
Chronic Exposure to Nitric Oxide Induces P53 Mutations and Malignant-like Features in Human Breast Epithelial Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
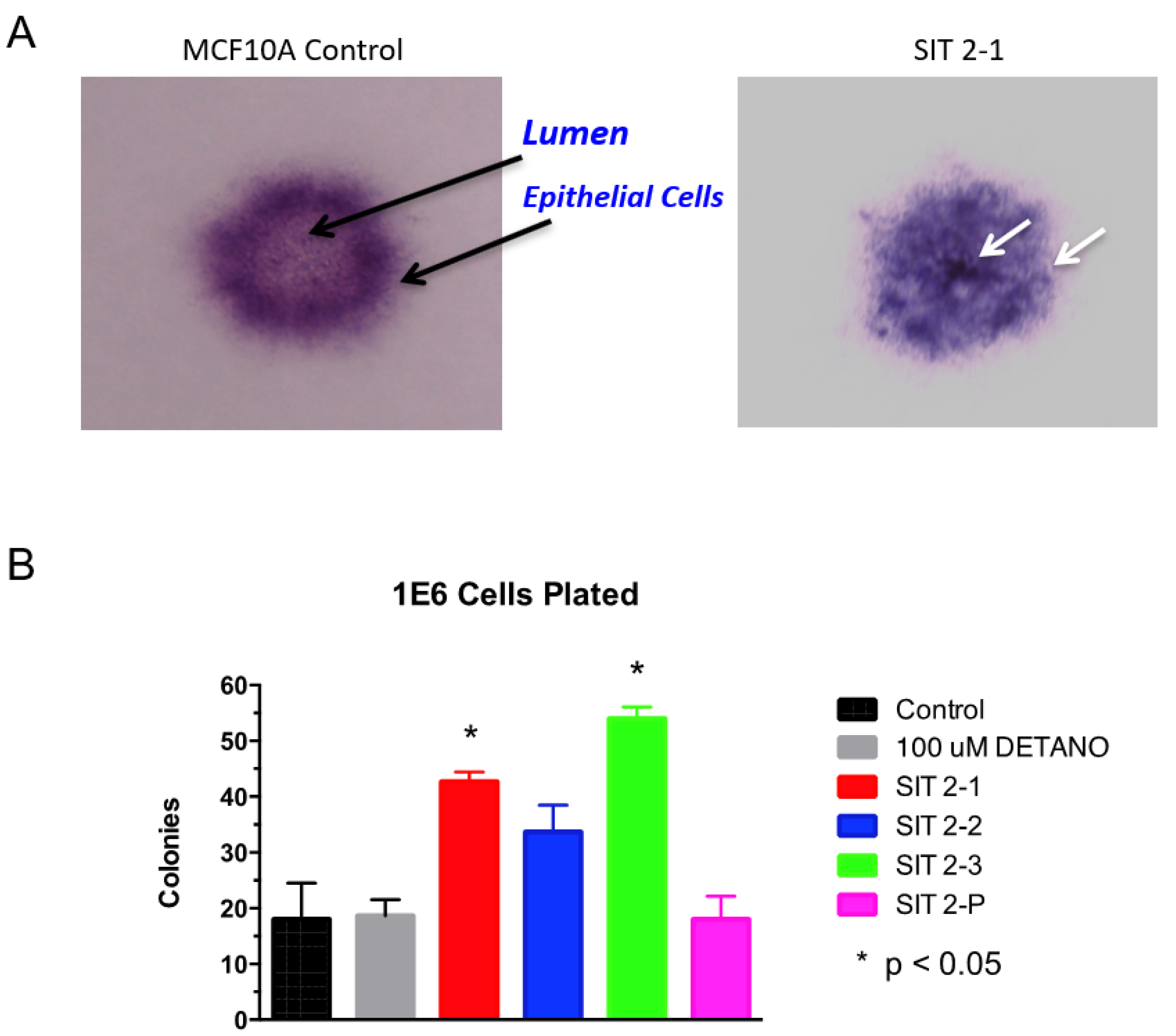
2.2. Cell Growth in Serum-Free Media Supplemented with Selenium, Insulin, and Transferrin (SIT)
2.3. Colony Formation in Soft Agar
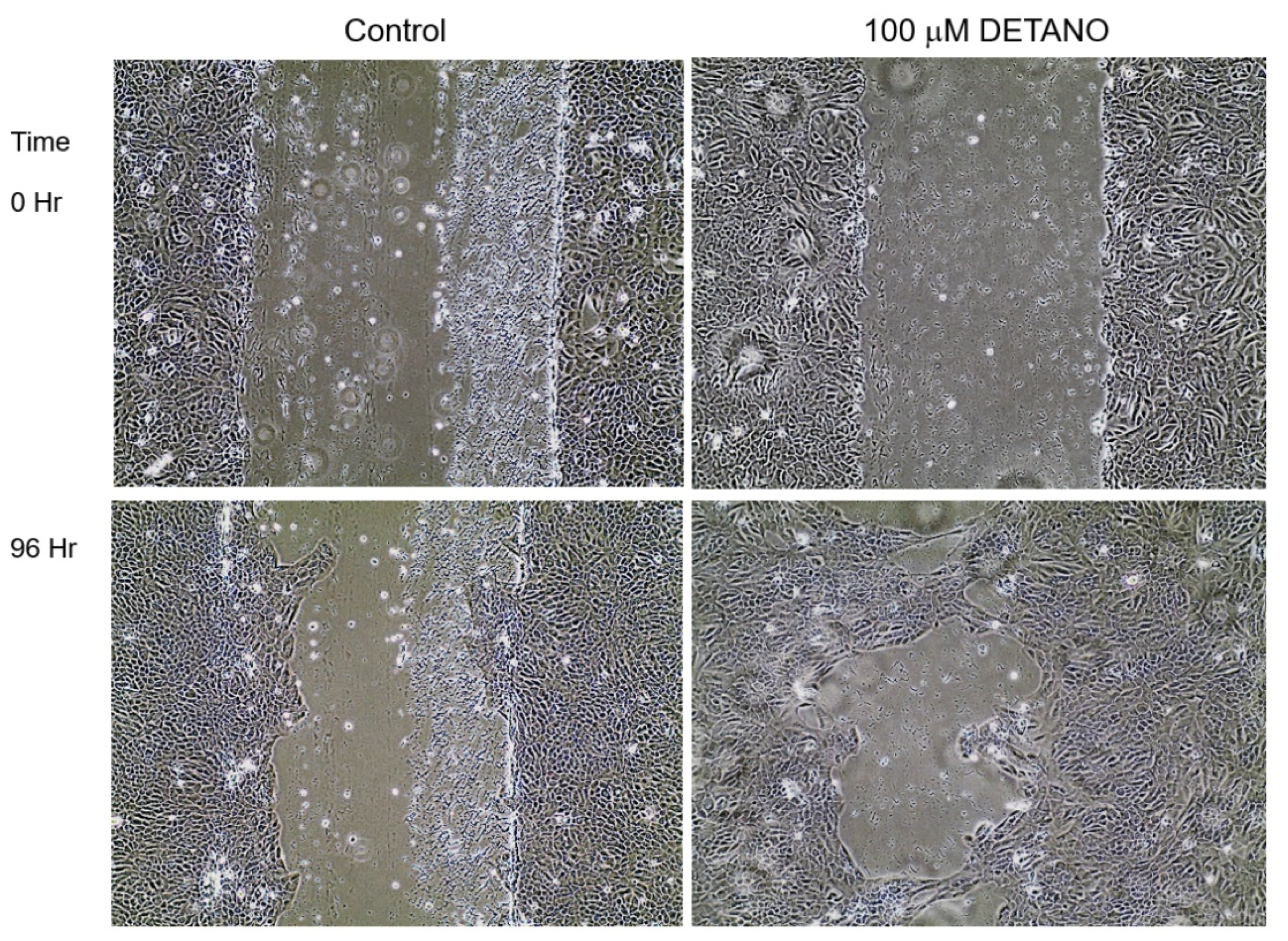
2.4. Scratch Test
2.5. Karyotype Analysis
2.6. Spectral Karyotyping
2.7. TOPO TA Cloning
2.8. RNA Transcript Expression Level Analyses
2.9. Protein Expression Analyses
2.10. Animal Study
2.11. Statistical Analysis
3. Results
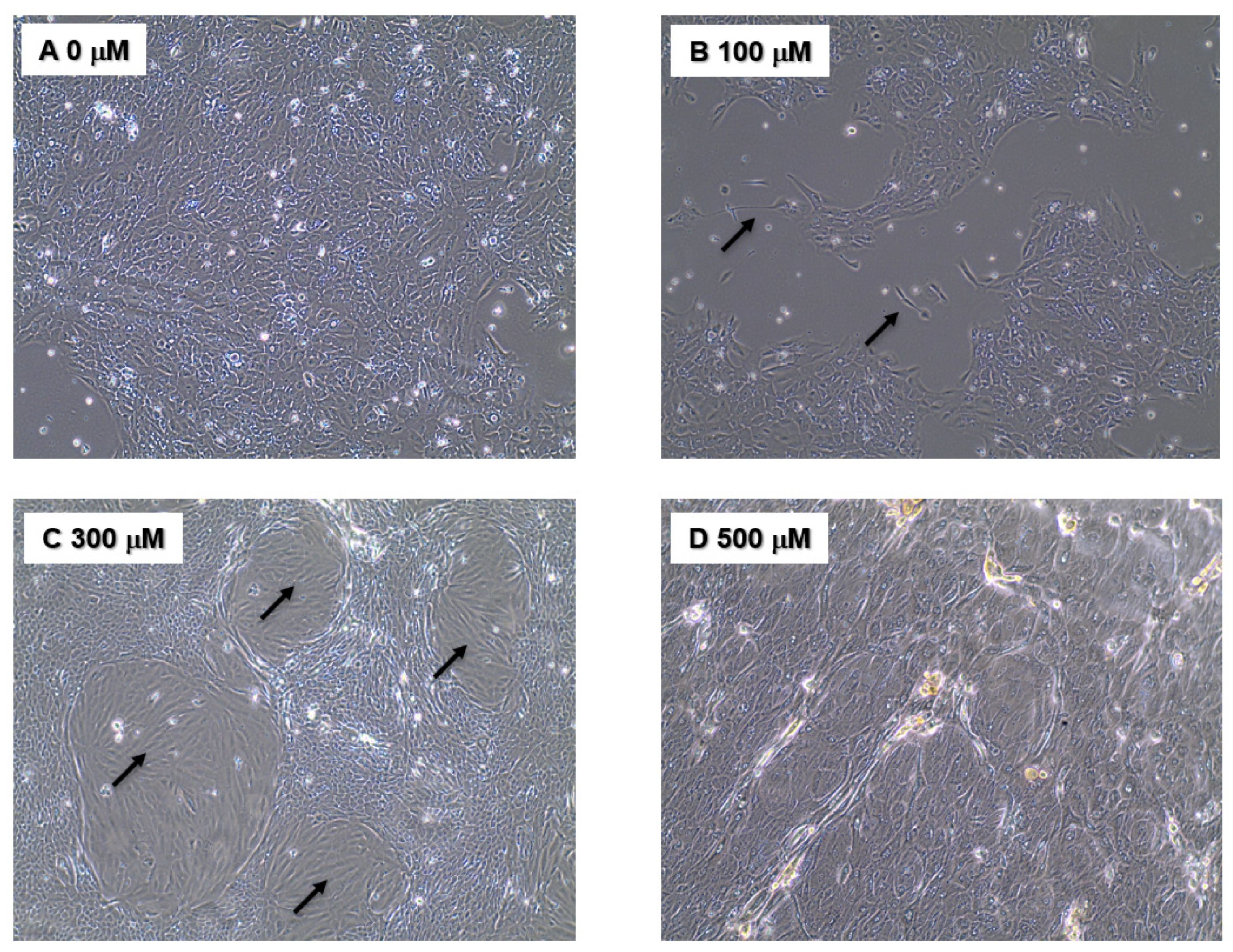
3.1. Phenotypic Assays
3.2. Cytogenetic Alterations
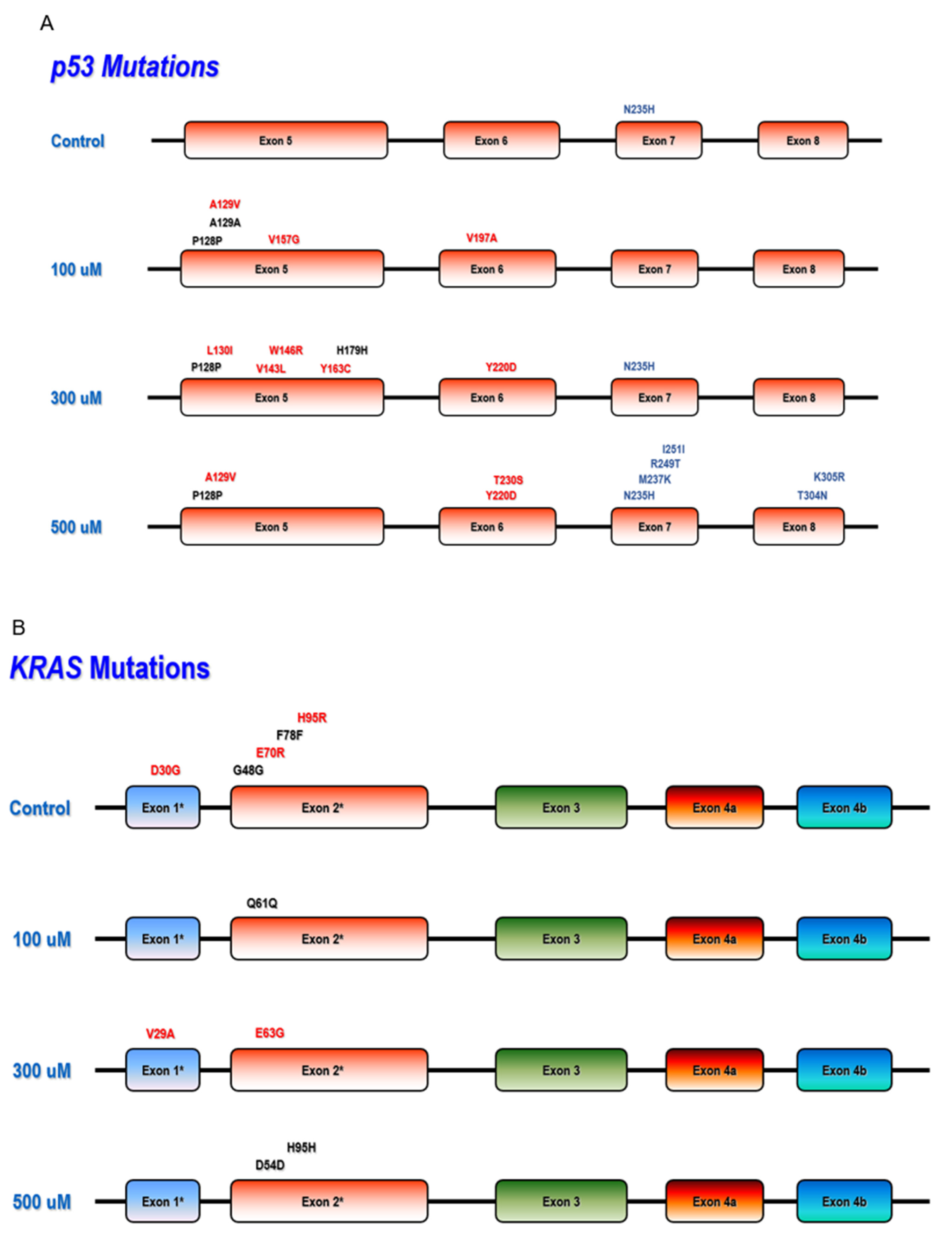
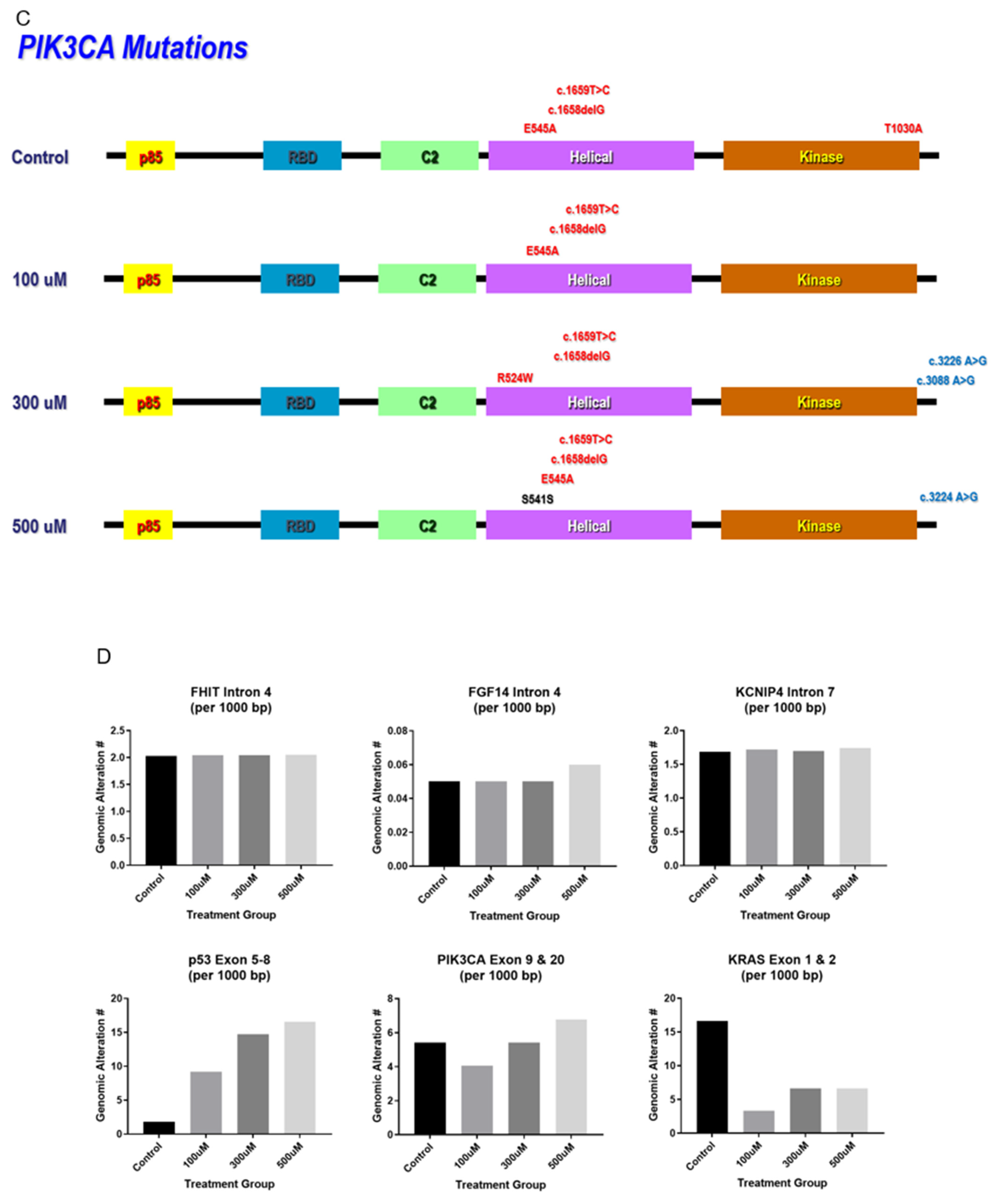
3.3. Mutation Detection in Hot Spot Regions of TP53, PI3KCA, and KRAS
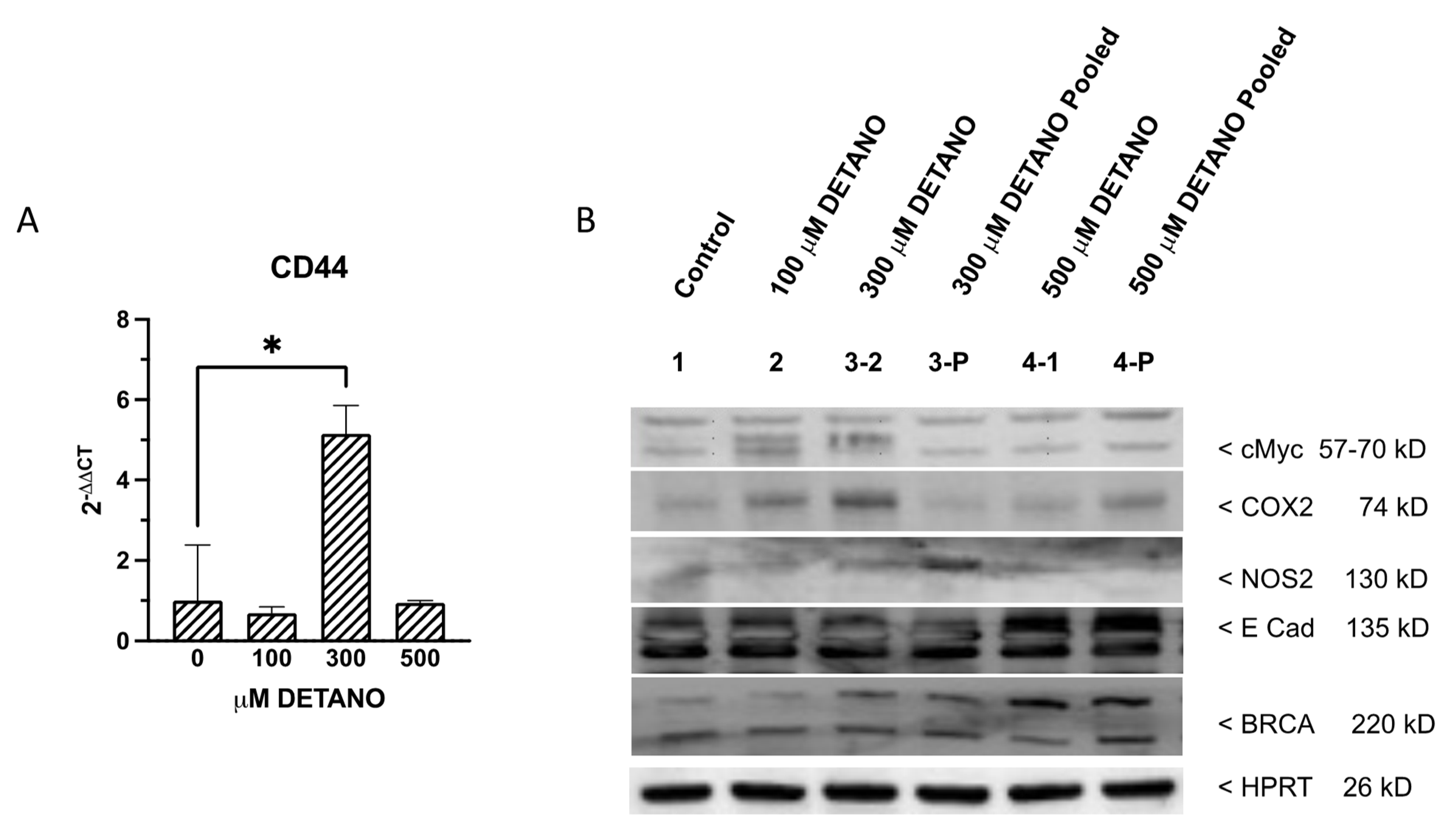
3.4. Cancer Stem Cell Marker Expression
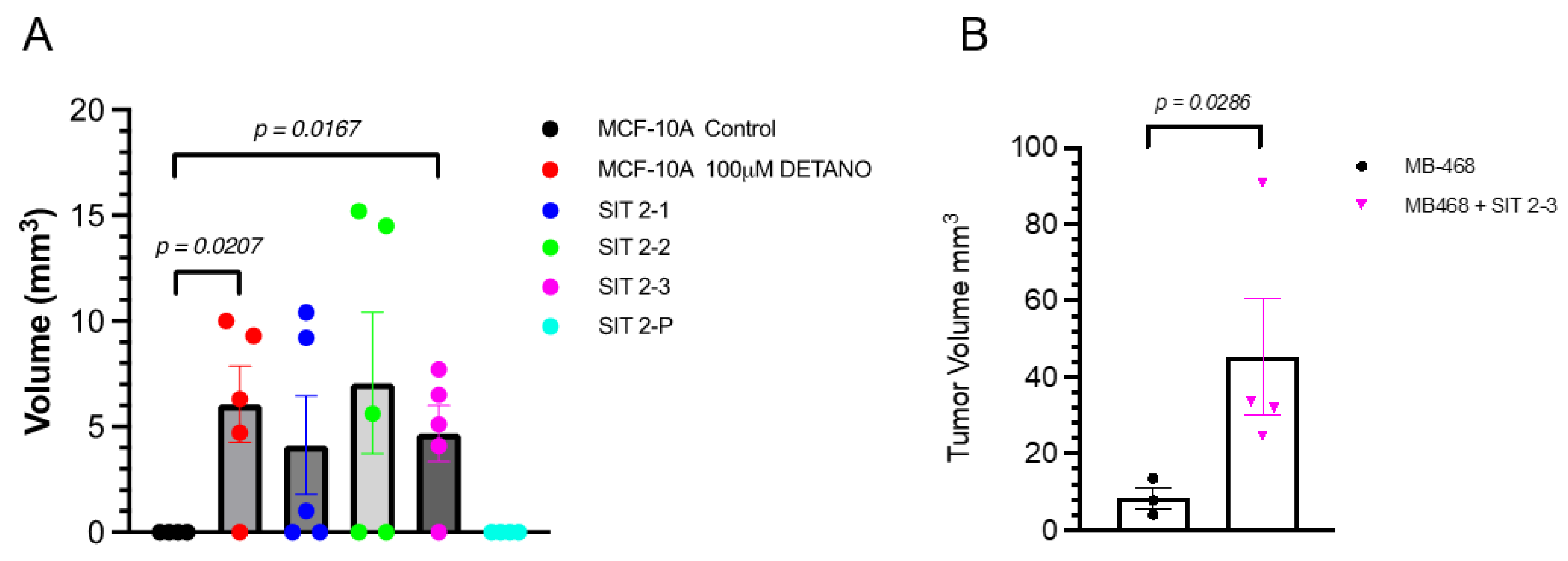
3.5. Growth in Nude Mice
4. Discussion
4.1. Chronic NO Exposure-Induced Phenotypic Alterations
4.2. Chronic NO Exposure-Induced Genomics Alterations
4.3. Chronic NO Exposure-Induced CD44 Stemness Biomarker Expression
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALDH1A1 | Aldehyde Dehydrogenase 1 Family Member A1 |
| ATM | Ataxia Telangiectasia Mutated |
| ATR | ATR Serine/Threonine Kinase |
| BRCA1 | Breast Cancer 1 |
| CCD | Charged Couple Device |
| CD133 | Cluster of Differentiation 133 |
| CD117 | Cluster of Differentiation 177 |
| CD31 | Cluster of Differentiation 31 |
| CD44 | Cluster of Differentiation 44 |
| Chr | Chromosome |
| COX | Cyclooxygenase |
| CSC | Cancer Stem Cell |
| Ct | Cycle Threshold |
| CXCR4 | C-X-C Motif Chemokine Receptor 4 |
| cMyc | MYC Proto-Oncogene, BHLH Transcription Factor |
| del() | chromosome deleted |
| der() | derivative chromosome |
| DETANO | Diethylenetriamine/nitric oxide |
| dic() | dicentric chromosome |
| ER | Estrogen Receptor |
| FISH | Fluorescent In Situ Hybridization |
| FMO | Fluorescence Minus One |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| IKK | I-Kappa-B-Kinase Beta |
| iNOS | Inducible Nitric Oxide Synthase |
| KRAS | Kirsten Rat Sarcoma Viral Proto-Oncogene |
| MAPK | Mitogen-Activated Protein Kinase 1 |
| MEK | MAPK/ERK Kinase 1 |
| NANOG | Nanog Homeobox |
| NFκB | Nuclear Factor Kappa B Subunit 1 |
| NGS | Next Generation Sequencing |
