
Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids
 , ,
, ,
Abstract
:1. Introduction
2. The Genetics of ASD
- Chromatin remodeling: mutations in genes that encode regulators of chromatin remodeling and gene transcription that influence neuronal connectivity and synaptic plasticity (i.e., MECP2, MEF2C, HDAC4, CHD8, and CTNNB1) [35];
- Protein synthesis: The levels of synaptic proteins can be influenced by neuronal activity through global and local mechanisms, and in ASD, the regulation of synaptic proteins occurs in an unregulated manner. For example, the mammalian target of rapamycin (mTOR) pathway controls global mRNA translation, and its deregulation may increase the risk of ASD in addition to causing associated diseases, increased cell proliferation, and loss of autophagy. Consequently, suppression of the mTOR pathway, such as NF1, PTEN, and SynGAP1, causes an increase in translation in neurons and synapses. Mutations in the FMRP complex, EIF4E, and CYFIP1 also increase the risk of autism [36,37];
- Protein degradation: The ubiquitin-proteasome system, which participates in protein degradation and regulation of synapse composition and is encoded by the UBE3A gene (which encodes ubiquitin ligase), is mutated in patients with Angelman syndrome and is duplicated in the maternal 15q11 chromosome in individuals with ASD [38,39];
- Synaptic functions: Many proteins encoded by autism risk genes are implicated in neuronal connectivity, glutaminergic pathways (GRIN2B), gamma-aminobutyric acid (GABA)ergic pathways (GABRA3 and GABRB3), glycinergic neurotransmission (GLRA2), neuritogenesis (CNTN), neuronal conduction (CNTNAP2), ion permeability (CACNA1, CACNA2D3, SCN2A), synaptic activity (NRXNs and NLGNs), dendritic structure, and neurotransmitter receptors. For example, SHANK deletions, duplications, and coding mutations (SHANK1, SHANK2, and SHANK3) are already well described in patients with ASD; however, they reduce actin accumulation, leading to cellular and dendritic structural dysmorphology as well as axonal outgrowth [40].
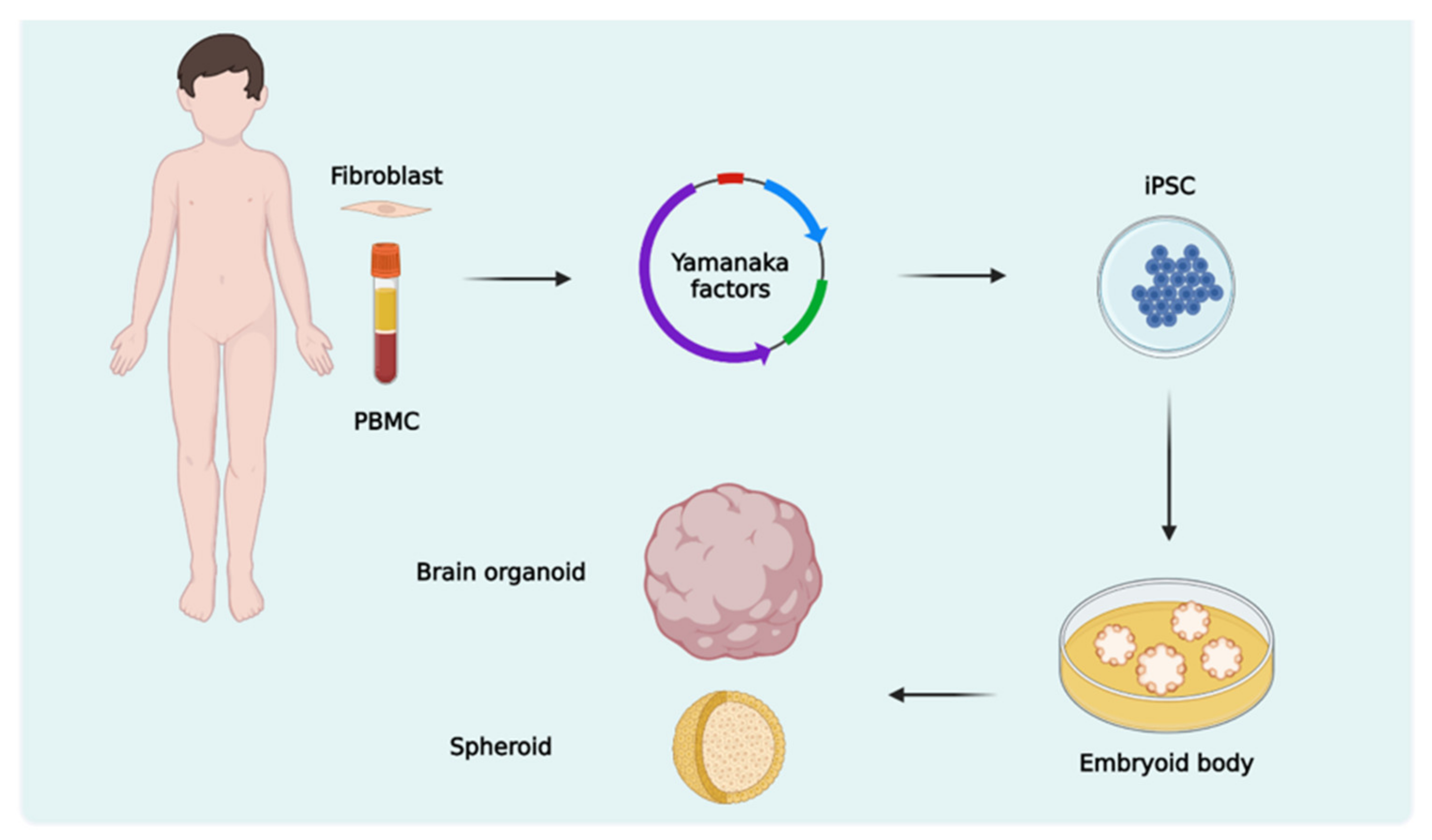
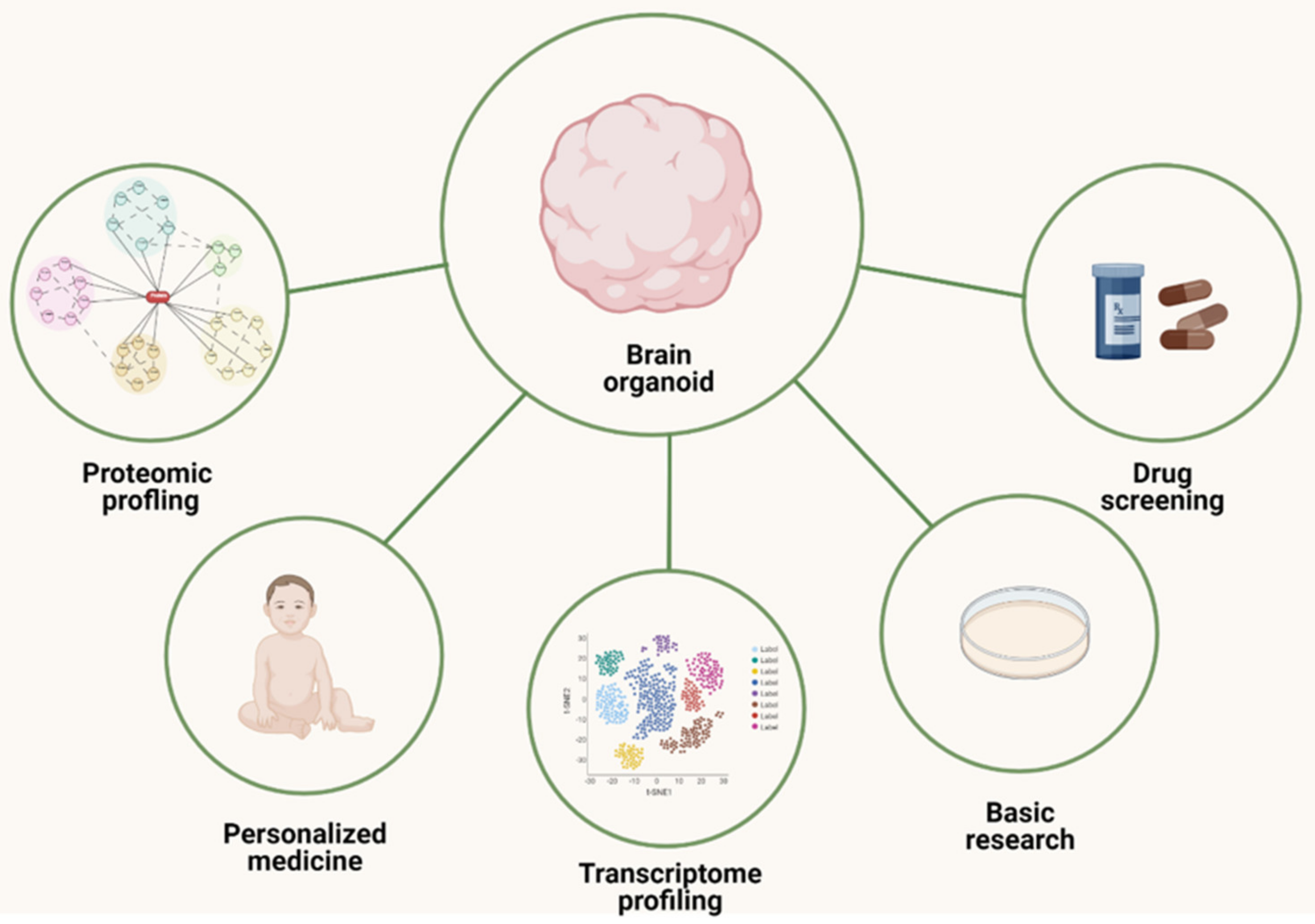
3. Brain Organoids
3.1. Protocols and Types of Brain Organoids
3.2. ASD Studies with Brain Organoid Models

{kind=link}
{kind=link}
| Ref. | Brain Organoid Type | Aims/Methods | Main Findings |
|---|---|---|---|
| Jong et al., 2021 [70] | Forebrain | Studied the effects of a protein-truncating homozygous mutation in CNTNAP2 on embryonic cortical development using ASD patients-derived iPSCs | CNTNAP2 is most highly expressed in several types of excitatory neurons and leads to cortical overgrowth |
| Lim et al., 2022 [97] | Cerebral | Performed RNA sequencing on samples comprising organoids from CRISPR-edited iPSCs and described a framework (Orgo-Seq) to integrate bulk RNA and single-cell RNA sequence data | YPEL3, KCTD13, and INO80E genes can be associated as driver genes linked with ASD |
