
Platelet and HIV Interactions and Their Contribution to Non-AIDS Comorbidities
 ,
,  , ,
, ,
Abstract
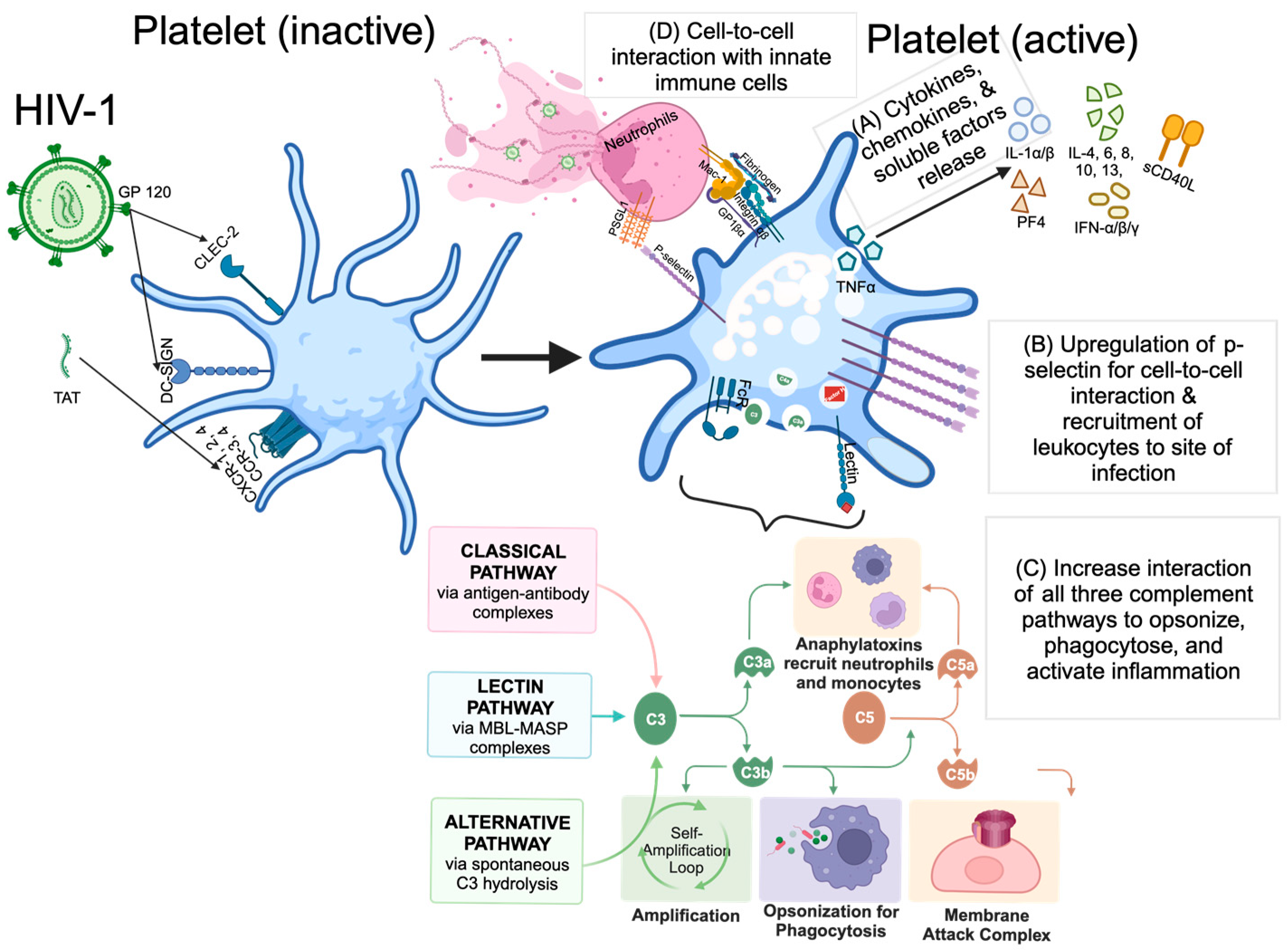
:1. Introduction
2. Role of Platelets in HIV Infection
2.1. Platelet and HIV Interaction
2.2. Role of Platelets in Modulating Innate and Adaptive Immune Clearance of HIV
2.3. Role of Platelets in Sheltering HIV and Viral Persistence
2.4. Role of Platelet Activation in the Pathophysiology of HIV
3. HIV Complications Due to Platelet Dysfunction
3.1. Thrombocytopenia
3.2. Thrombotic Microangiopathies
3.3. Venous Thromboembolism
3.4. Cardiovascular Disease
4. Effects of ART on Platelet Count and Activity in HIV-1 Infection
4.1. Effect of ART Initiation on Platelets in HIV-1 Infection
4.2. Adverse Effects of ART on Platelets
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wandeler, G.; Johnson, L.F.; Egger, M. Trends in life expectancy of HIV-positive adults on antiretroviral therapy across the globe: Comparisons with general population. Curr. Opin. HIV AIDS 2016, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Teeraananchai, S.; Kerr, S.J.; Amin, J.; Ruxrungtham, K.; Law, M.G. Life expectancy of HIV-positive people after starting combination antiretroviral therapy: A meta-analysis. HIV Med. 2017, 18, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.V.; Stelzle, D.; Lee, K.K.; Beck, E.J.; Alam, S.; Clifford, S.; Longenecker, C.T.; Strachan, F.; Bagchi, S.; Whiteley, W.; et al. Global Burden of Atherosclerotic Cardiovascular Disease in People Living With HIV: Systematic Review and Meta-Analysis. Circulation 2018, 138, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Serrao, R.; Pinero, C.; Velez, J.; Coutinho, D.; Maltez, F.; Lino, S.; Sarmento, E.C.R.; Tavares, A.P.; