
miRNA-Induced Downregulation of IPMK in Macrophages Mediates Lipopolysaccharide-Triggered TLR4 Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Line Generation
2.2. Immunoblotting and Immunoprecipitation
2.3. RNA Isolation and RT-qPCR
2.4. Multiplex Immunoassay
2.5. UTR Reporter Assays
2.6. Statistical Analysis
3. Results
3.1. Expression of Macrophage IPMK Is Dynamically Regulated by LPS
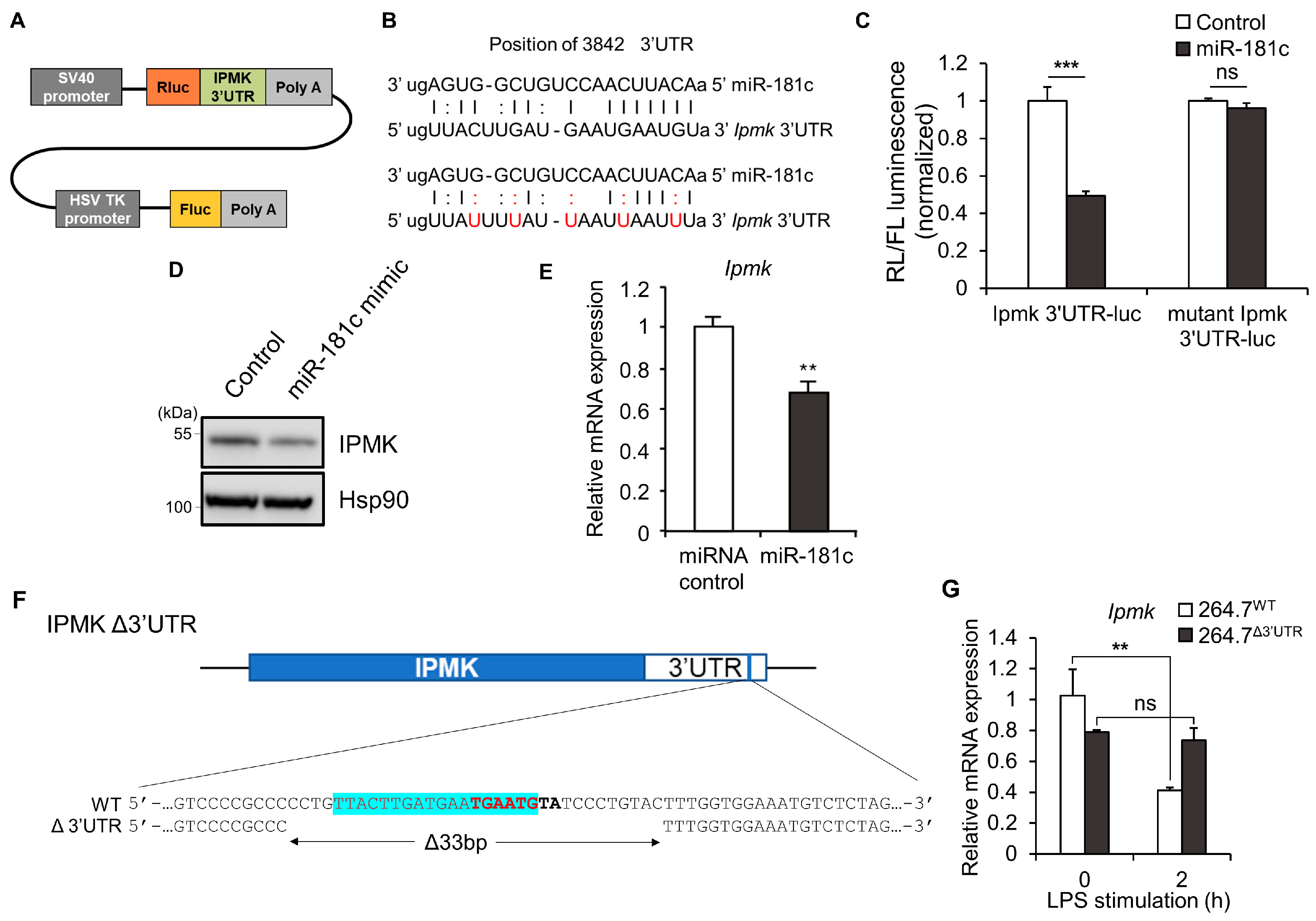
3.2. Generation of Mutant Macrophages in Which IPMK Expression Is Not Sensitive to LPS
3.3. TLR4 Signaling and Cytokine Expression Are Reduced in LPS-Stimulated 264.7Δ3′UTR Macrophages
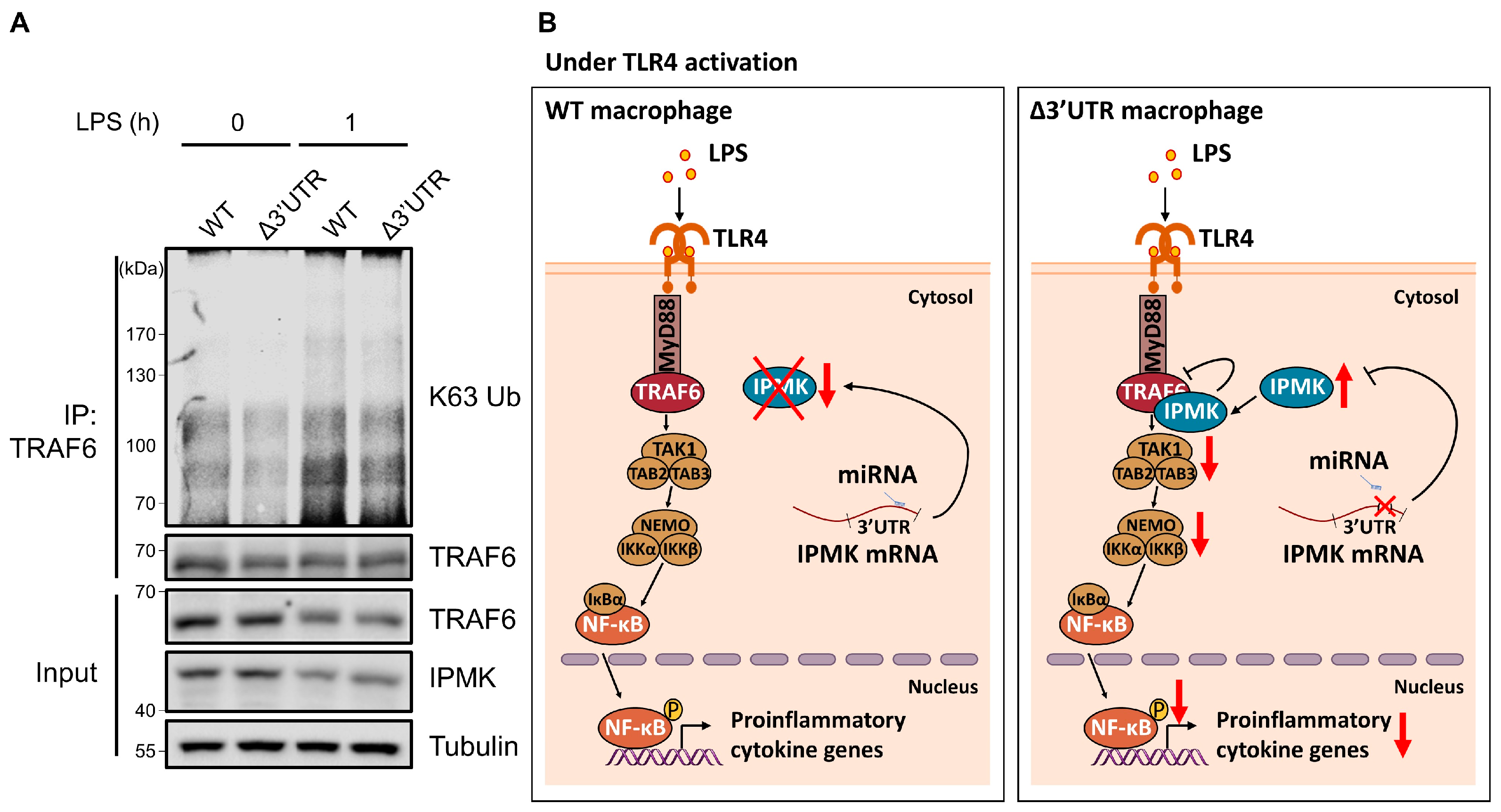
3.4. The Activation of TRAF6 Was Decreased in LPS-Stimulated 264.7Δ3′UTR Macrophages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Lee, B.; Park, S.J.; Hong, S.; Kim, K.; Kim, S. Inositol Polyphosphate Multikinase Signaling: Multifaceted Functions in Health and Disease. Mol. Cells 2021, 44, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ahn, H.; Kim, M.G.; Lee, H.; Kim, S. The Expanding Significance of Inositol Polyphosphate Multikinase as a Signaling Hub. Mol. Cells 2017, 40, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Kim, S.; Snyder, S.H. Inositol Pyrophosphates as Mammalian Cell Signals. Sci. Signal. 2011, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Maag, D.; Maxwell, M.J.; Hardesty, D.A.; Boucher, K.L.; Choudhari, N.; Hanno, A.G.; Ma, J.F.; Snowman, A.S.; Pietropaoli, J.W.; Xu, R.; et al. Inositol Polyphosphate Multikinase Is a Physiologic PI3-Kinase That Activates Akt/PKB. Proc. Natl. Acad. Sci. USA 2011, 108, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, E.; Min, H.; Kim, M.G.; Eisenbeis, V.B.; Dutta, A.K.; Pavlovic, I.; Jessen, H.J.; Kim, S.; Seong, R.H. Inositol Polyphosphates Promote T Cell-Independent Humoral Immunity via the Regulation of Bruton’s Tyrosine Kinase. Proc. Natl. Acad. Sci. USA 2019, 116, 12952–12957. [Google Scholar] [CrossRef]
- Watson, P.J.; Fairall, L.; Santos, G.M.; Schwabe, J.W.R. Structure of HDAC3 Bound to Co-Repressor and Inositol Tetraphosphate. Nature 2012, 481, 335–340. [Google Scholar] [CrossRef]