| Mariani et al., 2015 [61] | Telencephalic | Produced organoids from ASD patients-derived iPSCs, recapitulating transcriptional programs present in mid-fetal human cortical development | Overexpression of FOXG1 is associated with the overproduction of GABAergic neurons |
| Mellios et al., 2018 [92] | Cerebral | Characterize the effects on human neurogenesis and neuronal differentiation brought about by MeCP2 deficiency using ASD patients-derived iPSCs | Organoids exhibited increased ventricular area and alterations in GABAergic interneuron differentiation |
| Meng et al., 2022 [87] | Forebrain | Identified mechanisms by which valproic acid (VPA) contributed to ASD risk in humans, using hFOS from healthy donor-derived hiPSC lines; | VPA affected the expression of genes enriched in neural development, synaptic transmission, calcium, and potassium signaling pathways, which have been implicated in ASD |
| Modafferi et al., 2021 [72] | Brainspheroids | Identified potential synergy between a mutation in CHD8 and environmental exposure to the pesticide chlorpyrifos, using brain organoids produced from CRISPR-edited iPSCs | Identified metabolic perturbations and disruption of neurotransmitter systems involved in ASD |
| Paulsen et al., 2022 [81] | Cortical | Analyzed abnormalities that resulted from haploinsufficiency in three ASD risk genes: SUV420H, ARID1B, and CHD8 by single-cell RNA sequencing, with both patient-derived and CRISPR-edited iPSCs | Different ASD risk genes converge on a phenotype of asynchronous neuronal development |
| Pearson et al., 2022 [98] | Cerebral | Analyzed the differential methylation profile of a regulatory region of the GAD1 gene, using ASD patients-derived iPSCs | In the ASD group, GAD1 is subject to differential methylation patterns that may indicate variable epigenetic regulation |
| Trujillo et al., 2021 [91] | Cortical | Evaluated the therapeutic efficacy of two pharmacological compounds in reversing MECP2-KO phenotypes, using CRISPR-edited iPSCs | The compounds increased gene expression of specific neurotransmitter markers, such as cholinergic, GABAergic, and glutamatergic signaling, reverting neuropathological phenotypes and networks |
| Urresti et al., 2021 [99] | Cortical | Investigated the impact of mutations in the 16p11.2 region on neurodevelopmental processes, using ASD patients-derived iPSCs | Dysregulation on neuronal maturation, migration, synaptic processes, and morphology, resulting in defects in neurogenesis |
| Villa et al., 2022 [86] | Cerebral | Analyzed alterations in CHD8, using CRISPR-edited iPSCs | CHD8 haploinsufficiency disrupted neurodevelopmental trajectories with an accelerated and delayed generation of inhibitory and excitatory neurons, respectively |
| Wang et al., 2017 [100] | Cerebral | RNA-seq was carried out on CHD8+/− and isogenic control (CHD8+/+) cerebral organoids, using CRISPR-edited iPSCs | CHD8 regulates the expression of other genes implicated in ASD: TCF4 and AUTS2 |
