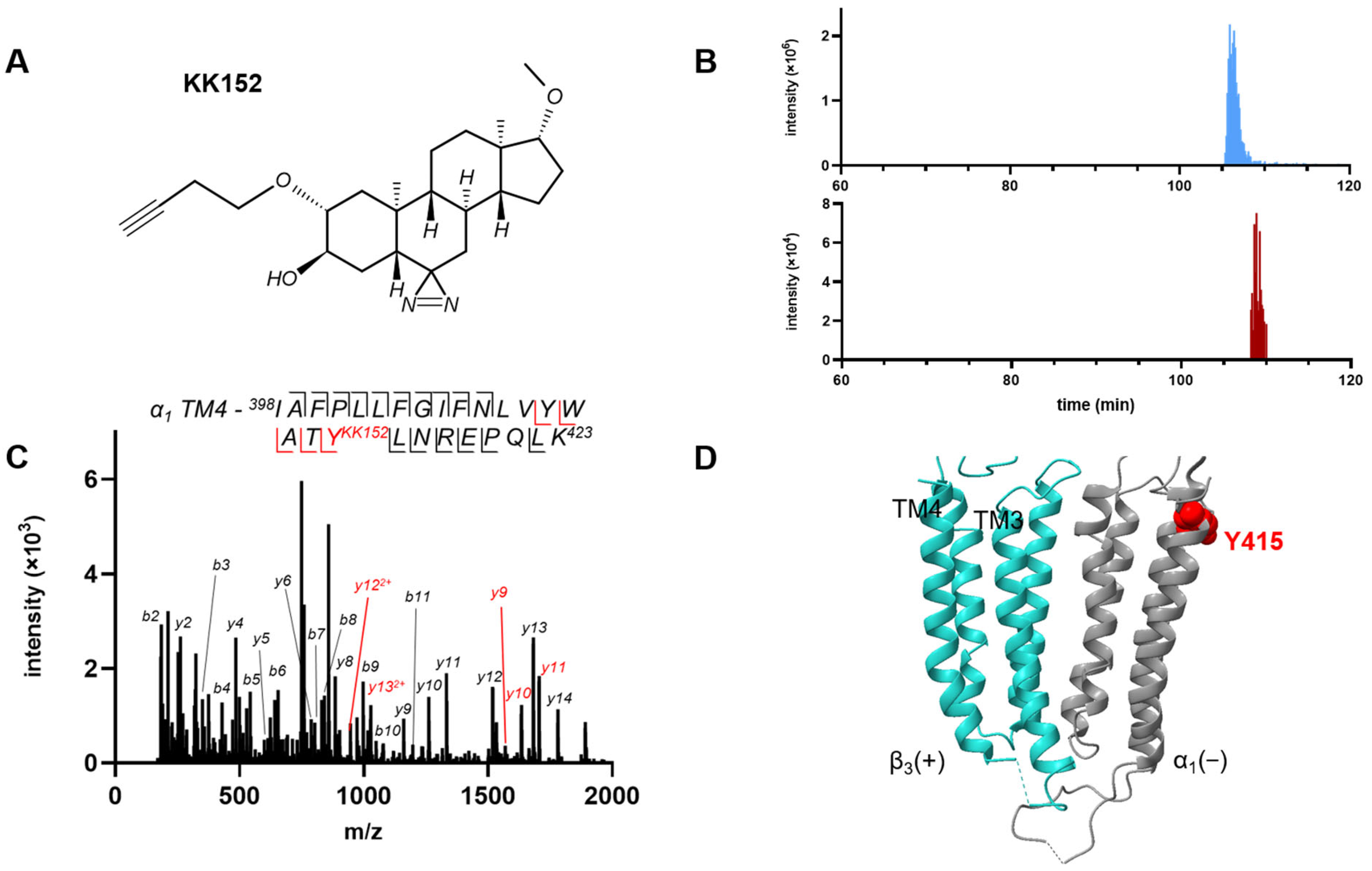
The syntheses of the neurosteroid photolabeling reagents KK123, KK152 and KK200 are detailed in previous reports [
22,
23]. For each photolabeling experiment, 10–20 mg of HEK cell membrane protein containing 100–150 pmoles of α
1β
3 GABA
A receptor was used. Frozen membranes were thawed and resuspended at a final concentration of 1.25 mg ml
−1 in a buffer containing 10 mM potassium phosphate, 100 mM potassium chloride (pH 7.5), and 1 mM GABA. For the photolabeling competition experiments, 3 μM KK123 or KK200 in the presence of 30 μM competitor (ALLO,
ent-ALLO, PREG, and
ent-PREG), or the same volume of ethanol for control group, was added to the membrane suspension and incubated on ice for 1 h. The samples were then irradiated in a quartz cuvette for 5 min, using a photoreactor emitting light with wavelengths >320 nm [
24]. Membranes were then collected by centrifugation at 20,000×
g at 4 °C for 30 min. The photolabeled membrane proteins were resuspended in lysis buffer containing 1% n-dodecyl-β-D-maltoside (DDM) (Anatrace, Maumee, OH, USA), 0.25% cholesteryl hemisuccinate (CHS) (Anatrace, Maumee, OH, USA), 50 mM Tris (pH 7.5), 150 mM NaCl, 2 mM CaCl
2, 5 mM KCl, 5 mM MgCl
2, 1 mM EDTA, 10% glycerol at a final concentration of 1 mg ml
−1. The membrane protein suspension was homogenized using a glass mortar and a Teflon pestle and incubated at 4 °C overnight. The protein lysate was centrifuged at 15,000×
g at 4 °C for 30 min and the supernatant was incubated with 0.5 mL anti-FLAG agarose (Sigma-Aldrich, St. Louis, MO, USA) at 4 °C for 2 h. The anti-FLAG agarose was then transferred to an empty column, followed by washing with 20 mL of washing buffer (50 mM triethylammonium bicarbonate and 0.02% DDM). GABA
A receptors were eluted with aliquots of 200 μg mL
−1 FLAG tag peptide and 100 μg ml
−1 3X FLAG (ApexBio) in the washing buffer. Pooled eluates (4 mL) containing GABA
A receptors were concentrated to 100 μL using 100 kDa cut-off centrifugal filters.
2.4. Middle-Down MS Analysis