- Millard, C.J.; Watson, P.J.; Celardo, I.; Gordiyenko, Y.; Cowley, S.M.; Robinson, C.V.; Fairall, L.; Schwabe, J.W.R. Class I HDACs Share a Common Mechanism of Regulation by Inositol Phosphates. Mol. Cell 2013, 51, 57–67. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.F.; Maag, D.; Maxwell, M.J.; Resnick, A.C.; Juluri, K.R.; Chakraborty, A.; Koldobskiy, M.A.; Cha, S.H.; Barrow, R.; et al. Amino Acid Signaling to MTOR Mediated by Inositol Polyphosphate Multikinase. Cell Metab. 2011, 13, 215–221. [Google Scholar] [CrossRef]
- Kim, E.; Tyagi, R.; Lee, J.-Y.; Park, J.; Kim, Y.-R.; Beon, J.; Chen, P.Y.; Cha, J.Y.; Snyder, S.H.; Kim, S. Inositol Polyphosphate Multikinase Is a Coactivator for Serum Response Factor-Dependent Induction of Immediate Early Genes. Proc. Natl. Acad. Sci. USA 2013, 110, 19938–19943. [Google Scholar] [CrossRef]
- Xu, R.; Sen, N.; Paul, B.D.; Snowman, A.M.; Rao, F.; Vandiver, M.S.; Xu, J.; Snyder, S.H. Inositol Polyphosphate Multikinase Is a Coactivator of P53-Mediated Transcription and Cell Death. Sci. Signal. 2013, 6, ra22. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Snyder, S.H. Gene Transcription by P53 Requires Inositol Polyphosphate Multikinase as a Co-Activator. Cell Cycle 2013, 12, 1819–1820. [Google Scholar] [CrossRef]
- Kim, E.; Beon, J.; Lee, S.; Park, S.J.; Ahn, H.; Kim, M.G.; Park, J.E.; Kim, W.; Yuk, J.-M.; Kang, S.-J.; et al. Inositol Polyphosphate Multikinase Promotes Toll-like Receptor–Induced Inflammation by Stabilizing TRAF6. Sci. Adv. 2017, 3, e1602296. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like Receptors: Critical Proteins Linking Innate and Acquired Immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef]
- Kawai, T.; Adachi, O.; Ogawa, T.; Takeda, K.; Akira, S. Unresponsiveness of MyD88-Deficient Mice to Endotoxin. Immunity 1999, 11, 115–122. [Google Scholar] [CrossRef]
- Gottipati, S.; Rao, N.L.; Fung-Leung, W.P. IRAK1: A Critical Signaling Mediator of Innate Immunity. Cell. Signal. 2008, 20, 269–276. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Lamothe, B.; Besse, A.; Campos, A.D.; Webster, W.K.; Wu, H.; Darnay, B.G. Site-Specific Lys-63-Linked Tumor Necrosis Factor Receptor-Associated Factor 6 Auto-Ubiquitination Is a Critical Determinant of IκB Kinase Activation. J. Biol. Chem. 2007, 282, 4102–4112. [Google Scholar] [CrossRef]
