
Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases



Abstract
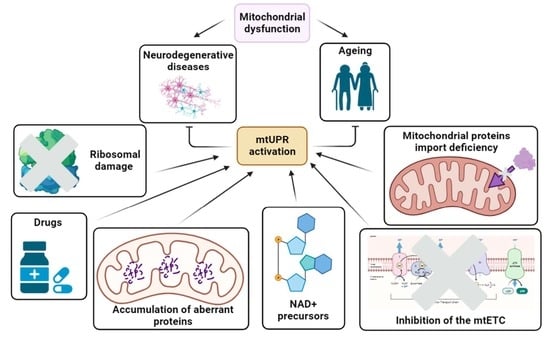
:
1. Introduction
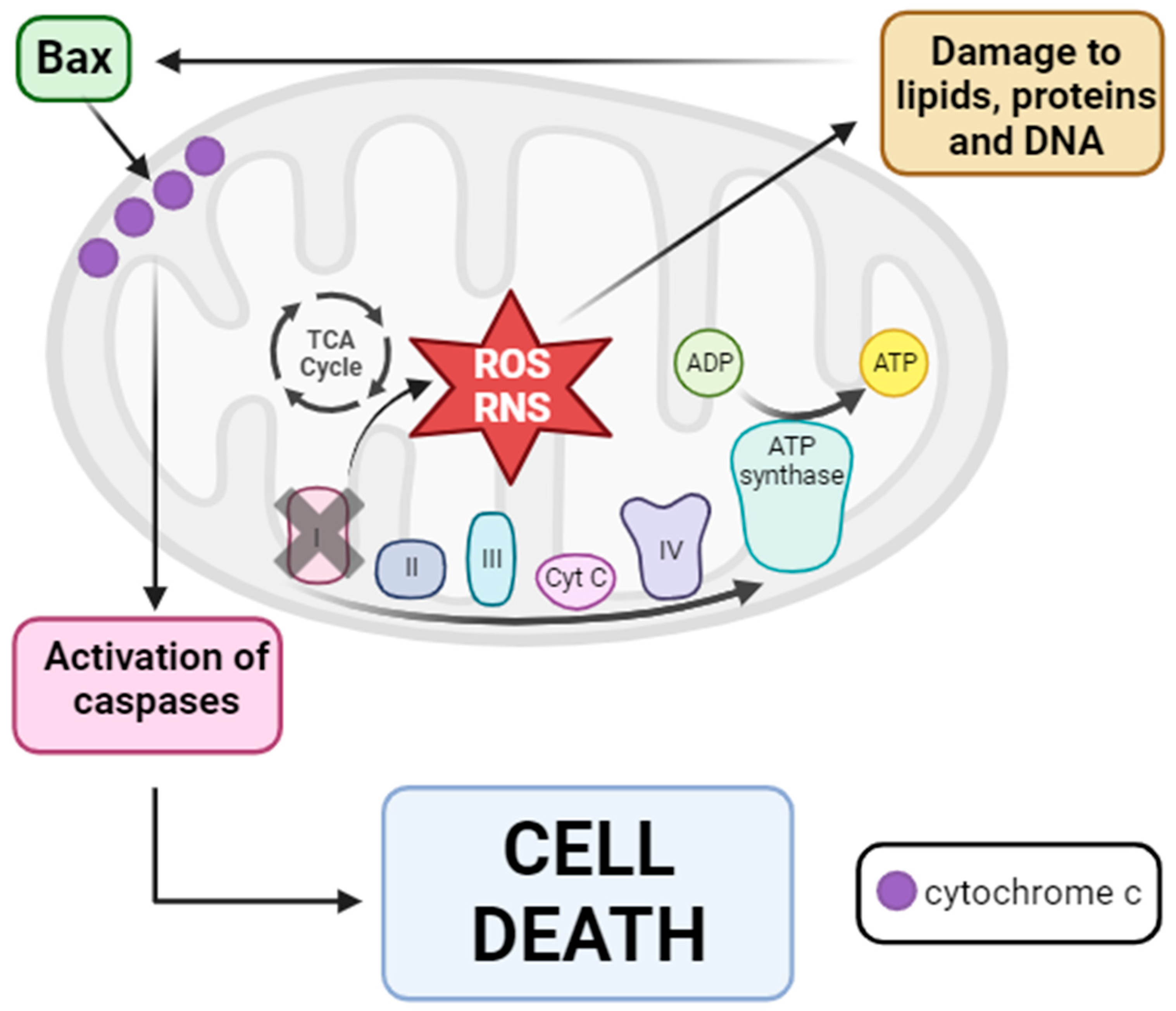
2. Mitochondrial Quality Control
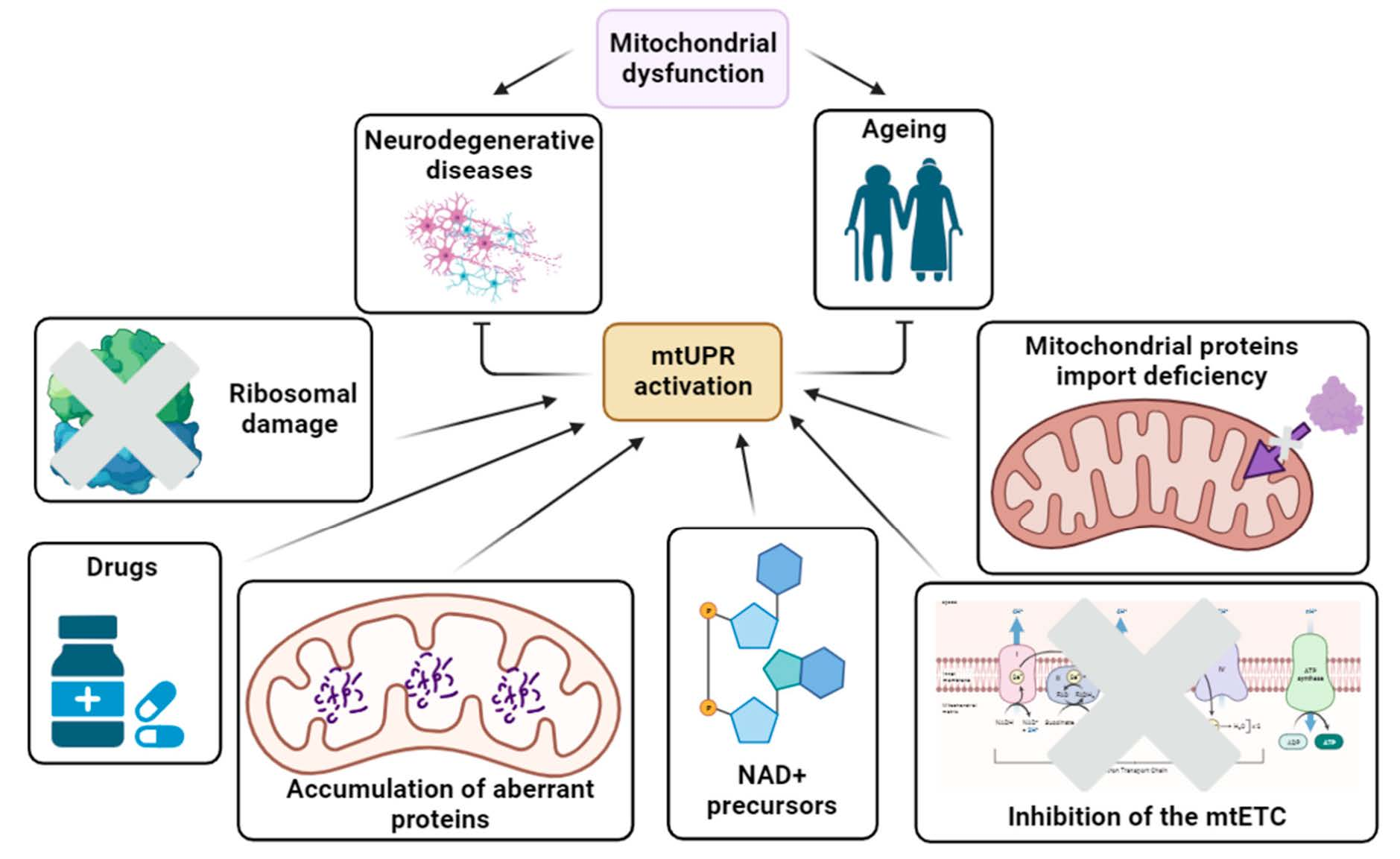
2.1. Mitochondrial Biogenesis
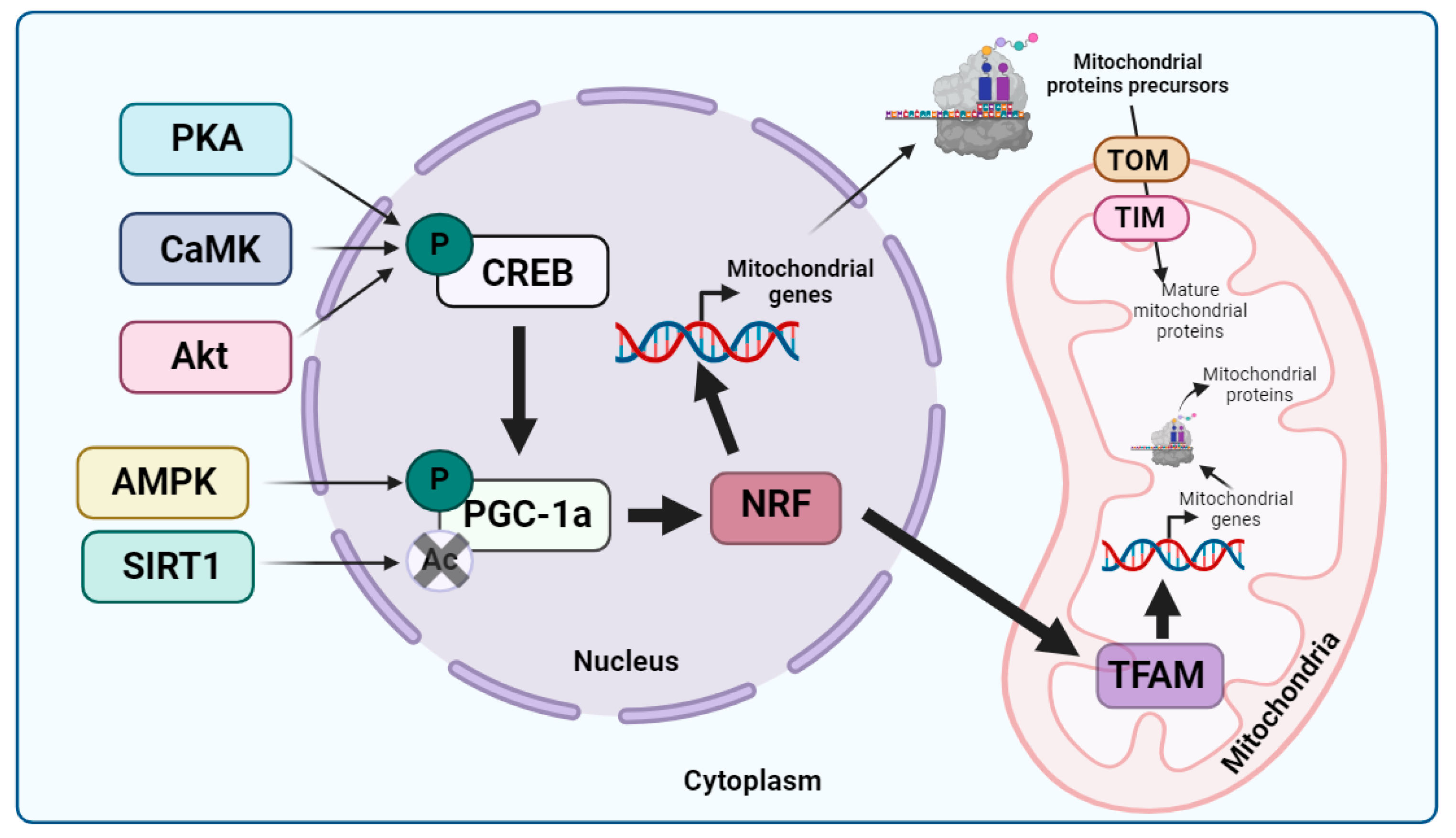
Cytosolic Quality Control of Mitochondrial Protein Import
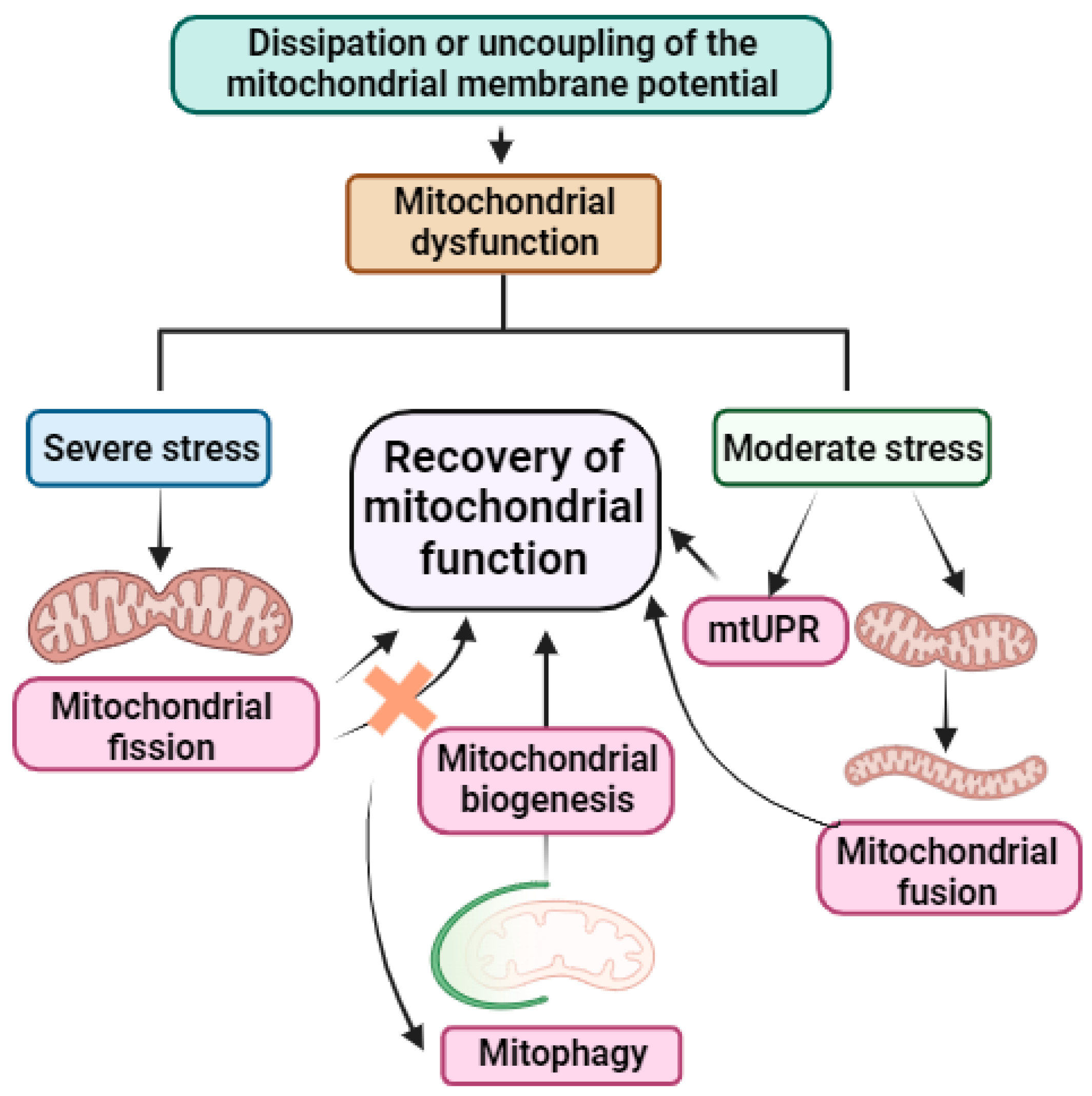
2.2. Mitochondrial Dynamics
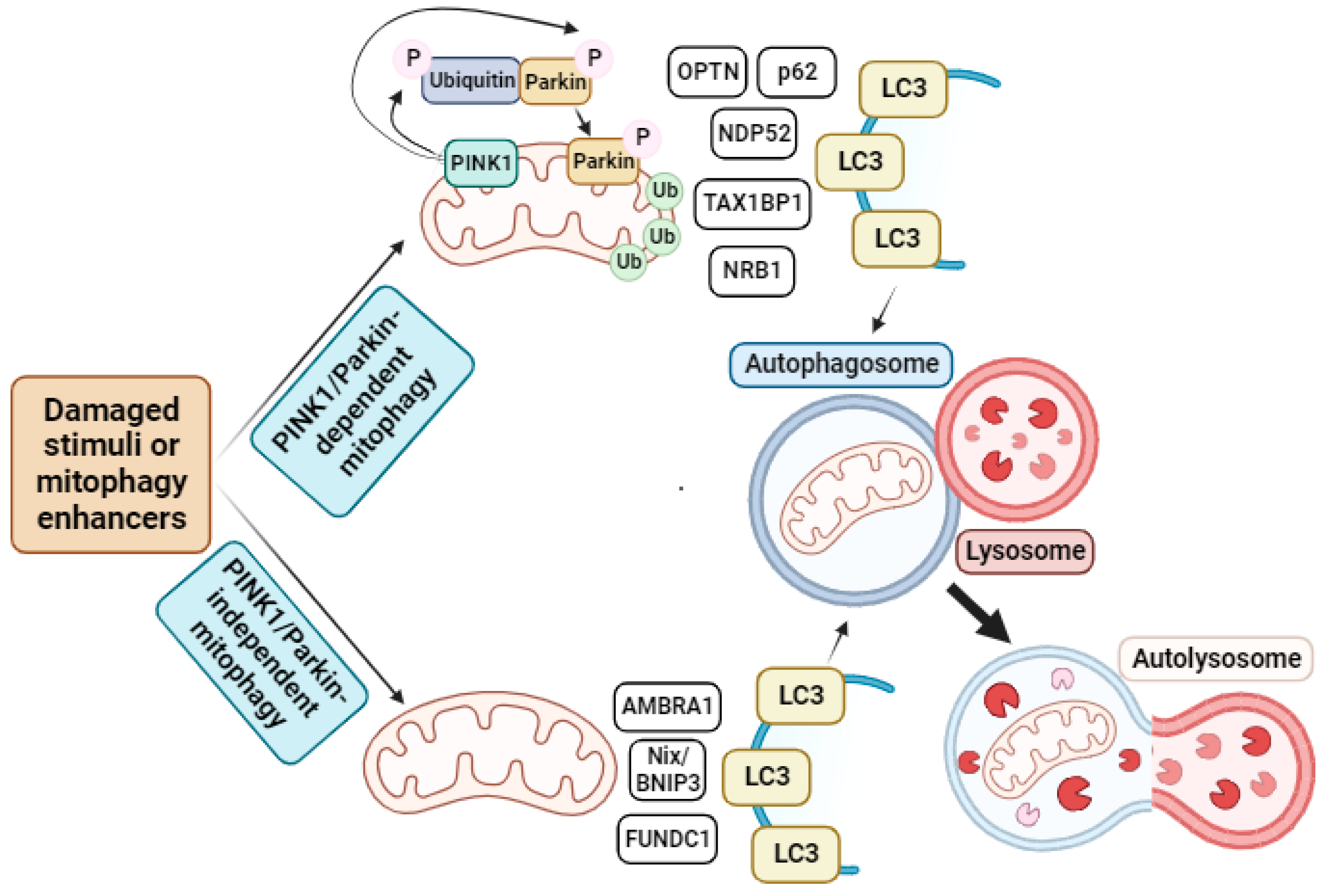
2.3. Mitophagy
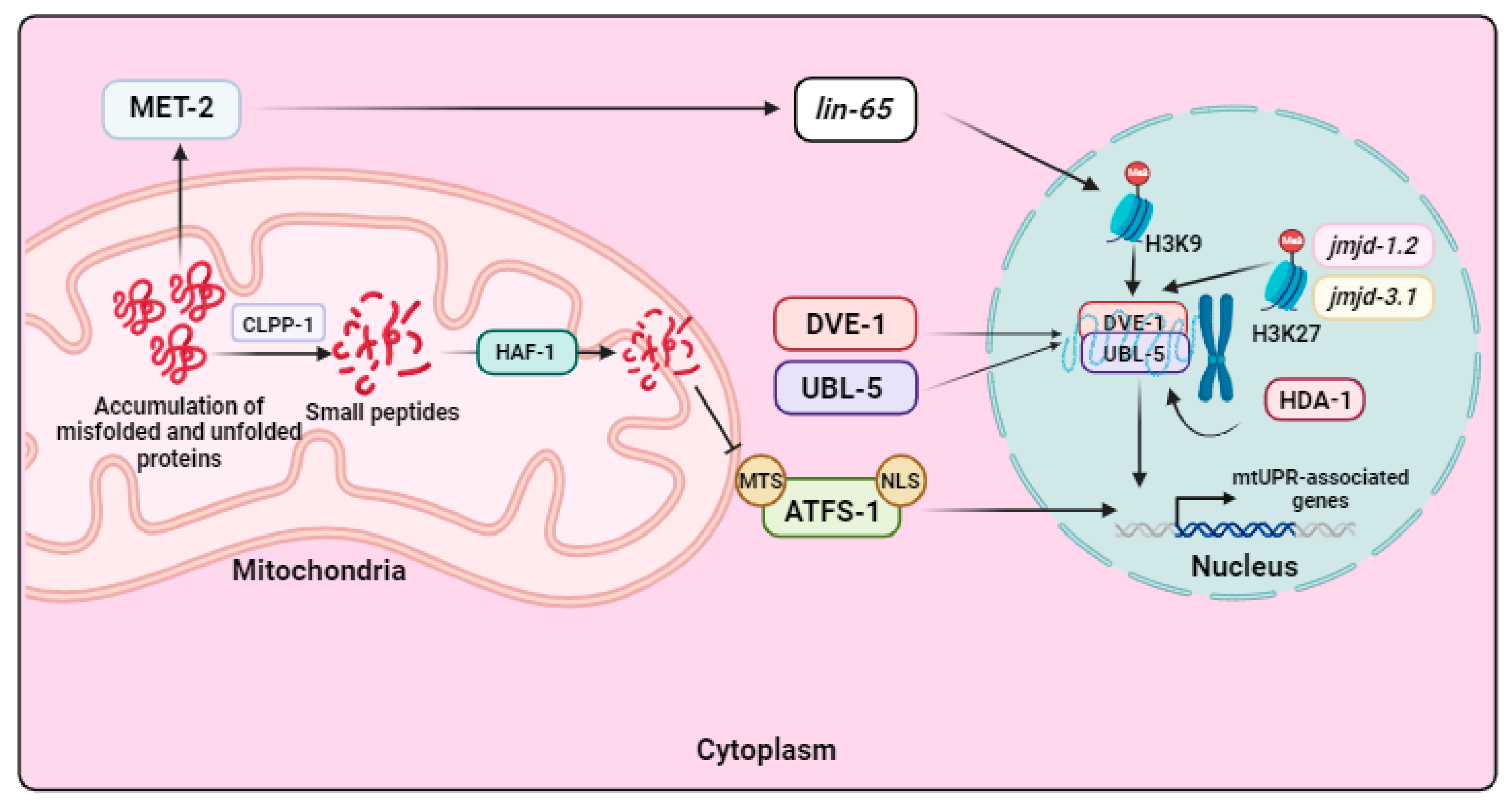
2.4. Mitochondrial Unfolded Protein Response (mtUPR)
3. mtUPR as a Therapeutic Target
3.1. Primary Mitochondrial Diseases
3.2. Cardiac and Metabolic Disorders
3.3. Cancer
3.4. Ageing and Neurodegenerative Diseases
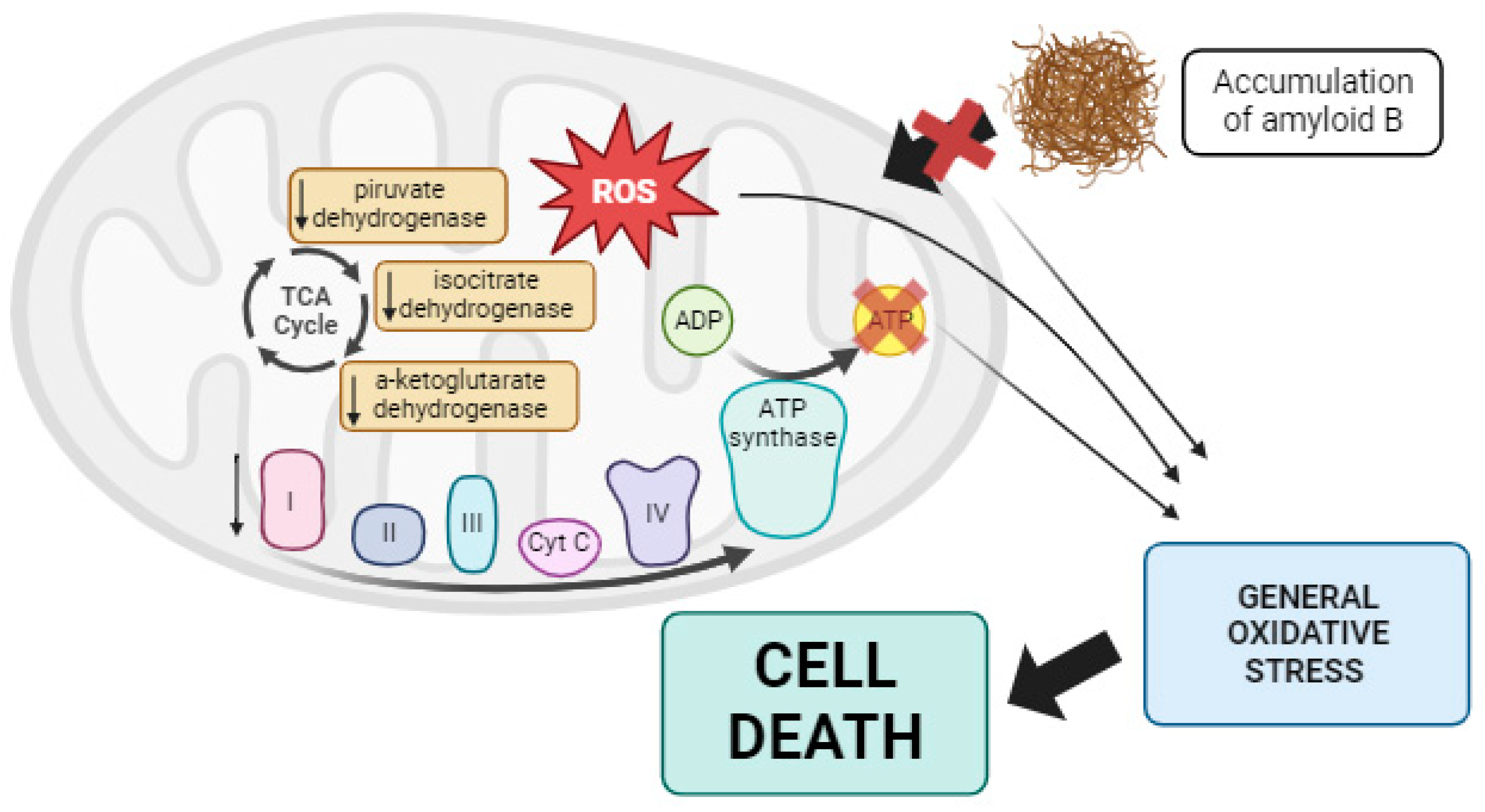
3.4.1. Alzheimer’s Disease
3.4.2. Parkinson’s Disease
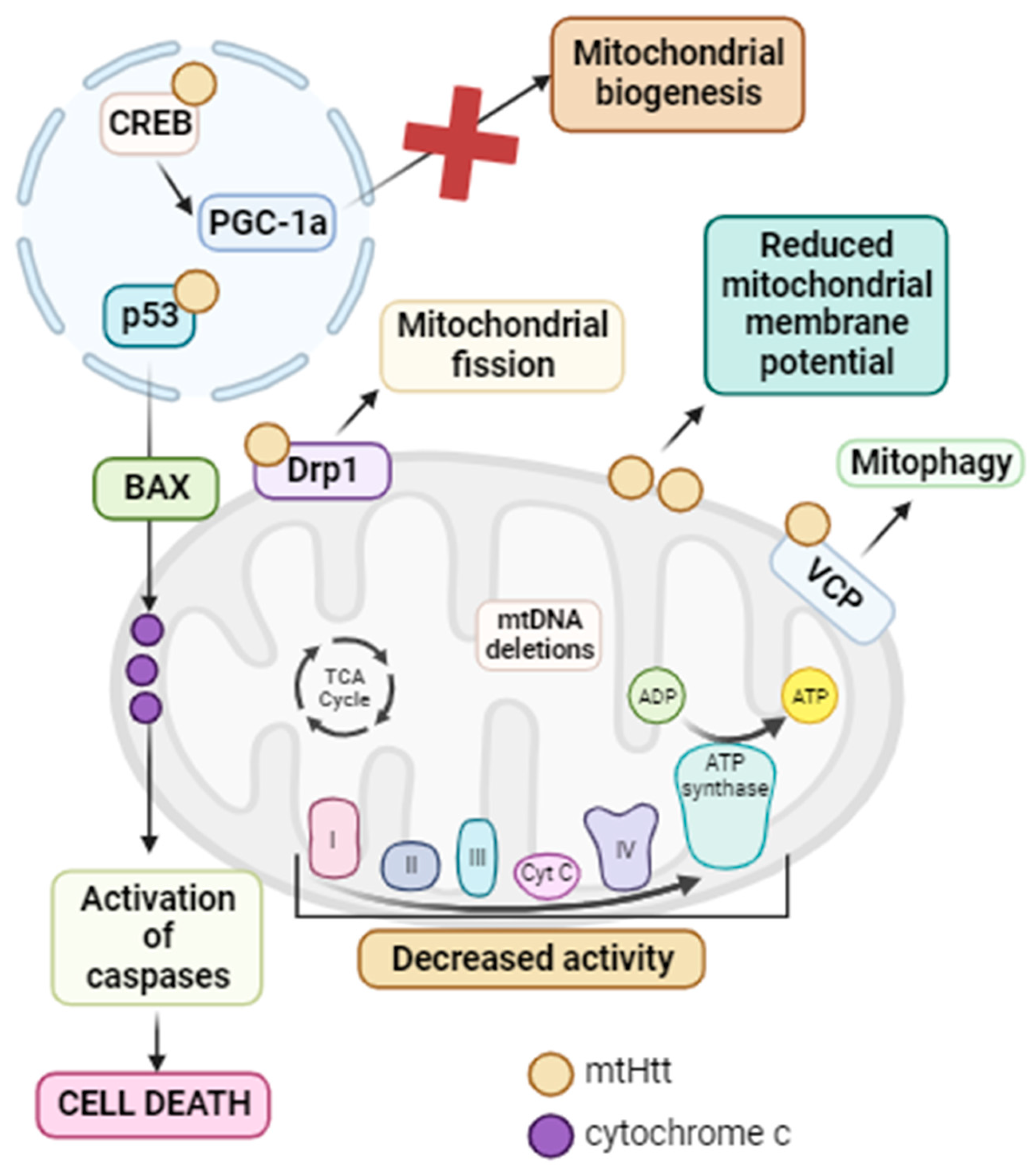
3.4.3. Huntington’s Disease
3.4.4. Amyotrophic Lateral Sclerosis
3.4.5. Friedreich’s Ataxia (FA)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid β |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| AMBRA1 | autophagy and beclin 1 regulator 1 |
| AMPK | AMP-activated kinase |
| APP | amyloid precursor protein |
| ATF4 | Activating Transcription Factor 4 |
| ATF5 | Activating Transcription Factor 5 |
| ATFS-1 | activating transcription factor associated with stress-1 |
| BNIP3 | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 |
| C9orf72 | chromosome 9 open reading frame 72 |
| CaMK | calcium-calcium/calmodulin-dependent protein kinase |
| CHCHD10 | coiled-coil-helix-coiled-coil-helix domain containing 10 |
| CHOP | C/EBP Homologous Protein |
| CLPP-1 | caseinolytic protease P |
| CREB | cAMP response element-binding protein |
| DELE1 | DAP3 Binding Cell Death Enhancer 1 |
| Drp1 | dynamin-related protein 1 |
| DVE-1 | homeodomain-containing protein transcription factor “defective proventiculus” |
| eif2α | eukaryotic translation initiation factor 2α |
| ER | endoplasmic reticulum |
| ERα | estrogen receptor α |
| FA | Friedreich’s Ataxia |
| FIS1 | mitochondrial fission 1 protein |
| FUNDC1 | Fun14 domain-containing protein 1 |
| FUS | fused in sarcoma/translocated in liposarcoma |
| HAF-1 | inner-membrane-spanning ATP-binding casette transporter |
| HD | Huntington’s Disease |
| HSF1 | heat shock transcription factor 1 |
| IMM | inner mitochondrial membrane |
| IMS | intermembrane space |
| ISR | Integrated Stress Response |
| LC3 | microtubule-associated protein 1A/1B light chain 3 |
| LonP1 | Lon protease-like 1 |
| MATR3 | matrin 3 |
| MDVs | mitochondrial-derived vesicles |
| MFF | mitochondrial fission factor |
| Mfn1 | mitofusin 1 |
| Mfn2 | mitofusin 2 |
| MID49 | mitochondrial dynamics protein 49 |
| MID51 | mitochondrial dynamics protein 51 |
| mPOS | mitochondrial precursor overaccumulation stress |
| mtDNA | mitochondrial DNA |
| mtETC | mitochondrial electron transport chain |
| MTS | mitochondrial targeting signal |
| mtUPR | mitochondrial unfolded protein response |
| nDNA | nuclear DNA |
| NLS | nuclear localization signal |
| NRF1 | nuclear respiratory factor 1 |
| NRF2 | nuclear respiratory factor 2 |
| OMM | outer mitochondrial membrane |
| Opa1 | Optic Atrophy 1 |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| PD | Parkinson’s Disease |
| PGC-1α | peroxisome proliferator-activated receptor γ co-activator 1α |
| PINK1 | PTEN-induced kinase 1 |
| PITRM1 | pitrylisin metallopeptidase 1 |
| PKA | protein kinase A |
| PLEKHM1 | pleckstrin homology domain-containing family M member 1 |
| ROS | reactive oxygen species |
| SIMH | stress-induced mitochondrial hyperfusion |
| SLP-2 | stromatin-like protein 2 |
| SOD1 | superoxide dismutase 1 |
| SOD2 | superoxide dismutase 2 |
| SSBP1 | single-stranded DNA binding protein 1 |
| TAX1BP1 | Tax1-binding protein 1 |
| TCA | tricarboxylic acid |
| TDP-43 | TAR DNA binding protein 43 |
| TFAM | mitochondrial transcription factor A |
| TIM | translocase of the inner membrane |
| TIMMDC1 | IMM-domain containing 1 |
| TOM | translocase of the outer membrane |
| UBL-5 | ubiquitin-like protein 5 |
| UPRam | Unfolded Protein Response activated by mistargeting of proteins |
| UPRER | Unfolded Protein Response of the endoplasmic reticulum |
| UV-C | ultraviolet C |
| VDAC1 | voltage/dependent anion channel 1 |
References
- Roger, A.J.; Munoz-Gomez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef]
- Deshpande, O.A.; Mohiuddin, S.S. Biochemistry, Oxidative Phosphorylation. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chandel, N.S. Mitochondria. Cold Spring Harb. Perspect. Biol. 2021, 13, a040543. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLlife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Muhlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef] [PubMed]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef]
- Guda, P.; Guda, C.; Subramaniam, S. Reconstruction of pathways associated with amino acid metabolism in human mitochondria. Genom. Proteom. Bioinform. 2007, 5, 166–176. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Suhm, T.; Kaimal, J.M.; Dawitz, H.; Peselj, C.; Masser, A.E.; Hanzen, S.; Ambrozic, M.; Smialowska, A.; Bjorck, M.L.; Brzezinski, P.; et al. Mitochondrial Translation Efficiency Controls Cytoplasmic Protein Homeostasis. Cell Metab. 2018, 27, 1309–1322.e1306. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W. The Significance of Mitochondrial Dysfunction in Cancer. Int. J. Mol. Sci. 2020, 21, 5598. [Google Scholar] [CrossRef]