The purified α1β3 GABAA receptors (100 μL) were reduced with 5 mM tris(2-carboxyethyl)phosphine for 30 min, followed by alkylation with 5 mM N-ethylmaleimide (NEM) for 45 min in the dark. The NEM was quenched with 5 mM dithiothreitol (DTT) for 15 min. These three steps were carried out at room temperature. Samples were then digested with 8 μg of trypsin for 7 days at 4 °C to obtain maximal recovery of TMD peptides. The digestions were terminated by adding formic acid to a final concentration of 1%, followed directly by LC-MS analysis on an Orbitrap Elite mass spectrometer. 20 μL samples were injected into a home-packed PLRP-S (Agilent, Santa Clara, CA, USA) column (10 cm × 75 μm, 300 Å), separated with a 135 min gradient from 10% to 90% acetonitrile, and introduced to the mass spectrometer at 800 nL min−1 with a nanospray source. MS acquisition was set as a MS1 Orbitrap scan (resolution of 60,000) followed by top 20 MS2 Orbitrap scans (resolution of 15,000) using data-dependent acquisition, and exclusion of singly charged precursors. Fragmentation was performed using high-energy dissociation with normalized energy of 35%. Analysis of datasets was performed using Xcalibur (Thermo Fisher Scientific) to manually search for TM1, TM2, TM3 or TM4 tryptic peptides with or without neurosteroid photolabeling modifications. Photolabeling efficiency was estimated by generating extracted chromatograms of unlabeled and labeled peptides, determining the area under the curve, and calculating the abundance of labeled peptide/(unlabeled + labeled peptide). Analysis of statistical significance comparing the photolabeling efficiency of KK123 and KK200 for α1β3 GABAAR was determined using one-way ANOVA with Bonferroni’s multiple comparisons test (GraphPad Prism version 9.4.0 for Windows, GraphPad Software, San Diego, CA, USA). MS2 spectra of photolabeled TMD peptides were analyzed by manual assignment of fragment ions with and without photolabeling modification. Fragment ions were accepted based on the presence of a monoisotopic mass within 20 ppm mass accuracy. In addition to manual analysis, PEAKS (Bioinformatics Solutions Inc., Waterloo, ON, Canada) database searches were performed for datasets of photolabeled α1β3 GABAAR. Search parameters were set for a precursor mass accuracy of 20 ppm, fragment ion accuracy of 0.1 Da, up to three missed cleavages on either end of the peptide, false discovery rate of 0.1%, and variable modifications of methionine oxidation, cysteine alkylation with NEM and DTT, and neurosteroid analogue photolabeling reagents on any amino acid.