- Takaesu, G.; Kishida, S.; Hiyama, A.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Ninomiya-Tsuji, J.; Matsumoto, K. TAB2, a Novel Adaptor Protein, Mediates Activation of TAK1 MAPKKK by Linking TAK1 to TRAF6 in the IL-1 Signal Transduction Pathway. Mol. Cell 2000, 5, 649–658. [Google Scholar] [CrossRef]
- Cheung, P.C.F.; Nebreda, A.R.; Cohen, P. TAB3, a New Binding Partner of the Protein Kinase TAK1. Biochem. J. 2004, 378, 27–34. [Google Scholar] [CrossRef]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.-K.; Hong, M.; Shaito, A.; Chiu, Y.-H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 Activate the NF-ΚB Pathway through Binding to Polyubiquitin Chains. Mol. Cell 2004, 15, 535–548. [Google Scholar] [CrossRef]
- Mattiroli, F.; Sixma, T.K. Lysine-Targeting Specificity in Ubiquitin and Ubiquitin-like Modification Pathways. Nat. Struct. Mol. Biol. 2014, 21, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like Receptors and Innate Immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting Negative Regulation of Toll-like Receptor Signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of MRNA Translation and Stability by MicroRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Baltimore, D.; Boldin, M.P.; O’connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New Regulators of Immune Cell Development and Function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef]
- Xiao, C.; Rajewsky, K. MicroRNA Control in the Immune System: Basic Principles. Cell 2009, 136, 26–36. [Google Scholar] [CrossRef]
- Li, H.; Jiang, T.; Li, M.-Q.; Zheng, X.-L.; Zhao, G.-J. Transcriptional Regulation of Macrophages Polarization by MicroRNAs. Front. Immunol. 2018, 9, 1175. [Google Scholar] [CrossRef] [Green Version]