- Lenkiewicz, A.M.; Krakowczyk, M.; Bragoszewski, P. Cytosolic Quality Control of Mitochondrial Protein Precursors-The Early Stages of the Organelle Biogenesis. Int. J. Mol. Sci. 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial-derived vesicles: Recent insights. J. Cell Mol. Med. 2022, 26, 3323–3328. [Google Scholar] [CrossRef]
- Naresh, N.U.; Haynes, C.M. Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033944. [Google Scholar] [CrossRef]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Ploumi, C.; Daskalaki, I.; Tavernarakis, N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J. 2017, 284, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1alpha and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Zhao, Q.; Tian, Z.; Zhou, G.; Niu, Q.; Chen, J.; Li, P.; Dong, L.; Xia, T.; Zhang, S.; Wang, A. SIRT1-dependent mitochondrial biogenesis supports therapeutic effects of resveratrol against neurodevelopment damage by fluoride. Theranostics 2020, 10, 4822–4838. [Google Scholar] [CrossRef]
- Bouchez, C.; Devin, A. Mitochondrial Biogenesis and Mitochondrial Reactive Oxygen Species (ROS): A Complex Relationship Regulated by the cAMP/PKA Signaling Pathway. Cells 2019, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R. The role of calcium and calcium/calmodulin-dependent kinases in skeletal muscle plasticity and mitochondrial biogenesis. Proc. Nutr. Soc. 2004, 63, 279–286. [Google Scholar] [CrossRef]
- Mehta, S.L.; Mendelev, N.; Kumari, S.; Andy Li, P. Overexpression of human selenoprotein H in neuronal cells enhances mitochondrial biogenesis and function through activation of protein kinase A, protein kinase B, and cyclic adenosine monophosphate response element-binding protein pathway. Int. J. Biochem. Cell Biol. 2013, 45, 604–611. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J. Biol. Chem. 2006, 281, 324–333. [Google Scholar] [CrossRef]
- Ahting, U.; Thieffry, M.; Engelhardt, H.; Hegerl, R.; Neupert, W.; Nussberger, S. Tom40, the pore-forming component of the protein-conducting TOM channel in the outer membrane of mitochondria. J. Cell Biol. 2001, 153, 1151–1160. [Google Scholar] [CrossRef]
- Schendzielorz, A.B.; Schulz, C.; Lytovchenko, O.; Clancy, A.; Guiard, B.; Ieva, R.; van der Laan, M.; Rehling, P. Two distinct membrane potential-dependent steps drive mitochondrial matrix protein translocation. J. Cell Biol. 2017, 216, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Craig, E.A.; Zufall, N.; von Ahsen, O.; Pfanner, N.; Voos, W. The protein import motor of mitochondria: Unfolding and trapping of preproteins are distinct and separable functions of matrix Hsp70. Cell 1999, 97, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Meisinger, C.; Vogtle, F.N. Processing of mitochondrial presequences. Biochim. Biophys. Acta 2012, 1819, 1098–1106. [Google Scholar] [CrossRef]
- Brix, J.; Rudiger, S.; Bukau, B.; Schneider-Mergener, J.; Pfanner, N. Distribution of binding sequences for the mitochondrial import receptors Tom20, Tom22, and Tom70 in a presequence-carrying preprotein and a non-cleavable preprotein. J. Biol. Chem. 1999, 274, 16522–16530. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial protein import: Common principles and physiological networks. Biochim. Biophys. Acta 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Grevel, A.; Pfanner, N.; Becker, T. Coupling of import and assembly pathways in mitochondrial protein biogenesis. Biol. Chem. 2019, 401, 117–129. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef]
- Bragoszewski, P.; Gornicka, A.; Sztolsztener, M.E.; Chacinska, A. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol. Cell Biol. 2013, 33, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, X.J. A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 2015, 524, 481–484. [Google Scholar] [CrossRef]
- Rolland, S.G.; Schneid, S.; Schwarz, M.; Rackles, E.; Fischer, C.; Haeussler, S.; Regmi, S.G.; Yeroslaviz, A.; Habermann, B.; Mokranjac, D.; et al. Compromised Mitochondrial Protein Import Acts as a Signal for UPR(mt). Cell Rep. 2019, 28, 1659–1669.e5. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Nelson, G.M.; Watson, J.L.; Morf, J.; Dalglish, M.; Luh, L.M.; Weber, A.; Bertolotti, A. Protein Stability Buffers the Cost of Translation Attenuation following eIF2alpha Phosphorylation. Cell Rep. 2020, 32, 108154. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, M.; Arnoult, D.; Chen, H.; Chan, D.C.; Smith, C.L.; Youle, R.J. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 2004, 164, 493–499. [Google Scholar] [CrossRef]
- van der Bliek, A.M. Fussy mitochondria fuse in response to stress. EMBO J. 2009, 28, 1533–1534. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, J.; Saunders, J.M.; Moraes, V.W.R.; Madhavan, A.; Madrazo, N.; Anthony, M.C.; Wiseman, R.L. The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Rep. 2018, 22, 2827–2836. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Frohlich, C.; Grabiger, S.; Schwefel, D.; Faelber, K.; Rosenbaum, E.; Mears, J.; Rocks, O.; Daumke, O. Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 2013, 32, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Ding, H.; Jiang, N.; Liu, H.; Liu, X.; Liu, D.; Zhao, F.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Response of mitochondrial fusion and fission protein gene expression to exercise in rat skeletal muscle. Biochim. Biophys. Acta 2010, 1800, 250–256. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, K.; Li, J.; Cui, L.; Dong, J.; Li, J.; Meng, X.; Zhu, G.; Wang, H. PINK1/Parkin-mediated mitophagy enhances the survival of Staphylococcus aureus in bovine macrophages. J. Cell Mol. Med. 2023, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Sulkshane, P.; Ram, J.; Thakur, A.; Reis, N.; Kleifeld, O.; Glickman, M.H. Ubiquitination and receptor-mediated mitophagy converge to eliminate oxidation-damaged mitochondria during hypoxia. Redox Biol. 2021, 45, 102047. [Google Scholar] [CrossRef]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Denk, D.; Petrocelli, V.; Conche, C.; Drachsler, M.; Ziegler, P.K.; Braun, A.; Kress, A.; Nicolas, A.M.; Mohs, K.; Becker, C.; et al. Expansion of T memory stem cells with superior anti-tumor immunity by Urolithin A-induced mitophagy. Immunity 2022, 55, 2059–2073.e2058. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Hindle, A.; Kshirsagar, S.; Reddy, P.H. Are mitophagy enhancers therapeutic targets for Alzheimer’s disease? Biomed. Pharmacother. 2022, 149, 112918. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef]
- Liu, K.; Jing, M.J.; Liu, C.; Yan, D.Y.; Ma, Z.; Wang, C.; Deng, Y.; Liu, W.; Xu, B. Effect of trehalose on manganese-induced mitochondrial dysfunction and neuronal cell damage in mice. Basic. Clin. Pharmacol. Toxicol. 2019, 125, 536–547. [Google Scholar] [CrossRef]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Chuang, Y.C.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef]
- Martin-Maestro, P.; Sproul, A.; Martinez, H.; Paquet, D.; Gerges, M.; Noggle, S.; Starkov, A.A. Autophagy Induction by Bexarotene Promotes Mitophagy in Presenilin 1 Familial Alzheimer’s Disease iPSC-Derived Neural Stem Cells. Mol. Neurobiol. 2019, 56, 8220–8236. [Google Scholar] [CrossRef]
- Wang, N.; Wang, H.; Pan, Q.; Kang, J.; Liang, Z.; Zhang, R. The Combination of beta-Asarone and Icariin Inhibits Amyloid-beta and Reverses Cognitive Deficits by Promoting Mitophagy in Models of Alzheimer’s Disease. Oxid. Med. Cell Longev. 2021, 2021, 7158444. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Z.; Wang, H.; Zhang, D.; Han, T.; Chen, H.; Chen, J.; Chen, Z.; Xie, Y.; Wang, L.; et al. Melatonin promotes peripheral nerve repair through Parkin-mediated mitophagy. Free Radic. Biol. Med. 2022, 185, 52–66. [Google Scholar] [CrossRef]
- Cen, X.; Xu, X.; Xia, H. Targeting MCL1 to induce mitophagy is a potential therapeutic strategy for Alzheimer disease. Autophagy 2021, 17, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Ai, R.; Zhuang, X.X.; Anisimov, A.; Lu, J.H.; Fang, E.F. A synergized machine learning plus cross-species wet-lab validation approach identifies neuronal mitophagy inducers inhibiting Alzheimer disease. Autophagy 2022, 18, 939–941. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55.e46. [Google Scholar] [CrossRef]
- Quiles, J.M.; Gustafsson, Å.B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Wanet, A.; De Pauw, A.; Rommelaere, G.; Arnould, T.; Renard, P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J. Cell Physiol. 2012, 227, 2297–2310. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial unfolded protein response: An emerging pathway in human diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef]
- Haynes, C.M.; Petrova, K.; Benedetti, C.; Yang, Y.; Ron, D. ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 2007, 13, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Yang, Y.; Blais, S.P.; Neubert, T.A.; Ron, D. The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 2010, 37, 529–540. [Google Scholar] [CrossRef]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef]
- Benedetti, C.; Haynes, C.M.; Yang, Y.; Harding, H.P.; Ron, D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 2006, 174, 229–239. [Google Scholar] [CrossRef]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.W.; Peng, Q.; Dong, M.; Gao, K.; Li, Y.; Li, Y.; Li, C.Y.; Liu, Y. Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 2020, 11, 4639. [Google Scholar] [CrossRef]
- Tian, Y.; Garcia, G.; Bian, Q.; Steffen, K.K.; Joe, L.; Wolff, S.; Meyer, B.J.; Dillin, A. Mitochondrial Stress Induces Chromatin Reorganization to Promote Longevity and UPR(mt). Cell 2016, 165, 1197–1208. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Berendzen, K.M.; Durieux, J.; Shao, L.W.; Tian, Y.; Kim, H.E.; Wolff, S.; Liu, Y.; Dillin, A. Neuroendocrine Coordination of Mitochondrial Stress Signaling and Proteostasis. Cell 2016, 166, 1553–1563.e1510. [Google Scholar] [CrossRef]
- Cilleros-Holgado, P.; Gomez-Fernandez, D.; Pinero-Perez, R.; Reche-Lopez, D.; Alvarez-Cordoba, M.; Munuera-Cabeza, M.; Talaveron-Rey, M.; Povea-Cabello, S.; Suarez-Carrillo, A.; Romero-Gonzalez, A.; et al. mtUPR Modulation as a Therapeutic Target for Primary and Secondary Mitochondrial Diseases. Int. J. Mol. Sci. 2023, 24, 1482. [Google Scholar] [CrossRef]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Horibe, T.; Hoogenraad, N.J. The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2007, 2, e835. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Al-Furoukh, N.; Ianni, A.; Nolte, H.; Holper, S.; Kruger, M.; Wanrooij, S.; Braun, T. ClpX stimulates the mitochondrial unfolded protein response (UPRmt) in mammalian cells. Biochim. Biophys. Acta 2015, 1853, 2580–2591. [Google Scholar] [CrossRef]
- Kang, Z.; Chen, F.; Wu, W.; Liu, R.; Chen, T.; Xu, F. UPR(mt) and coordinated UPR(ER) in type 2 diabetes. Front. Cell Dev. Biol. 2022, 10, 974083. [Google Scholar] [CrossRef] [PubMed]
- Mesbah Moosavi, Z.S.; Hood, D.A. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef]
- Anderson, N.S.; Haynes, C.M. Folding the Mitochondrial UPR into the Integrated Stress Response. Trends Cell Biol. 2020, 30, 428–439. [Google Scholar] [CrossRef]
- Fessler, E.; Krumwiede, L.; Jae, L.T. DELE1 tracks perturbed protein import and processing in human mitochondria. Nat. Commun. 2022, 13, 1853. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Fujimoto, M.; Takii, R.; Takaki, E.; Hayashida, N.; Nakai, A. Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat. Commun. 2015, 6, 6580. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713. [Google Scholar] [CrossRef]
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor. Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Watroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Ma, Y.; Chen, L.; Cai, G.; Chen, Z.; Zhang, S.; Ye, Q. A New Vision of Mitochondrial Unfolded Protein Response to the Sirtuin Family. Curr. Neuropharmacol. 2020, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S. Emerging therapeutic roles for NAD(+) metabolism in mitochondrial and age-related disorders. Clin. Transl. Med. 2016, 5, 25. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Papa, L.; Germain, D. Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 2011, 124, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, J.E.; Horibe, T.; Hoogenraad, N.J. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2007, 2, e874. [Google Scholar] [CrossRef]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef]
- Tao, R.; Vassilopoulos, A.; Parisiadou, L.; Yan, Y.; Gius, D. Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid. Redox Signal 2014, 20, 1646–1654. [Google Scholar] [CrossRef]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef]
- Wang, Y.; Jasper, H.; Toan, S.; Muid, D.; Chang, X.; Zhou, H. Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury. Redox Biol. 2021, 45, 102049. [Google Scholar] [CrossRef]
- Lin, Y.F.; Schulz, A.M.; Pellegrino, M.W.; Lu, Y.; Shaham, S.; Haynes, C.M. Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 2016, 533, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fan, Y.; Zong, R.; Tan, K. The mitochondrial unfolded protein response: A multitasking giant in the fight against human diseases. Ageing Res. Rev. 2022, 81, 101702. [Google Scholar] [CrossRef]
- Yi, H.S. Implications of Mitochondrial Unfolded Protein Response and Mitokines: A Perspective on Fatty Liver Diseases. Endocrinol. Metab. 2019, 34, 39–46. [Google Scholar] [CrossRef]
- Yi, H.S.; Chang, J.Y.; Shong, M. The mitochondrial unfolded protein response and mitohormesis: A perspective on metabolic diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [PubMed]
- Nargund, A.M.; Fiorese, C.J.; Pellegrino, M.W.; Deng, P.; Haynes, C.M. Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 2015, 58, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Luo, S.; Chen, W.; He, L.; Liu, D.; Wang, X.; Xiao, L.; Sun, L. Mitochondrial unfolded protein response (mtUPR) and diseases. Curr. Med. Chem. 2023. E-pub Ahead of Print. [Google Scholar] [CrossRef]
- Lu, H.; Wang, X.; Li, M.; Ji, D.; Liang, D.; Liang, C.; Liu, Y.; Zhang, Z.; Cao, Y.; Zou, W. Mitochondrial Unfolded Protein Response and Integrated Stress Response as Promising Therapeutic Targets for Mitochondrial Diseases. Cells 2022, 12, 20. [Google Scholar] [CrossRef]
- Perry, E.A.; Bennett, C.F.; Luo, C.; Balsa, E.; Jedrychowski, M.; O’Malley, K.E.; Latorre-Muro, P.; Ladley, R.P.; Reda, K.; Wright, P.M.; et al. Tetracyclines promote survival and fitness in mitochondrial disease models. Nat. Metab. 2021, 3, 33–42. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; et al. UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 2022, 17, 204. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Romero-Gonzalez, A.; Gomez-Fernandez, D.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; et al. Pterostilbene in Combination With Mitochondrial Cofactors Improve Mitochondrial Function in Cellular Models of Mitochondrial Diseases. Front. Pharmacol. 2022, 13, 862085. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Talaveron-Rey, M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Sanchez-Alcazar, J.A. Mitochondria and Antibiotics: For Good or for Evil? Biomolecules 2021, 11, 1050. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, Z.; Yan, X.; Liu, J.; Li, F.; Liu, P.; Li, J.; Wei, Y.; Sun, Y.E.; Zhang, Y.; et al. ATF5, a putative therapeutic target for the mitochondrial DNA 3243A > G mutation-related disease. Cell Death Dis. 2021, 12, 701. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, X.; Zheng, S.; Zhu, A.; Wang, J. The Mitochondrial Unfolded Protein Response: A Novel Protective Pathway Targeting Cardiomyocytes. Oxid. Med. Cell Longev. 2022, 2022, 6430342. [Google Scholar] [CrossRef]
- Wang, Y.T.; Lim, Y.; McCall, M.N.; Huang, K.T.; Haynes, C.M.; Nehrke, K.; Brookes, P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H472–H478. [Google Scholar] [CrossRef] [PubMed]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef]
- Xu, M.; Xue, R.Q.; Lu, Y.; Yong, S.Y.; Wu, Q.; Cui, Y.L.; Zuo, X.T.; Yu, X.J.; Zhao, M.; Zang, W.J. Choline ameliorates cardiac hypertrophy by regulating metabolic remodelling and UPRmt through SIRT3-AMPK pathway. Cardiovasc. Res. 2019, 115, 530–545. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tan, Y.; Zhang, Z.; Feng, P.; Ding, W.; Wang, Q.; Liang, H.; Duan, W.; Wang, X.; Yu, S.; et al. Novel PGC-1alpha/ATF5 Axis Partly Activates UPR(mt) and Mediates Cardioprotective Role of Tetrahydrocurcumin in Pathological Cardiac Hypertrophy. Oxid. Med. Cell Longev. 2020, 2020, 9187065. [Google Scholar] [CrossRef]
- Sreekumar, R.; Halvatsiotis, P.; Schimke, J.C.; Nair, K.S. Gene expression profile in skeletal muscle of type 2 diabetes and the effect of insulin treatment. Diabetes 2002, 51, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef] [PubMed]
- Wardelmann, K.; Blumel, S.; Rath, M.; Alfine, E.; Chudoba, C.; Schell, M.; Cai, W.; Hauffe, R.; Warnke, K.; Flore, T.; et al. Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 2019, 21, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Chung, K.; Lee, H.; Lee, K.; Lim, J.H.; Song, J. Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef]
- Wu, G.; Xiong, Q.; Wei, X.; Wang, Y.; Hu, X.; He, G.; Liu, L.; Lai, Q.; Dai, Z.; Anushesh, D.; et al. Mitochondrial unfolded protein response gene CLPP changes mitochondrial dynamics and affects mitochondrial function. PeerJ 2019, 7, e7209. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Lauritzen, H.P.; Ussar, S.; Christensen, J.H.; Mori, M.A.; Bross, P.; Kahn, C.R. Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Investig. 2013, 123, 4667–4680. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef]
- Ghosh, J.C.; Seo, J.H.; Agarwal, E.; Wang, Y.; Kossenkov, A.V.; Tang, H.Y.; Speicher, D.W.; Altieri, D.C. Akt phosphorylation of mitochondrial Lonp1 protease enables oxidative metabolism and advanced tumor traits. Oncogene 2019, 38, 6926–6939. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s disease: From genetic variants to the distinct pathological mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Luk, J.M.; Lee, N.P.; Peng, J.; Leng, X.; Guan, X.Y.; Lau, G.K.; Beretta, L.; Fan, S.T. Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol. Cell. Proteom. 2008, 7, 315–325. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xiao, H.; Cai, Y.; Zhang, N. ATF5 and HIF1α cooperatively activate HIF1 signaling pathway in esophageal cancer. Cell Commun. Signal. 2021, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Qian, D.; Wang, B.; Hu, M.; Lu, J.; Qi, Y.; Liu, D.X. ATF5 is overexpressed in epithelial ovarian carcinomas and interference with its function increases apoptosis through the downregulation of Bcl-2 in SKOV-3 Cells. Int. J. Gynecol. Pathol. 2012, 31, 532–537. [Google Scholar] [CrossRef]
- Kim, W.; Ryu, J.; Kim, J.E. CCAR2/DBC1 and Hsp60 positively regulate expression of survivin in neuroblastoma cells. Int. J. Mol. Sci. 2019, 20, 131. [Google Scholar] [CrossRef]
- Luo, J.; Zeng, B.; Tao, C.; Lu, M.; Ren, G. ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 2020, 2020, e8754. [Google Scholar] [CrossRef]
- Tang, H.; Li, J.; Liu, X.; Wang, G.; Luo, M.; Deng, H. Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. Sci. Rep. 2016, 6, 28388. [Google Scholar] [CrossRef] [PubMed]
- Feldheim, J.; Kessler, A.F.; Schmitt, D.; Wilczek, L.; Linsenmann, T.; Dahlmann, M.; Monoranu, C.M.; Ernestus, R.I.; Hagemann, C.; Löhr, M. Expression of activating transcription factor 5 (ATF5) is increased in astrocytomas of different WHO grades and correlates with survival of glioblastoma patients. OncoTargets Ther. 2018, 11, 8673–8684. [Google Scholar] [CrossRef]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. Heat shock protein 60 overexpression is associated with the progression and prognosis in gastric cancer. PLoS ONE 2014, 9, e107507. [Google Scholar] [CrossRef]
- Kumar, S.; O’Malley, J.; Chaudhary, A.K.; Inigo, J.R.; Yadav, N.; Kumar, R.; Chandra, D. Hsp60 and IL-8 axis promotes apoptosis resistance in cancer. Br. J. Cancer 2019, 121, 934–943. [Google Scholar] [CrossRef]
- Guo, J.; Li, X.; Zhang, W.; Chen, Y.; Zhu, S.; Chen, L.; Xu, R.; Lv, Y.; Wu, D.; Guo, M.; et al. HSP60-regulated Mitochondrial Proteostasis and Protein Translation Promote Tumor Growth of Ovarian Cancer. Sci. Rep. 2019, 9, 6792. [Google Scholar] [CrossRef] [PubMed]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.; Wang, Z.; Coyaud, E.; Voisin, V.; Gronda, M.; Jitkova, Y.; Mattson, R.; Hurren, R.; Babovic, S.; Maclean, N.; et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2015, 27, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Losi, L.; De Biasi, S.; Nasi, M.; Tartaro, D.L.; Pecorini, S.; Patergnani, S.; Pinton, P.; De Gaetano, A.; Carnevale, G.; et al. LonP1 differently modulates mitochondrial function and bioenergetics of primary versus metastatic colon cancer cells. Front. Oncol. 2018, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.S.; Porter, J.W. Mechanism of fatty acid synthesis. Life Sci. 1977, 20, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Kao, T.Y.; Chiu, Y.C.; Fang, W.C.; Cheng, C.W.; Kuo, C.Y.; Juan, H.F.; Wu, S.H.; Lee, A.Y.L. Mitochondrial Lon regulates apoptosis through the association with Hsp60-mtHsp70 complex. Cell Death Dis. 2015, 6, e1642. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial metabolism in major neurological diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef]
- Starenki, D.; Sosonkina, N.; Hong, S.K.; Lloyd, R.V.; Park, J.I. Mortalin (GRP75/HSPA9) promotes survival and proliferation of thyroid carcinoma cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef]