2.5. Molecular Docking and Binding Energy Calculations
The molecular coordinates of ALLO (PubChem CID: 92786) and PREG (PubChem CID: 31402) were obtained from PubChem. The structures of
ent-ALLO and
ent-PREG were generated by inverting the chiral configurations of all the chiral centers in the structures of ALLO and PREG. These structures were then energy minimized using UFF force field and Steepest Descent algorithm in the Avogadro software [
25] to obtain the coordinates of
ent-ALLO and
ent-PREG. The docking template of α
1β
3γ
2 was generated using the CryoEM structure (PDB: 6HUO) [
26]. The ligands bound to the protein were deleted and the structure was energy minimized in Chimera ver. 1.16 [
27]. DockPrep was used to add hydrogens and charges to the protein structure. The interface between β
3-TM3 and α
1-TM1 was used for grid generation of size 20 × 18 × 29 Å encompassing the neurosteroid binding site. Docking was performed using AutoDock Vina [
28] in the Chimera software to obtain the binding energies and binding poses of ALLO, PREG,
ent-ALLO and
ent-PREG.
4. Discussion
In this study we determined the molecular basis for the enantioselective action of ALLO and the relative lack of enantioselectivity of PREG as GABA
A-PAMs. The data show that the enantioselectivity of ALLO is based on differential binding affinity and efficacy of the ALLO enantiomers in the β
3/α
1 intersubunit neurosteroid binding site on GABA
A receptors. The evidence in support of this conclusion includes: (1) Electrophysiological concentration-response data showing that
ent-ALLO is at least 20-fold less potent and less efficacious than ALLO as a GABA
A-PAM; (2) Mutagenesis data showing that the modest PAM effects of
ent-ALLO are prevented by a mutation (α
1(Q242L)) known to prevent neurosteroid action in the β
3/α
1 intersubunit site [
8]; and (3) Photolabeling/mass spectrometry results showing that both
ent-ALLO and ALLO bind to the α
1 and β
3 intrasubunit neurosteroid binding sites, but that
ent-ALLO binds with very low affinity to the β
3/α
1 intersubunit site.
These data also explain the lack of diasteroselectivity between ALLO and its 5β-epimer PREG and the minimal enantioselectivity of PREG. No significant differences in binding to the intrasubunit or intersubunit neurosteroid binding sites were observed between ALLO, PREG and ent-PREG. All three compounds had equal efficacy as GABA-PAMs and their PAM effects were prevented by the α1(Q242L) mutation, indicating that their PAM actions are largely mediated by binding to the intersubunit site.
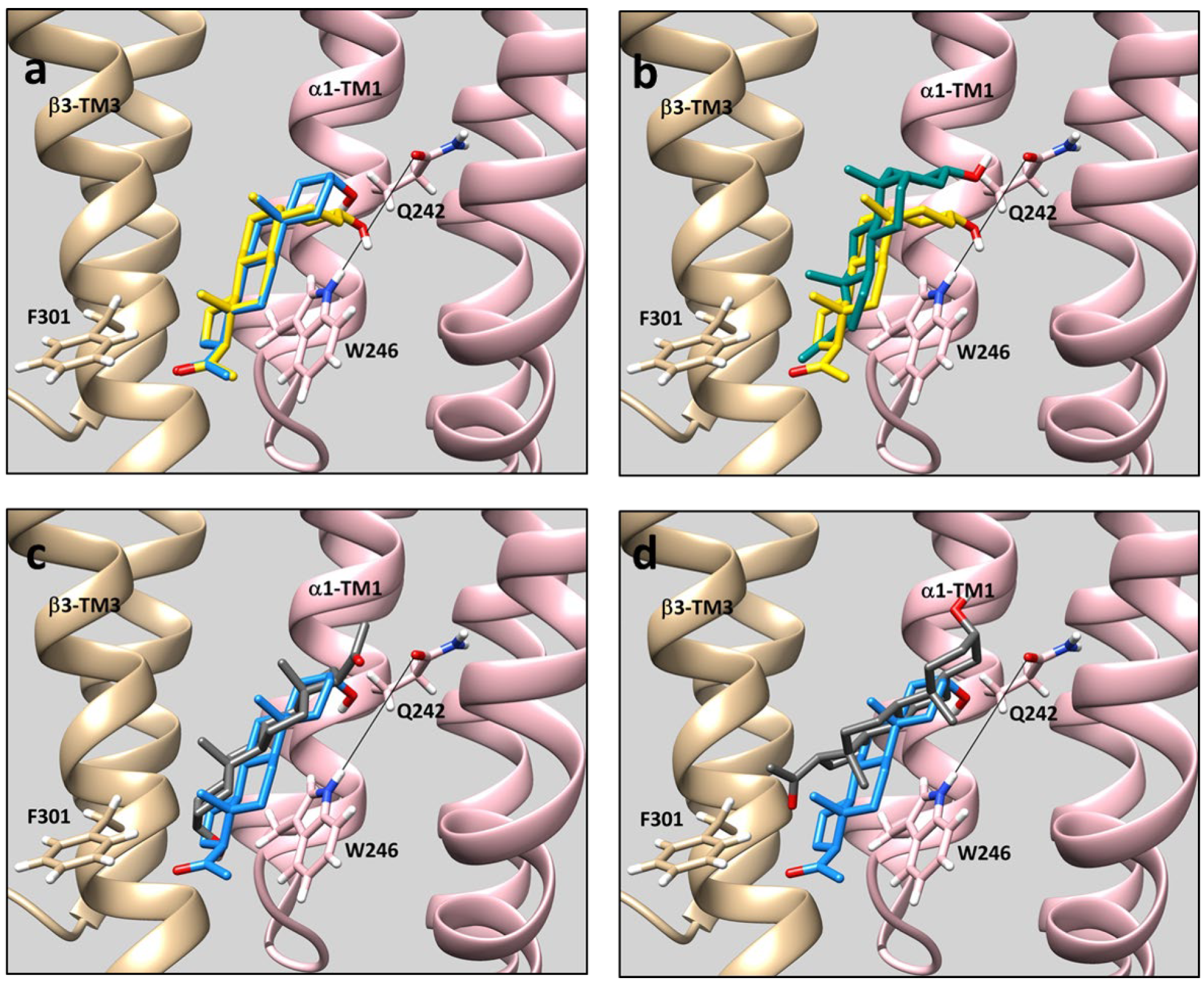
Rigid body docking provides some insight into the complex pattern of diastereoselective and enantioselective binding and GABA
A-PAM activity of the ALLO and PREG enantiomeric pairs. Despite the marked difference in configuration between ALLO and PREG (
cis vs.
trans A,B-ring fusion), their energetically preferred poses in the intersubunit site are closely aligned, with their 18 and 19 methyl (“rough surface”) and 17-methyl ketone groups superimposed and their 3-hydroxy groups positioned to form coordinated hydrogen bonds with α
1(Q242) and α
1(W246) (
Figure 3a) [
12]. This explains their lack of diasteroselective GABA
A-PAM activity.