| Wang et al., 2022 [94] | Telencephalon | Investigated telencephalic development under normal and autism-associated SHANK3 deficiency, using CRISPR-edited iPSCs | Neurons in organoids with a hemizygous deletion of the autism gene, SHANK3, exhibit intrinsic and excitatory synaptic deficits |
| Wegscheid et al., 2021 [101] | Cerebral | Defined molecular and cellular causes for the neurodevelopmental abnormalities in patients with NF1 mutation, using ASD patients-derived iPSCs | Neural stem cell proliferation and neuronal maturation abnormalities were observed and caused by reduced cytokine receptor-like factor 3 (CRLF3) expression and impaired RhoA signaling |
| Zhang et al., 2020 [96] | Cerebral | Model human neurodevelopmental dysregulation using RAB39b mutant cerebral organoids, using CRISPR-edited iPSCs | RAB39b mutations result in over-proliferation and differentiation deficits of neural progenitor cells (NPCs) |
4. Advances, Limitations, and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5-TR, 5th ed.; American Psychiatric Association Publishing: Washington, DC, USA, 2022. [Google Scholar]
- Maenner, M.J.; Shaw, K.A.; Bakian, A.V.; Bilder, D.A.; Durkin, M.S.; Esler, A.; Furnier, S.M.; Hallas, L.; Hall-Lande, J.; Hudson, A.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2018. MMWR Surveill. Summ. 2021, 70, 1–16. [Google Scholar] [CrossRef]
- Forsberg, S.L.; Ilieva, M.; Maria Michel, T. Epigenetics and cerebral organoids: Promising directions in autism spectrum disorders. Transl. Psychiatry 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef] [Green Version]
- Willsey, H.R.; Willsey, A.J.; Wang, B.; State, M.W. Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat. Rev. Neurosci. 2022, 23, 323–341. [Google Scholar] [CrossRef]
- Hulbert, S.W.; Jiang, Y.H. Cellular and Circuitry Bases of Autism: Lessons Learned from the Temporospatial Manipulation of Autism Genes in the Brain. Neurosci. Bull. 2017, 33, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brant, B.; Stern, T.; Shekhidem, H.A.; Mizrahi, L.; Rosh, I.; Stern, Y.; Ofer, P.; Asleh, A.; Umanah, G.K.E.; Jada, R.; et al. IQSEC2 mutation associated with epilepsy, intellectual disability, and autism results in hyperexcitability of patient-derived neurons and deficient synaptic transmission. Mol. Psychiatry 2021, 26, 7498–7508. [Google Scholar] [CrossRef]
- Wolff, J.J.; Jacob, S.; Elison, J.T. The journey to autism: Insights from neuroimaging studies of infants and toddlers. Dev. Psychopathol. 2018, 30, 479–495. [Google Scholar] [CrossRef]
- Varghese, M.; Keshav, N.; Jacot-Descombes, S.; Warda, T.; Wicinski, B.; Dickstein, D.L.; Harony-Nicolas, H.; De Rubeis, S.; Drapeau, E.; Buxbaum, J.D.; et al. Autism spectrum disorder: Neuropathology and animal models. Acta Neuropathol. 2017, 134, 537–566. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.X.; Pek, N.M.Q.; Soh, B.S. Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2018, 19, 936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adegbola, A.; Bury, L.A.; Fu, C.; Zhang, M.; Wynshaw-Boris, A. Concise Review: Induced Pluripotent Stem Cell Models for Neuropsychiatric Diseases. Stem Cells Transl. Med. 2017, 6, 2062–2070. [Google Scholar] [CrossRef]