- Curtale, G.; Rubino, M.; Locati, M. MicroRNAs as Molecular Switches in Macrophage Activation. Front. Immunol. 2019, 10, 799. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-ΚB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Boucher, A.; Klopfenstein, N.; Hallas, W.M.; Skibbe, J.; Appert, A.; Jang, S.H.; Pulakanti, K.; Rao, S.; Cowden Dahl, K.D.; Dahl, R. The MiR-23a∼ 27a∼ 24-2 MicroRNA Cluster Promotes Inflammatory Polarization of Macrophages. J. Immunol. 2021, 206, 540–553. [Google Scholar] [CrossRef]
- Wang, D.; He, S.; Liu, B.; Liu, C. MiR-27-3p Regulates TLR2/4-Dependent Mouse Alveolar Macrophage Activation by Targetting PPARγ. Clin. Sci. 2018, 132, 943–958. [Google Scholar] [CrossRef]
- Cai, M.; Shi, Y.; Zheng, T.; Hu, S.; Du, K.; Ren, A.; Jia, X.; Chen, S.; Wang, J.; Lai, S. Mammary Epithelial Cell Derived Exosomal MiR-221 Mediates M1 Macrophage Polarization via SOCS1/STATs to Promote Inflammatory Response. Int. Immunopharmacol. 2020, 83, 106493. [Google Scholar] [CrossRef]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The Kinase Akt1 Controls Macrophage Response to Lipopolysaccharide by Regulating MicroRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef]
- Lee, H.; Park, S.J.; Hong, S.; Lim, S.-W.; Kim, S. Deletion of IP6K1 in Mice Accelerates Tumor Growth by Dysregulating the Tumor-Immune Microenvironment. Animal Cells Syst. (Seoul) 2022, 26, 19–27. [Google Scholar] [CrossRef]
- Ahn, H.; Roh, J.S.; Lee, S.; Beon, J.; Lee, B.; Sohn, D.H.; Kim, S. Myeloid IPMK Promotes the Resolution of Serum Transfer-Induced Arthritis in Mice. Animal Cells Syst. (Seoul) 2021, 25, 219–226. [Google Scholar] [CrossRef]
- Park, J.; Bae, S.; Kim, J.-S. Cas-Designer: A Web-Based Tool for Choice of CRISPR-Cas9 Target Sites. Bioinformatics 2015, 31, 4014–4016. [Google Scholar] [CrossRef]
- Bae, S.; Park, J.; Kim, J.-S. Cas-OFFinder: A Fast and Versatile Algorithm That Searches for Potential off-Target Sites of Cas9 RNA-Guided Endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Kuersten, S.; Goodwin, E.B. The Power of the 3′ UTR: Translational Control and Development. Nat. Rev. Genet. 2003, 4, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of MiRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting Effective MicroRNA Target Sites in Mammalian MRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates That Thousands of Human Genes Are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X. MiRDB: An Online Database for Prediction of Functional MicroRNA Targets. Nucleic Acids Res. 2019, 48, D127–D131. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X. Prediction of Functional MicroRNA Targets by Integrative Modeling of MicroRNA Binding and Target Expression Data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Liu, P.; Qi, X.; Bian, C.; Yang, F.; Lin, X.; Zhou, S.; Xie, C.; Zhao, X.; Yi, T. MicroRNA-18a Inhibits Ovarian Cancer Growth via Directly Targeting TRIAP1 and IPMK. Oncol. Lett. 2017, 13, 4039–4046. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, H.; Wang, B.; Ao, Q.; Shi, J.; He, Z. MicroRNA-23b Alleviates Neuroinflammation and Brain Injury in Intracerebral Hemorrhage by Targeting Inositol Polyphosphate Multikinase. Int. Immunopharmacol. 2019, 76, 105887. [Google Scholar] [CrossRef]
- Wang, S.; Ma, Q.; Xie, Z.; Shen, Y.; Zheng, B.; Jiang, C.; Yuan, P.; An, Q.; Fan, S.; Jie, Z. An Antioxidant Sesquiterpene Inhibits Osteoclastogenesis Via Blocking IPMK/TRAF6 and Counteracts OVX-Induced Osteoporosis in Mice. J. Bone Miner. Res. 2021, 36, 1850–1865. [Google Scholar] [CrossRef]
- Yokoyama, J.S.; Wang, Y.; Schork, A.J.; Thompson, W.K.; Karch, C.M.; Cruchaga, C.; McEvoy, L.K.; Witoelar, A.; Chen, C.-H.; Holland, D.; et al. Association Between Genetic Traits for Immune-Mediated Diseases and Alzheimer Disease. JAMA Neurol. 2016, 73, 691–697. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Kim, E.; Kim, S. miRNA-Induced Downregulation of IPMK in Macrophages Mediates Lipopolysaccharide-Triggered TLR4 Signaling. Biomolecules 2023, 13, 332. https://doi.org/10.3390/biom13020332
Lee H, Kim E, Kim S. miRNA-Induced Downregulation of IPMK in Macrophages Mediates Lipopolysaccharide-Triggered TLR4 Signaling. Biomolecules. 2023; 13(2):332. https://doi.org/10.3390/biom13020332
Chicago/Turabian StyleLee, Haein, Eunha Kim, and Seyun Kim. 2023. "miRNA-Induced Downregulation of IPMK in Macrophages Mediates Lipopolysaccharide-Triggered TLR4 Signaling" Biomolecules 13, no. 2: 332. https://doi.org/10.3390/biom13020332