| NO | Nitric Oxide |
| NOS2 | Nitric Oxide Synthase 2 |
| NOS3 | Nitric Oxide Synthase 3 |
| OCT4 | Octamer-Binding Protein 4 |
| PARP | Poly (ADP-Ribose) Polymerase |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha |
| PR | Progesterone Receptor |
| SIT | Selenium, Insulin, and Transferrin |
| SKY | Spectral Karyotyping |
| t() | chromosome translocation |
| TKI | Tyrosine Kinase Inhibitors |
| TP53 | Tumor Protein P53 |
| WAF1 | Wild-Type P53-Activated Fragment 1 |
References
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hanbauer, I.; Krishna, M.C.; DeGraff, W.; Gamson, J.; Mitchell, J.B. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc. Natl. Acad. Sci. USA 1993, 90, 9813–9817. [Google Scholar] [CrossRef] [PubMed]
- Hofseth, L.J.; Saito, S.; Hussain, S.P.; Espey, M.G.; Miranda, K.M.; Araki, Y.; Jhappan, C.; Higashimoto, Y.; He, P.; Linke, S.P.; et al. Nitric oxide-induced cellular stress and p53 activation in chronic inflammation. Proc. Natl. Acad. Sci. USA 2003, 100, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.E.; Hofseth, L.J.; Hussain, S.P.; Harris, C.C. Nitric oxide and p53 in cancer-prone chronic inflammation and oxyradical overload disease. Environ. Mol. Mutagen. 2004, 44, 3–9. [Google Scholar] [CrossRef]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.J.; Swenberg, J.A.; Marrogi, A.J.; et al. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Zingarelli, B.; Szabo, C.; Salzman, A.L. Reduced oxidative and nitrosative damage in murine experimental colitis in the absence of inducible nitric oxide synthase. Gut 1999, 45, 199–209. [Google Scholar] [CrossRef]
- Glynn, S.A.; Boersma, B.J.; Dorsey, T.H.; Yi, M.; Yfantis, H.G.; Ridnour, L.A.; Martin, D.N.; Switzer, C.H.; Hudson, R.S.; Wink, D.A.; et al. Increased NOS2 predicts poor survival in estrogen receptor-negative breast cancer patients. J. Clin. Investig. 2010, 120, 3843–3854. [Google Scholar] [CrossRef]
- Dave, B.; Granados-Principal, S.; Zhu, R.; Benz, S.; Rabizadeh, S.; Soon-Shiong, P.; Yu, K.D.; Shao, Z.; Li, X.; Gilcrease, M.; et al. Targeting RPL39 and MLF2 reduces tumor initiation and metastasis in breast cancer by inhibiting nitric oxide synthase signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 8838–8843. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Liu, Y.; Guevara, M.L.; Blanco, E.; Choi, D.S.; Qian, W.; Patel, T.; Rodriguez, A.A.; Cusimano, J.; Weiss, H.L.; et al. Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 2015, 17, 25. [Google Scholar] [CrossRef]