- Sun, J.; Che, S.L.; Piao, J.J.; Xu, M.; Chen, L.Y.; Lin, Z.H. Mortalin overexpression predicts poor prognosis in early stage of non–small cell lung cancer. Tumor Biol. 2017, 39, 1010428317695918. [Google Scholar] [CrossRef]
- Liu, L.X.; Lu, J.C.; Zeng, H.Y.; Cai, J.B.; Zhang, P.F.; Guo, X.J.; Huang, X.Y.; Dong, R.Z.; Zhang, C.; Kang, Q.; et al. Mortalin stabilizes CD151-depedent tetraspanin-enriched microdomains and implicates in the progression of hepatocellular carcinoma. J. Cancer 2019, 10, 6199–6206. [Google Scholar] [CrossRef]
- Vocka, M.; Langer, D.; Fryba, V.; Petrtyl, J.; Hanus, T.; Kalousova, M.; Zima, T.; Petruzelka, L. Novel serum markers HSP60, CHI3L1, and IGFBP-2 in metastatic colorectal cancer. Oncol. Lett. 2019, 18, 6284–6292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Zheng, C.; Gao, J.; Fu, Q.; Hu, N.; Shao, X.; Zhou, Y.; Xiong, J.; Nie, K.; et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth article. Cell Death Dis. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, L.; Yang, Y.; Han, Y.; Wang, Y.; Liu, W.; Zuo, J. Oncogenic role of mortalin contributes to ovarian tumorigenesis by activating the MAPK–ERK pathway. J. Cell. Mol. Med. 2016, 20, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Meng, W.; Zhou, Z.; Li, Y.; Zhou, B.; Wang, R.; Zhan, L. Overexpression of activating transcription factor 5 in human rectal cancer. Exp. Ther. Med. 2011, 2, 827–831. [Google Scholar] [CrossRef]
- Hua, Z.Y.; Hansen, J.N.; He, M.; Dai, S.K.; Choi, Y.; Fulton, M.D.; Lloyd, S.M.; Szemes, M.; Sen, J.; Ding, H.F.; et al. PRMT1 promotes neuroblastoma cell survival through ATF5. Oncogenesis 2020, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, S.A.; Eidelman, A.S.; Ewer, J.C.; Ricca, J.M.; Serrano, A.; Tucker, K.C.; Vail, C.M.; Kurt, R.A. A role for HMGB1, HSP60 and Myd88 in growth of murine mammary carcinoma in vitro. Cell. Immunol. 2013, 282, 136–145. [Google Scholar] [CrossRef]
- Nukuda, A.; Endoh, H.; Yasuda, M.; Mizutani, T.; Kawabata, K.; Haga, H. Role of ATF5 in the invasive potential of diverse human cancer cell lines. Biochem. Biophys. Res. Commun. 2016, 474, 509–514. [Google Scholar] [CrossRef]
- Monaco, S.E.; Angelastro, J.M.; Szabolcs, M.; Greene, L.A. The transcription factor ATF5 is widely expressed in carcinomas, and interference with its function selectively kills neoplastic, but not nontransformed, breast cell lines. Int. J. Cancer 2007, 120, 1883–1890. [Google Scholar] [CrossRef]
- Greene, L.A.; Lee, H.Y.; Angelastro, J.M. The transcription factor ATF5: Role in neurodevelopment and neural tumors. J. Neurochem. 2009, 108, 11–22. [Google Scholar] [CrossRef]
- Wadhwa, R.; Takano, S.; Kaur, K.; Deocaris, C.C.; Pereira-Smith, O.M.; Reddel, R.R.; Kaul, S.C. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 2006, 118, 2973–2980. [Google Scholar] [CrossRef]
- Yoon, A.R.; Wadhwa, R.; Kaul, S.C.; Yun, C.O. Why is Mortalin a Potential Therapeutic Target for Cancer? Front. Cell Dev. Biol. 2022, 10, 1219. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.R.; Chen, H.H.; Ko, C.H.; Chan, M.H. Neuroprotective activity of honokiol and magnolol in cerebellar granule cell damage. Eur. J. Pharmacol. 2006, 537, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.U.; Trumpff, C.; Yang, H.S.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol. Neurodegener. 2021, 16, 75. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Mancuso, M.; Siciliano, G.; Filosto, M.; Murri, L. Mitochondrial dysfunction and Alzheimer’s disease: New developments. J. Alzheimers Dis. 2006, 9, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.; Schiavi, A.; Ventura, N. Mitochondrial autophagy promotes healthy aging. Cell Cycle 2016, 15, 1805–1806. [Google Scholar] [CrossRef]
- Yuan, Y.; Cruzat, V.F.; Newsholme, P.; Cheng, J.; Chen, Y.; Lu, Y. Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 2016, 155, 10–21. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef]
- Hashimoto, T.; Horikawa, M.; Nomura, T.; Sakamoto, K. Nicotinamide adenine dinucleotide extends the lifespan of Caenorhabditis elegans mediated by sir-2.1 and daf-16. Biogerontology 2010, 11, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Ozkurede, U.; Miller, R.A. Improved mitochondrial stress response in long-lived Snell dwarf mice. Aging Cell 2019, 18, e13030. [Google Scholar] [CrossRef] [PubMed]
- Baqri, R.M.; Pietron, A.V.; Gokhale, R.H.; Turner, B.A.; Kaguni, L.S.; Shingleton, A.W.; Kunes, S.; Miller, K.E. Mitochondrial chaperone TRAP1 activates the mitochondrial UPR and extends healthspan in Drosophila. Mech. Ageing Dev. 2014, 141–142, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Luce, K.; Osiewacz, H.D. Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat. Cell Biol. 2009, 11, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Uryu, K.; Leight, S.; Trojanoswki, J.Q.; Lee, V.M. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J. Neurosci. 2001, 21, 4183–4187. [Google Scholar] [CrossRef]
- Ohyagi, Y.; Yamada, T.; Nishioka, K.; Clarke, N.J.; Tomlinson, A.J.; Naylor, S.; Nakabeppu, Y.; Kira, J.; Younkin, S.G. Selective increase in cellular A beta 42 is related to apoptosis but not necrosis. Neuroreport 2000, 11, 167–171. [Google Scholar] [CrossRef]