- Castelbaum, L.; Sylvester, C.M.; Zhang, Y.; Yu, Q.; Constantino, J.N. On the Nature of Monozygotic Twin Concordance and Discordance for Autistic Trait Severity: A Quantitative Analysis. Behav. Genet. 2020, 50, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Ercument Cicek, A.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Iossifov, I.; O’roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Zhao, D.; Lachman, H.M.; Zheng, D. Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl. Psychiatry 2018, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, T.; Klei, L.; Sanders, S.J.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.; Manaa, D.; Pawitan, Y.; Reichert, J.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trost, B.; Thiruvahindrapuram, B.; Chan, A.J.; Engchuan, W.; Higginbotham, E.J.; Howe, J.L.; Loureiro, L.O.; Reuter, M.S.; Roshandel, D.; Whitney, J.; et al. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022, 185, 4409–4427.e18. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Sampaio, G.L.; Martins, G.L.S.; Paredes, B.D.; Nonaka, C.K.V.; da Silva, K.N.; Rossi, E.A.; Dos Santos, R.R.; Soares, M.B.P.; de Freitas Souza, B.S. Generation of an induced pluripotent stem cell line from a patient with autism spectrum disorder and SCN2A haploinsufficiency. Stem Cell Res. 2019, 39, 101488. [Google Scholar] [CrossRef]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, É.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015, 23, 1505–1512. [Google Scholar] [CrossRef]
- Spratt, P.W.; Ben-Shalom, R.; Keeshen, C.M.; Burke Jr, K.J.; Clarkson, R.L.; Sanders, S.J.; Bender, K.J. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 2019, 103, 673–685.e5. [Google Scholar] [CrossRef]
- Yi, F.; Danko, T.; Botelho, S.C.; Patzke, C.; Pak, C.; Wernig, M.; Südhof, T.C. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 2016, 352, aaf2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugsley, K.; Scherer, S.W.; Bellgrove, M.A.; Hawi, Z. Environmental exposures associated with elevated risk for autism spectrum disorder may augment the burden of deleterious de novo mutations among probands. Mol. Psychiatry 2022, 27, 710–730. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [Green Version]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef]
- Sahin, M.; Sur, M. Genes, circuits, and precision therapies for autism and related neurodevelopmental disorders. Science 2015, 350, aab3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, B.D.; Osterweil, E.K.; Bear, M.F. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011, 480, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Gabel, H.W.; Hemberg, M.; Hutchinson, A.N.; Sadacca, L.A.; Ebert, D.H.; Harmin, D.A.; Greenberg, R.S.; Verdine, V.K.; Zhou, Z.; et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron 2011, 72, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Budimirovic, D.B.; Kaufmann, W.E. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 2011, 33, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, P.L.; Hanayama, R.; Bloodgood, B.L.; Mardinly, A.R.; Lipton, D.M.; Flavell, S.W.; Kim, T.K.; Griffith, E.C.; Waldon, Z.; Maehr, R.; et al. The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 2010, 40, 704–716. [Google Scholar] [CrossRef] [Green Version]
- Mabb, A.M.; Ehlers, M.D. Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell Dev. Biol. 2010, 26, 179–210. [Google Scholar] [CrossRef] [Green Version]
- Sassone-Corsi, P.; Christen, Y. A Time for Metabolism and Hormones; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Dumas, G.; Goubran-Botros, H.; Matondo, M.; Pagan, C.; Boulègue, C.; Chaze, T.; Chamot-Rooke, J.; Maronde, E.; Bourgeron, T. Mass-spectrometry analysis of the human pineal proteome during night and day and in autism. J. Pineal Res. 2021, 70, e12713. [Google Scholar] [CrossRef]
- Khalid, M.; Raza, H.; MDriessen, T.; JLee, P.; Tejwani, L.; Sami, A.; Nawaz, M.; Mehmood Baig, S.; Lim, J.; Kaukab Raja, G. Genetic Risk of Autism Spectrum Disorder in a Pakistani Population. Genes 2020, 11, 1206. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhou, Y.; Wang, K. Multiplex gene and phenotype network to characterize shared genetic pathways of epilepsy and autism. Sci. Rep. 2021, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- Howland, J.G.; Greenshaw, A.J.; Winship, I.R. Practical Aspects of Animal Models of Psychiatric Disorders. Can. J Psychiatry 2019, 64, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Bendriem, R.M.; Wu, W.W.; Shen, R.F. 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Cheffer, A.; Flitsch, L.J.; Krutenko, T.; Röderer, P.; Sokhranyaeva, L.; Iefremova, V.; Hajo, M.; Peitz, M.; Schwarz, M.K.; Brüstle, O. Human stem cell-based models for studying autism spectrum disorder-related neuronal dysfunction. Mol. Autism 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; Martín, J.M.; Eggan, K.C. Mind the translational gap: Using iPS cell models to bridge from genetic discoveries to perturbed pathways and therapeutic targets. Mol. Autism 2021, 12, 10. [Google Scholar] [CrossRef]
- Kim, J.; Sullivan, G.J.; Park, I.H. How well do brain organoids capture your brain? iScience 2021, 24, 102063. [Google Scholar] [CrossRef]
- Eichmüller, O.L.; Knoblich, J.A. Human cerebral organoids—A new tool for clinical neurology research. Nat. Rev. Neurol. 2022, 18, 661–680. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willsey, H.R.; Exner, C.R.; Xu, Y.; Everitt, A.; Sun, N.; Wang, B.; Dea, J.; Schmunk, G.; Zaltsman, Y.; Teerikorpi, N.; et al. Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 2021, 109, 788–804.e8. [Google Scholar] [CrossRef] [PubMed]