- Ekmekcioglu, S.; Ellerhorst, J.A.; Prieto, V.G.; Johnson, M.M.; Broemeling, L.D.; Grimm, E.A. Tumor iNOS predicts poor survival for stage III melanoma patients. Int. J. Cancer 2006, 119, 861–866. [Google Scholar] [CrossRef]
- Wang, J.; He, P.; Gaida, M.; Yang, S.; Schetter, A.J.; Gaedcke, J.; Ghadimi, B.M.; Ried, T.; Yfantis, H.; Lee, D.; et al. Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer. Oncotarget 2016, 7, 52993–53004. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Glynn, S.A.; Cheng, R.Y.; Ridnour, L.A.; Green, J.E.; Ambs, S.; Wink, D.A. S-Nitrosylation of EGFR and Src Activates an Oncogenic Signaling Network in Human Basal-Like Breast Cancer. Mol. Cancer Res. 2012, 10, 1203–1215. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Ridnour, L.A.; Cheng, R.; Heinecke, J.; Burke, A.; Glynn, S.; Ambs, S.; Wink, D.A. S-Nitrosation Mediates Multiple Pathways That Lead to Tumor Progression in Estrogen Receptor-Negative Breast Cancer. Onco Ther. 2012, 3, 117–124. [Google Scholar] [CrossRef]
- Heinecke, J.L.; Ridnour, L.A.; Cheng, R.Y.; Switzer, C.H.; Lizardo, M.M.; Khanna, C.; Glynn, S.A.; Hussain, S.P.; Young, H.A.; Ambs, S.; et al. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, 6323–6328. [Google Scholar] [CrossRef]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ambs, S.; Hussain, S.P.; Harris, C.C. Interactive effects of nitric oxide and the p53 tumor suppressor gene in carcinogenesis and tumor progression. FASEB J. 1997, 11, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Ridnour, L.A.; Barasch, K.M.; Windhausen, A.N.; Dorsey, T.H.; Lizardo, M.M.; Yfantis, H.G.; Lee, D.H.; Switzer, C.H.; Cheng, R.Y.; Heinecke, J.L.; et al. Nitric oxide synthase and breast cancer: Role of TIMP-1 in NO-mediated Akt activation. PLoS ONE 2012, 7, e44081. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Windhausen, A.N.; Isenberg, J.S.; Yeung, N.; Thomas, D.D.; Vitek, M.P.; Roberts, D.D.; Wink, D.A. Nitric oxide regulates matrix metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 16898–16903. [Google Scholar] [CrossRef] [Green Version]