- Busciglio, J.; Pelsman, A.; Wong, C.; Pigino, G.; Yuan, M.; Mori, H.; Yankner, B.A. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 2002, 33, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Delanty, N. Oxidative injury in diseases of the central nervous system: Focus on Alzheimer’s disease. Am. J. Med. 2000, 109, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Haroutunian, V.; Zhang, H.; Park, L.C.; Shi, Q.; Lesser, M.; Mohs, R.C.; Sheu, R.K.; Blass, J.P. Mitochondrial damage in Alzheimer’s disease varies with apolipoprotein E genotype. Ann. Neurol. 2000, 48, 297–303. [Google Scholar] [CrossRef]
- Sorbi, S.; Bird, E.D.; Blass, J.P. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann. Neurol. 1983, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res. Brain Res. Rev. 2005, 49, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S499–S512. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef]
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 18725. [Google Scholar] [CrossRef]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for Mitochondrial UPR Gene Activation in Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Regitz, C.; Fitzenberger, E.; Mahn, F.L.; Dussling, L.M.; Wenzel, U. Resveratrol reduces amyloid-beta (Abeta(1)(-)(4)(2))-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 2016, 55, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2021, 26, 5733–5750. [Google Scholar] [CrossRef] [PubMed]
- Darreh-Shori, T.; Rezaeianyazdi, S.; Lana, E.; Mitra, S.; Gellerbring, A.; Karami, A.; Bogdanovic, N.; Lithner, C.U.; Winblad, B.; Behbahani, H. Increased Active OMI/HTRA2 Serine Protease Displays a Positive Correlation with Cholinergic Alterations in the Alzheimer’s Disease Brain. Mol. Neurobiol. 2019, 56, 4601–4619. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ding, M.; Xie, Z.; Liu, X.; Yang, H.; Jin, S.; Xu, S.; Zhu, Z.; Wang, Y.; Wang, D.; et al. Activation of Mitochondrial Unfolded Protein Response in SHSY5Y Expressing APP Cells and APP/PS1 Mice. Front. Cell Neurosci. 2019, 13, 568. [Google Scholar] [CrossRef]
- Romani, M.; Sorrentino, V.; Oh, C.M.; Li, H.; de Lima, T.I.; Zhang, H.; Shong, M.; Auwerx, J. NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 2021, 34, 108660. [Google Scholar] [CrossRef]
- Jagadeesan, A.J.; Murugesan, R.; Vimala Devi, S.; Meera, M.; Madhumala, G.; Vishwanathan Padmaja, M.; Ramesh, A.; Banerjee, A.; Sushmitha, S.; Khokhlov, A.N.; et al. Current trends in etiology, prognosis and therapeutic aspects of Parkinson’s disease: A review. Acta Biomed. 2017, 88, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Muller, T.; Bornemann, A.; et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef]
- Tanner, C.M.; Kamel, F.; Ross, G.W.; Hoppin, J.A.; Goldman, S.M.; Korell, M.; Marras, C.; Bhudhikanok, G.S.; Kasten, M.; Chade, A.R.; et al. Rotenone, paraquat, and Parkinson’s disease. Environ. Health Perspect. 2011, 119, 866–872. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The rotenone model of Parkinson’s disease: Genes, environment and mitochondria. Parkinsonism Relat. Disord. 2003, 9 (Suppl. S2), S59–S64. [Google Scholar] [CrossRef]
- Spivey, A. Rotenone and paraquat linked to Parkinson’s disease: Human exposure study supports years of animal studies. Environ. Health Perspect. 2011, 119, A259. [Google Scholar] [CrossRef]
- Vos, M. Mitochondrial Complex I deficiency: Guilty in Parkinson’s disease. Signal Transduct. Target. Ther. 2022, 7, 136. [Google Scholar] [CrossRef] [PubMed]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Eve, D.J.; Nisbet, A.P.; Kingsbury, A.E.; Hewson, E.L.; Daniel, S.E.; Lees, A.J.; Marsden, C.D.; Foster, O.J. Basal ganglia neuronal nitric oxide synthase mRNA expression in Parkinson’s disease. Brain Res. Mol. Brain Res. 1998, 63, 62–71. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Tieu, K.; Ischiropoulos, H.; Przedborski, S. Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 2003, 55, 329–335. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Pham, M.T.; Mitchell, M.B.; Yeom, S.G.; Jordan, S.; Chang, A.; Sopher, B.L.; La Spada, A.R. 4E-BP1 Protects Neurons from Misfolded Protein Stress and Parkinson’s Disease Toxicity by Inducing the Mitochondrial Unfolded Protein Response. J. Neurosci. 2020, 40, 8734–8745. [Google Scholar] [CrossRef]
- Liu, M.; Yu, S.; Wang, J.; Qiao, J.; Liu, Y.; Wang, S.; Zhao, Y. Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in Drosophila melanogaster PINK1 model of Parkinson’s disease. J. Ethnopharmacol. 2020, 247, 112213. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Marcus, R. What Is Huntington Disease? JAMA 2023, 330, 1014. [Google Scholar] [CrossRef]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef]
- Gu, M.; Gash, M.T.; Mann, V.M.; Javoy-Agid, F.; Cooper, J.M.; Schapira, A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996, 39, 385–389. [Google Scholar] [CrossRef]
- Milakovic, T.; Johnson, G.V. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J. Biol. Chem. 2005, 280, 30773–30782. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P. Effects of chronic MPTP and 3-nitropropionic acid in nonhuman primates. Curr. Opin. Neurol. 1995, 8, 469–473. [Google Scholar] [CrossRef]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef]
- Horton, T.M.; Graham, B.H.; Corral-Debrinski, M.; Shoffner, J.M.; Kaufman, A.E.; Beal, M.F.; Wallace, D.C. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 1995, 45, 1879–1883. [Google Scholar] [CrossRef]
- Bossy-Wetzel, E.; Petrilli, A.; Knott, A.B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008, 31, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Liu, F.; Liu, C.; Jin, B.; Jiang, Y.; Tang, M.; Qi, X.; Guo, X. Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1428–1435. [Google Scholar] [CrossRef]