- St Clair, D.; Johnstone, M. Using mouse transgenic and human stem cell technologies to model genetic mutations associated with schizophrenia and autism. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelava, I.; Lancaster, M.A. Stem Cell Models of Human Brain Development. Cell Stem Cell 2016, 18, 736–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trujillo, C.A.; Muotri, A.R. Brain Organoids and the Study of Neurodevelopment. Trends Mol. Med. 2018, 24, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Ciarpella, F.; Zamfir, R.G.; Campanelli, A.; Ren, E.; Pedrotti, G.; Bottani, E.; Borioli, A.; Caron, D.; Di Chio, M.; Dolci, S.; et al. Murine cerebral organoids develop network of functional neurons and hippocampal brain region identity. iScience 2021, 24, 103438. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Tetzlaff, W.; Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992, 12, 4565–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamies, D.; Barrera, P.; Block, K.; Makri, G.; Kumar, A.; Wiersma, D.; Smirnova, L.; Zhang, C.; Bressler, J.; Christian, K.M.; et al. A human brain microphysiological system derived from induced pluripotent stem cells to study neurological diseases and toxicity. ALTEX 2017, 34, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Song, H.; Ming, G.L. Brain organoids: Advances, applications and challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569.e7. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef] [PubMed]
- Pașca, S.P.; Arlotta, P.; Bateup, H.S.; Camp, J.G.; Cappello, S.; Gage, F.H.; Knoblich, J.A.; Kriegstein, A.R.; Lancaster, M.A.; Ming, G.L.; et al. A nomenclature consensus for nervous system organoids and assembloids. Nature 2022, 609, 907–910. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- de Jong, J.O.; Llapashtica, C.; Genestine, M.; Strauss, K.; Provenzano, F.; Sun, Y.; Zhu, H.; Cortese, G.P.; Brundu, F.; Brigatti, K.W.; et al. Cortical overgrowth in a preclinical forebrain organoid model of CNTNAP2-associated autism spectrum disorder. Nat. Commun. 2021, 12, 4087. [Google Scholar] [CrossRef]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modafferi, S.; Zhong, X.; Kleensang, A.; Murata, Y.; Fagiani, F.; Pamies, D.; Hogberg, H.T.; Calabrese, V.; Lachman, H.; Hartung, T.; et al. Gene-Environment Interactions in Developmental Neurotoxicity: A Case Study of Synergy between Chlorpyrifos and CHD8 Knockout in Human BrainSpheres. Environ. Health Perspect. 2021, 129, 077001. [Google Scholar] [CrossRef] [PubMed]
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Klaus, J.; Kanton, S.; Kyrousi, C.; Ayo-Martin, A.C.; Di Giaimo, R.; Riesenberg, S.; O’Neill, A.C.; Camp, J.G.; Tocco, C.; Santel, M.; et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat. Med. 2019, 25, 561–568. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, A.C.; Kyrousi, C.; Klaus, J.; Leventer, R.J.; Kirk, E.P.; Fry, A.; Pilz, D.T.; Morgan, T.; Jenkins, Z.A.; Drukker, M.; et al. A Primate-Specific Isoform of PLEKHG6 Regulates Neurogenesis and Neuronal Migration. Cell Rep. 2018, 25, 2729–2741.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcez, P.P.; Loiola, E.C.; Madeiro da Costa, R.; Higa, L.M.; Trindade, P.; Delvecchio, R.; Nascimento, J.M.; Brindeiro, R.; Tanuri, A.; Rehen, S.K. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova, M.F.; Buxhoeveden, D.; Gomez, J. Disruption in the inhibitory architecture of the cell minicolumn: Implications for autism. Neuroscientist 2003, 9, 496–507. [Google Scholar] [CrossRef]