- Wydooghe, E.; Heras, S.; Dewulf, J.; Piepers, S.; Van den Abbeel, E.; De Sutter, P.; Vandaele, L.; Van Soom, A. Replacing serum in culture medium with albumin and insulin, transferrin and selenium is the key to successful bovine embryo development in individual culture. Reprod. Fertil. Dev. 2014, 26, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.R.; Disteche, C.M. Chromosome banding techniques. Curr. Protoc. Hum. Genet 2001, 4.2.1–4.2.36. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003, 30, 256–268. [Google Scholar] [CrossRef]
- Camara-Sanchez, P.; Diaz-Riascos, Z.V.; Garcia-Aranda, N.; Gener, P.; Seras-Franzoso, J.; Giani-Alonso, M.; Royo, M.; Vazquez, E.; Schwartz, S., Jr.; Abasolo, I. Selectively Targeting Breast Cancer Stem Cells by 8-Quinolinol and Niclosamide. Int. J. Mol. Sci. 2022, 23, 11760. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Buonaguro, L.; Buonaguro, F.M. Mutations of the TP53 gene in adenocarcinoma and squamous cell carcinoma of the cervix: A systematic review. Gynecol. Oncol. 2013, 128, 442–448. [Google Scholar] [CrossRef]
- Stephenson Clarke, J.R.; Douglas, L.R.; Duriez, P.J.; Balourdas, D.I.; Joerger, A.C.; Khadiullina, R.; Bulatov, E.; Baud, M.G.J. Discovery of Nanomolar-Affinity Pharmacological Chaperones Stabilizing the Oncogenic p53 Mutant Y220C. ACS Pharmacol. Transl. Sci. 2022, 5, 1169–1180. [Google Scholar] [CrossRef]
- Mayani, H.; Chavez-Gonzalez, A.; Vazquez-Santillan, K.; Contreras, J.; Guzman, M.L. Cancer Stem Cells: Biology and Therapeutic Implications. Arch Med. Res. 2022, 53, 770–784. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Sawa, T.; Ohshima, H. Nitrative DNA damage in inflammation and its possible role in carcinogenesis. Nitric Oxide 2006, 14, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Torquato, S. Evolution and morphology of microenvironment-enhanced malignancy of three-dimensional invasive solid tumors. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2013, 87, 052707. [Google Scholar] [CrossRef] [PubMed]
- Mousavikhamene, Z.; Sykora, D.J.; Mrksich, M.; Bagheri, N. Morphological features of single cells enable accurate automated classification of cancer from non-cancer cell lines. Sci. Rep. 2021, 11, 24375. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, E.; Castle, J.; Quirk, A.; Taylor, C.D.L.; Xu, W.; Prasad, A. Cellular morphological features are predictive markers of cancer cell state. Comput. Biol. Med. 2020, 126, 104044. [Google Scholar] [CrossRef] [PubMed]