ent-PREG, in its preferred pose, is rotated 180° on its short axis in comparison to PREG. In this pose the methyl groups and methyl ketone groups of
ent-PREG and PREG are aligned and the 3-hydroxy group of
ent-PREG is positioned to form a hydrogen bond with α
1(Q242) (
Figure 3b). These poses explain the modest degree of enantioselectivity of PREG as a GABA
A-PAM. In contrast,
ent-ALLO in its lowest energy pose, is rotated 180° on its long axis in comparison to ALLO, PREG and
ent-PREG. While the methyl groups of ALLO and
ent-ALLO align in this pose, the
ent-ALLO 3-hydroxy group is proximal to β
3(F301), and is not positioned to form a hydrogen bond with either α
1(Q242) or α
1(W246) (
Figure 3c). The lowest energy pose in which the 3-hydroxyl group of
ent-ALLO points toward α
1(Q242) (
Figure 3d) is energetically unfavorable both because the 3-hydroxy group is not positioned to form a hydrogen bond with Q242 and/or W246 and because the 18 and 19 methyl groups point toward W246, potentially interfering with steroid ring-tryptophan stacking interactions.
While docking studies provide inference about binding, the β
3-α
1 intersubunit site is on the protein surface and bound neurosteroids interact with both protein and surrounding membrane lipid. The contribution of hydrogen binding to total binding energy is likely to be greater in the nonpolar environment of the lipid membrane than in the
in vacuo conditions modeled in docking algorithms [
29]. As such, the absence of
ent-ALLO hydrogen bonding to Q242 or W246 may reduce binding energy more than is predicted by docking studies. It is also not clear if
ent-ALLO dominantly binds in orientation #1 (
Figure 3d), orientation #2 (
Figure 3c) or both. However, the absence of hydrogen bonding in either orientation is likely to contribute to low affinity and low efficacy.
The finding that
ent-ALLO binds in the opposite orientation from ALLO in the β
3-α
1 intersubunit site is consistent with our previous observation that mutations in the intersubunit binding site that interfere with neurosteroid action change steroid orientation in the binding site. In a wild-type ELIC-α
1 GABA
A chimeric receptor, KK200 (photolabeling moiety on the D-ring) labeled residue Y309 at the bottom of TM3, whereas in receptors with the Q242L or W246L mutations, it labeled residue F298 in the middle of the TM3 helix [
30]. These data indicate that the 3-hydroxy group on the steroid A-ring is oriented to the center of the transmembrane domain in wild-type receptors, but to the cytoplasmic end of the transmembrane helices in the mutant receptors. Elimination of a hydrogen bonding interaction between the 3-hydroxy group of a neurosteroid and the Q242 and/or W246 residues, either because of mutation or steroid structure, may favor a steroid orientation in which the 3-hydroxyl group points to the cytoplasmic interface with the membrane, similar to its preferred orientation in a lipid bilayer [
31].
The results of this study elucidate the molecular interactions underlying ALLO enantioselectivity, but may also have some practical pharmacological implications. ALLO interacts with three binding sites on α
1β
3 GABA
A receptors, all of which mediate allosteric effects on channel function [
9,
15,
18]. The preferential interaction of
ent-ALLO with the intrasubunit sites suggests its potential utility as a site-selective ligand or as a scaffold for a site-selective neurosteroid ligand.
The enantiomers of ALLO were originally used to demonstrate that PAM neurosteroids act by binding to specific sites on the GABA
A receptor, rather than by perturbing the lipid membrane milieu in which the receptor resides [
5]. Consistent with this idea, our data demonstrate differential binding of ALLO and
ent-ALLO to the same site on a GABA
A receptor. However, our data for PREG and
ent-PREG show that an enantiomeric pair of ligands can also bind to the same site with near identical affinity and efficacy. Thus, the presence of enantioselectivity supports a direct protein binding interaction, but its absence does not refute it. There is an old pharmacologic “rule” (Pfeiffer’s Rule) that states that the ratio (eudismic ratio) of the potency of a ligand (eutomer) to its less active enantiomer (distomer) is proportional to the potency of the eutomer [
32]. ALLO and PREG bind with similar affinity to the β
3/α
1 intersubunit site on GABA
A receptors, producing a PAM effect of similar magnitude. However,
ent-ALLO binds to the intersubunit site with much lower affinity than
ent-PREG, producing a markedly smaller PAM effect. The difference in eudismic ratio between the ALLO and PREG enantiomeric pairs clearly contradicts Pfeiffer’s rule [
33].


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}