- Rubenstein, J.L. Annual Research Review: Development of the cerebral cortex: Implications for neurodevelopmental disorders. J. Child Psychol. Psychiatry 2011, 52, 339–355. [Google Scholar] [CrossRef]
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602, 268–273. [Google Scholar] [CrossRef]
- Wang, Z.J.; Rein, B.; Zhong, P.; Williams, J.; Cao, Q.; Yang, F.; Zhang, F.; Ma, K.; Yan, Z. Autism risk gene KMT5B deficiency in prefrontal cortex induces synaptic dysfunction and social deficits via alterations of DNA repair and gene transcription. Neuropsychopharmacology 2021, 46, 1617–1626. [Google Scholar] [CrossRef]
- Moffat, J.J.; Smith, A.L.; Jung, E.M.; Ka, M.; Kim, W.Y. Neurobiology of ARID1B haploinsufficiency related to neurodevelopmental and psychiatric disorders. Mol. Psychiatry 2022, 27, 476–489. [Google Scholar] [CrossRef]
- Weissberg, O.; Elliott, E. The Mechanisms of CHD8 in Neurodevelopment and Autism Spectrum Disorders. Genes 2021, 12, 1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Villa, C.E.; Cheroni, C.; Dotter, C.P.; López-Tóbon, A.; Oliveira, B.; Sacco, R.; Yahya, A.Ç.; Morandell, J.; Gabriele, M.; Tavakoli, M.R.; et al. CHD8 haploinsufficiency links autism to transient alterations in excitatory and inhibitory trajectories. Cell Rep. 2022, 39, 110615. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhang, W.; Wang, X.; Jiao, C.; Xu, S.; Liu, C.; Tang, B.; Chen, C. Human forebrain organoids reveal connections between valproic acid exposure and autism risk. Transl. Psychiatry 2022, 12, 130. [Google Scholar] [CrossRef]
- Tong, D.L.; Chen, R.G.; Lu, Y.L.; Li, W.K.; Zhang, Y.F.; Lin, J.K.; He, L.J.; Dang, T.; Shan, S.F.; Xu, X.H.; et al. The critical role of ASD-related gene CNTNAP3 in regulating synaptic development and social behavior in mice. Neurobiol. Dis. 2019, 130, 104486. [Google Scholar] [CrossRef]
- Hali, S.; Kim, J.; Kwak, T.H.; Lee, H.; Shin, C.Y.; Han, D.W. Modelling monogenic autism spectrum disorder using mouse cortical organoids. Biochem. Biophys. Res. Commun. 2020, 521, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological reversal of synaptic and network pathology in human MECP2-KO neurons and cortical organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Yoo, T.; Yoo, Y.E.; Kang, H.; Kim, E. Age, brain region, and gene dosage-differential transcriptomic changes in. Front. Mol. Neurosci. 2022, 15, 1017512. [Google Scholar] [CrossRef]
- Wang, Y.; Chiola, S.; Yang, G.; Russell, C.; Armstrong, C.J.; Wu, Y.; Spampanato, J.; Tarboton, P.; Ullah, H.M.; Edgar, N.U.; et al. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun. 2022, 13, 5688. [Google Scholar] [CrossRef]