- Grosser, S.; Lippoldt, J.; Oswald, L.; Merkel, M.; Sussman, D.M.; Renner, F.; Gottheil, P.; Morawetz, E.W.; Fuhs, T.; Xie, X.F.; et al. Cell and Nucleus Shape as an Indicator of Tissue Fluidity in Carcinoma. Phys. Rev. X 2021, 11, 011033. [Google Scholar] [CrossRef]
- Nicholson, J.M.; Cimini, D. Cancer karyotypes: Survival of the fittest. Front. Oncol. 2013, 3, 148. [Google Scholar] [CrossRef]
- Cowell, J.K.; LaDuca, J.; Rossi, M.R.; Burkhardt, T.; Nowak, N.J.; Matsui, S. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet. Cytogenet. 2005, 163, 23–29. [Google Scholar] [CrossRef]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Kennedy, M.C.; Lowe, S.W. Mutant p53: It’s not all one and the same. Cell Death Differ. 2022, 29, 983–987. [Google Scholar] [CrossRef]
- Alsner, J.; Sorensen, S.B.; Overgaard, J. TP53 mutation is related to poor prognosis after radiotherapy, but not surgery, in squamous cell carcinoma of the head and neck. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2001, 59, 179–185. [Google Scholar] [CrossRef]
- Leung, W.K.; To, K.F.; Ng, Y.P.; Lee, T.L.; Lau, J.Y.; Chan, F.K.; Ng, E.K.; Chung, S.C.; Sung, J.J. Association between cyclo-oxygenase-2 overexpression and missense p53 mutations in gastric cancer. Br. J. Cancer 2001, 84, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Ridanpaa, M.; Karjalainen, A.; Anttila, S.; Vainio, H.; Husgafvelpursiainen, K. Genetic alterations in p53 and k-ras in lung-cancer in relation to histopathology of the tumor and smoking history of the patient. Int. J. Oncol. 1994, 5, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Enomoto, T.; Inoue, M.; Tanizawa, O.; Ozaki, M.; Rice, J.M.; Nomura, T. Alteration of the p53 tumor suppressor gene occurs independently of K-ras activation and more frequently in serous adenocarcinomas than in other common epithelial tumors of the human ovary. Jpn. J. Cancer Res. Gann 1994, 85, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, H.; Nishikawa, R.; Fujimaki, T.; Nakagomi, T.; Matsutani, M.; Huang, H.J.; Cavenee, W.K. Analysis of the p300/CBP-Associated Factor (PCAF) gene in astrocytic tumors. J. Neuro-Oncol. 2000, 46, 17–22. [Google Scholar] [CrossRef]
- Lukas, J.; Gao, D.Q.; Keshmeshian, M.; Wen, W.H.; Tsao-Wei, D.; Rosenberg, S.; Press, M.F. Alternative and aberrant messenger RNA splicing of the mdm2 oncogene in invasive breast cancer. Cancer Res. 2001, 61, 3212–3219. [Google Scholar]
- Ueki, K.; Nishikawa, R.; Nakazato, Y.; Hirose, T.; Hirato, J.; Funada, N.; Fujimaki, T.; Hojo, S.; Kubo, O.; Ide, T.; et al. Correlation of histology and molecular genetic analysis of 1p, 19q, 10q, TP53, EGFR, CDK4, and CDKN2A in 91 astrocytic and oligodendroglial tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 196–201. [Google Scholar]
- Leroy, K.; Haioun, C.; Lepage, E.; Le Metayer, N.; Berger, F.; Labouyrie, E.; Meignin, V.; Petit, B.; Bastard, C.; Salles, G.; et al. p53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2002, 13, 1108–1115. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Sessa, F.; Renault, B.; Bonato, M.; Leone, B.E.; Solcia, E.; Ranzani, G.N. K-ras and p53 gene mutations in pancreatic cancer: Ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994, 54, 1556–1560. [Google Scholar]
- Kubo, Y.; Urano, Y.; Yoshimoto, K.; Iwahana, H.; Fukuhara, K.; Arase, S.; Itakura, M. p53 gene mutations in human skin cancers and precancerous lesions: Comparison with immunohistochemical analysis. J. Investig. Dermatol. 1994, 102, 440–444. [Google Scholar] [CrossRef]
- Mu, W.; Lu, H.M.; Chen, J.; Li, S.; Elliott, A.M. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J. Mol. Diagn. 2016, 18, 923–932. [Google Scholar] [CrossRef]
- Felley-Bosco, E.; Mirkovitch, J.; Ambs, S.; Mace, K.; Pfeifer, A.; Keefer, L.K.; Harris, C.C. Nitric oxide and ethylnitrosourea: Relative mutagenicity in the p53 tumor suppressor and hypoxanthine-phosphoribosyltransferase genes. Carcinogenesis 1995, 16, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, F.; Wang, Y.; Wu, Q.; Wang, B.; Yao, Y.; Zhang, Y.; Han-Zhang, H.; Ye, J.; Zhang, L.; et al. Mutations in exon 8 of TP53 are associated with shorter survival in patients with advanced lung cancer. Oncol. Lett. 2019, 18, 3159–3169. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; He, P.; Subleski, J.; Hofseth, L.J.; Trivers, G.E.; Mechanic, L.; Hofseth, A.B.; Bernard, M.; Schwank, J.; Nguyen, G.; et al. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res. 2008, 68, 7130–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, H.; Sasaki, J.; Matsumoto, M.; Suga, M.; Ando, Y.; Iggo, R.; Tada, M.; Saya, H.; Ando, M. Significant correlation of nitric oxide synthase activity and p53 gene mutation in stage I lung adenocarcinoma. Jpn. J. Cancer Res. 1998, 89, 696–702. [Google Scholar] [CrossRef]