- Hernandez, I.H.; Torres-Peraza, J.; Santos-Galindo, M.; Ramos-Moron, E.; Fernandez-Fernandez, M.R.; Perez-Alvarez, M.J.; Miranda-Vizuete, A.; Lucas, J.J. The neuroprotective transcription factor ATF5 is decreased and sequestered into polyglutamine inclusions in Huntington’s disease. Acta Neuropathol. 2017, 134, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.M.; Oliveira, A.; Oliveira, J.M.A.; Pinho, B.R. Stress response mechanisms in protein misfolding diseases: Profiling a cellular model of Huntington’s disease. Arch. Biochem. Biophys. 2023, 745, 109711. [Google Scholar] [CrossRef] [PubMed]
- Naia, L.; Carmo, C.; Campesan, S.; Fao, L.; Cotton, V.E.; Valero, J.; Lopes, C.; Rosenstock, T.R.; Giorgini, F.; Rego, A.C. Mitochondrial SIRT3 confers neuroprotection in Huntington’s disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic. Biol. Med. 2021, 163, 163–179. [Google Scholar] [CrossRef]
- Naia, L.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira-Sousa, S.I.; Caldeira, G.L.; Carmo, C.; Laco, M.N.; Hayden, M.R.; Oliveira, C.R.; Rego, A.C. Comparative Mitochondrial-Based Protective Effects of Resveratrol and Nicotinamide in Huntington’s Disease Models. Mol. Neurobiol. 2017, 54, 5385–5399. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Jin, J.; Cichewicz, R.H.; Hageman, S.A.; Ellis, T.K.; Xiang, L.; Peng, Q.; Jiang, M.; Arbez, N.; Hotaling, K.; et al. trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 2012, 287, 24460–24472. [Google Scholar] [CrossRef]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Rosen, D.R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Chia, R.; Chio, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e539. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, L.; Qiu, W.; Liu, Y.; Yang, F.; Chen, W.; Xu, R. Nicotinamide Riboside Enhances Mitochondrial Proteostasis and Adult Neurogenesis through Activation of Mitochondrial Unfolded Protein Response Signaling in the Brain of ALS SOD1(G93A) Mice. Int. J. Biol. Sci. 2020, 16, 284–297. [Google Scholar] [CrossRef]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, E.A. Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Riar, A.K.; Burstein, S.R.; Palomo, G.M.; Arreguin, A.; Manfredi, G.; Germain, D. Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 2017, 26, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Krasilnikova, M.M.; Humphries, C.L.; Shinsky, E.M. Friedreich’s ataxia: New insights. Emerg. Top. Life Sci. 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Stepanova, A.; Magrane, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| mtUPR Axis | Location of the Mitochondrial Stress | Markers | References |
|---|---|---|---|
| Transcriptional canonical mtUPR | Mitochondrial matrix | eif2α CHOP ATF5 ATF4 Hsp60 Hsp10 Hsp70 ClpP LonP1 | [120] [86] [98] [99] [85] [85] [85] [102] [120] |
| Translational canonical mtUPR | Mitochondrial matrix | MRPP3 | [108] |
| Sirtuin mtUPR | Mitochondrial matrix | SIRT1 SIRT3 FOXO3A MnSOD Catalase | [116] [121] [121] [115] [122] |
| IMS mtUPR | Intermembrane space | ERα NRF1 OMI | [119] [119] [123] |
| ALS-Related Gene | Pathogenic Mechanism | References |
|---|---|---|
| SOD1 | Antioxidant defense deficiency ROS accumulation Mitochondrial dysfunction Axonal transport disturbance Axonal neurodegeneration Apoptosis induction | [262] |
| C9orf72 | Decreased complex I assembly and activity Reduced OXPHOS ROS overproduction Mitochondrial dysfunction | [263] |
| TDP-43 | Increased Fis1 and Drp1 levels Decreased Mfn1 levels Mitochondrial network fragmentation Mitochondrial membrane potential loss Reduced mitochondrial ATP synthesis Mitochondrial dysfunction | [264] |
| OPTN | Decreased mitophagy Accumulation of damaged mitochondria Mitochondrial dysfunction | [265] |
| MATR3 | Interaction with TDP-43 and FUS Mitochondrial dysfunction | [266] |
| CHCHD10 | Mitochondrial dynamics alteration Cellular bioenergetics failure TDP-43 aggregates Mitochondrial dysfunction | [267,268] |
| FUS | Aggregates formation Decreased mitochondrial complexes mRNA transcription Reduced OXPHOS Increased ROS Mitochondrial dysfunction | [269] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilleros-Holgado, P.; Gómez-Fernández, D.; Piñero-Pérez, R.; Romero-Domínguez, J.M.; Reche-López, D.; López-Cabrera, A.; Álvarez-Córdoba, M.; Munuera-Cabeza, M.; Talaverón-Rey, M.; Suárez-Carrillo, A.; et al. Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases. Biomolecules 2023, 13, 1789. https://doi.org/10.3390/biom13121789
Cilleros-Holgado P, Gómez-Fernández D, Piñero-Pérez R, Romero-Domínguez JM, Reche-López D, López-Cabrera A, Álvarez-Córdoba M, Munuera-Cabeza M, Talaverón-Rey M, Suárez-Carrillo A, et al. Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases. Biomolecules. 2023; 13(12):1789. https://doi.org/10.3390/biom13121789
Chicago/Turabian StyleCilleros-Holgado, Paula, David Gómez-Fernández, Rocío Piñero-Pérez, Jose Manuel Romero-Domínguez, Diana Reche-López, Alejandra López-Cabrera, Mónica Álvarez-Córdoba, Manuel Munuera-Cabeza, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, and et al. 2023. "Mitochondrial Quality Control via Mitochondrial Unfolded Protein Response (mtUPR) in Ageing and Neurodegenerative Diseases" Biomolecules 13, no. 12: 1789. https://doi.org/10.3390/biom13121789