- Koss, D.J.; Bondarevaite, O.; Adams, S.; Leite, M.; Giorgini, F.; Attems, J.; Outeiro, T.F. RAB39B is redistributed in dementia with Lewy bodies and is sequestered within aβ plaques and Lewy bodies. Brain Pathol. 2021, 31, 120–132. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.F. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.T.; Chan, Y.; Dawes, P.; Guo, X.; Erdin, S.; Tai, D.J.; Liu, S.; Reichert, J.M.; Burns, M.J.; Chan, Y.K.; et al. Orgo-Seq integrates single-cell and bulk transcriptomic data to identify cell type specific-driver genes associated with autism spectrum disorder. Nat. Commun. 2022, 13, 3243. [Google Scholar] [CrossRef]
- Pearson, G.; Song, C.; Hohmann, S.; Prokhorova, T.; Sheldrick-Michel, T.M.; Knöpfel, T. DNA Methylation Profiles of GAD1 in Human Cerebral Organoids of Autism Indicate Disrupted Epigenetic Regulation during Early Development. Int. J. Mol. Sci. 2022, 23, 9188. [Google Scholar] [CrossRef]
- Urresti, J.; Zhang, P.; Moran-Losada, P.; Yu, N.K.; Negraes, P.D.; Trujillo, C.A.; Antaki, D.; Amar, M.; Chau, K.; Pramod, A.B.; et al. Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 2021, 26, 7560–7580. [Google Scholar] [CrossRef]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Wegscheid, M.L.; Anastasaki, C.; Hartigan, K.A.; Cobb, O.M.; Papke, J.B.; Traber, J.N.; Morris, S.M.; Gutmann, D.H. Patient-derived iPSC-cerebral organoid modeling of the 17q11.2 microdeletion syndrome establishes CRLF3 as a critical regulator of neurogenesis. Cell Rep. 2021, 36, 109315. [Google Scholar] [CrossRef]
- Sun, A.X.; Ng, H.H.; Tan, E.K. Translational potential of human brain organoids. Ann. Clin. Transl. Neurol. 2018, 5, 226–235. [Google Scholar] [CrossRef]
- Sun, X.Y.; Ju, X.C.; Li, Y.; Zeng, P.M.; Wu, J.; Zhou, Y.Y.; Shen, L.B.; Dong, J.; Chen, Y.J.; Luo, Z.G. Generation of vascularized brain organoids to study neurovascular interactions. eLife 2022, 11, e76707. [Google Scholar] [CrossRef]
- Shen, H.; Bocksteins, E.; Kondrychyn, I.; Snyders, D.; Korzh, V. Functional antagonism of voltage-gated K+ channel α-subunits in the developing brain ventricular system. Development 2016, 143, 4249–4260. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, J.L.d.S.; Araújo, C.d.A.; Rocha, C.A.G.; Costa-Ferro, Z.S.M.; Souza, B.S.d.F. Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids. Biomolecules 2023, 13, 260. https://doi.org/10.3390/biom13020260
Santos JLdS, Araújo CdA, Rocha CAG, Costa-Ferro ZSM, Souza BSdF. Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids. Biomolecules. 2023; 13(2):260. https://doi.org/10.3390/biom13020260
Chicago/Turabian StyleSantos, John Lenon de Souza, Cecília de Almeida Araújo, Clarissa Araújo Gurgel Rocha, Zaquer Suzana Munhoz Costa-Ferro, and Bruno Solano de Freitas Souza. 2023. "Modeling Autism Spectrum Disorders with Induced Pluripotent Stem Cell-Derived Brain Organoids" Biomolecules 13, no. 2: 260. https://doi.org/10.3390/biom13020260