- Schon, K.; Tischkowitz, M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res. Tr. 2018, 167, 417–423. [Google Scholar] [CrossRef]
- Vandal, G.; Geiling, B.; Dankort, D. Ras effector mutant expression suggest a negative regulator inhibits lung tumor formation. PLoS ONE 2014, 9, e84745. [Google Scholar] [CrossRef]
- Takahashi, M.; Mutoh, M.; Shoji, Y.; Kamanaka, Y.; Naka, M.; Maruyama, T.; Sugimura, T.; Wakabayashi, K. Transfection of K-rasAsp12 cDNA markedly elevates IL-1beta- and lipopolysaccharide-mediated inducible nitric oxide synthase expression in rat intestinal epithelial cells. Oncogene 2003, 22, 7667–7676. [Google Scholar] [CrossRef]
- Ohnishi, S.; Saito, H.; Suzuki, N.; Ma, N.; Hiraku, Y.; Murata, M.; Kawanishi, S. Nitrative and oxidative DNA damage caused by K-ras mutation in mice. Biochem. Biophys. Res. Commun. 2011, 413, 236–240. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Oshi, M.; Katsuta, E.; Yan, L.; Satyananda, V.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K.; Takabe, K. KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival. Am. J. Cancer Res. 2020, 10, 897–907. [Google Scholar]
- Kim, J.K.; Jeon, H.Y.; Kim, H. The molecular mechanisms underlying the therapeutic resistance of cancer stem cells. Arch. Pharmacal Res. 2015, 38, 389–401. [Google Scholar] [CrossRef]
- Karamboulas, C.; Ailles, L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim. Biophys. Acta 2013, 1830, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; Ouzounova, M.; Le Calvez-Kelm, F.; Lambert, M.P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Miyazono, K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef]
- Al-Othman, N.; Alhendi, A.; Ihbaisha, M.; Barahmeh, M.; Alqaraleh, M.; Al-Momany, B.Z. Role of CD44 in breast cancer. Breast Dis. 2020, 39, 1–13. [Google Scholar] [CrossRef]
- Dzobo, K.; Sinkala, M. Cancer Stem Cell Marker CD44 Plays Multiple Key Roles in Human Cancers: Immune Suppression/Evasion, Drug Resistance, Epithelial-Mesenchymal Transition, and Metastasis. OMICS 2021, 25, 313–332. [Google Scholar] [CrossRef]
- Glynn, S.A.; Prueitt, R.L.; Ridnour, L.A.; Boersma, B.J.; Dorsey, T.M.; Wink, D.A.; Goodman, J.E.; Yfantis, H.G.; Lee, D.H.; Ambs, S. COX-2 activation is associated with Akt phosphorylation and poor survival in ER-negative, HER2-positive breast cancer. BMC Cancer 2010, 10, 626. [Google Scholar] [CrossRef]
- Li, B.; Lu, Y.; Yu, L.; Han, X.; Wang, H.; Mao, J.; Shen, J.; Wang, B.; Tang, J.; Li, C.; et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-kappaB/COX-2 activation. Chem. Biol. Interact 2017, 277, 33–42. [Google Scholar] [CrossRef]
- Shen, W.; Zhang, X.; Du, R.; Gao, W.; Wang, J.; Bao, Y.; Yang, W.; Luo, N.; Li, J. Ibuprofen mediates histone modification to diminish cancer cell stemness properties via a COX2-dependent manner. Br. J. Cancer 2020, 123, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Hachim, M.Y.; Hachim, I.Y.; Dai, M.; Lo, C.; Raffa, F.A.; Ali, S.; Lebrun, J.J. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci. Rep. 2017, 7, 40258. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Basudhar, D.; Glynn, S.A.; Greer, M.; Somasundaram, V.; No, J.H.; Scheiblin, D.A.; Garrido, P.; Heinz, W.F.; Ryan, A.E.; Weiss, J.M.; et al. Coexpression of NOS2 and COX2 accelerates tumor growth and reduces survival in estrogen receptor-negative breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 13030–13035. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control: 47,XX, | dic(1;1)(q11;q11), | +der(1),-1,-3, | t(3;9)(p13;22),-5 | del(5),-8, | i(8)(q11;q11), | -9, t(9;3;5) |
| 100 μM: 47,XX, | dic(1;1)(q11;q11), | +der(1), | t(3;9)(p13;p22), | der(8), | i(8)(q11;q11), | t(5;3;9) |
| 2-1 100 μM: 47,XX, | +dic(1q;1q), | der(1) t(1;11), | der(3)t(9;3), | der(8), | der(9) | t(5;3;9) |
| 2-2 100 μM: 47,XX, | der(1),+(1q), | der(3), | der(8), | t(5;3;9) | ||
| 2-3 100 μM: 47,XX, | +dic(1q;1q), | der(1), | der(3)t(9;3), | der(8), | der(9) | t(5;3;9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, R.Y.S.; Burkett, S.; Ambs, S.; Moody, T.; Wink, D.A.; Ridnour, L.A. Chronic Exposure to Nitric Oxide Induces P53 Mutations and Malignant-like Features in Human Breast Epithelial Cells. Biomolecules 2023, 13, 311. https://doi.org/10.3390/biom13020311
Cheng RYS, Burkett S, Ambs S, Moody T, Wink DA, Ridnour LA. Chronic Exposure to Nitric Oxide Induces P53 Mutations and Malignant-like Features in Human Breast Epithelial Cells. Biomolecules. 2023; 13(2):311. https://doi.org/10.3390/biom13020311
Chicago/Turabian StyleCheng, Robert Y. S., Sandra Burkett, Stefan Ambs, Terry Moody, David A. Wink, and Lisa A. Ridnour. 2023. "Chronic Exposure to Nitric Oxide Induces P53 Mutations and Malignant-like Features in Human Breast Epithelial Cells" Biomolecules 13, no. 2: 311. https://doi.org/10.3390/biom13020311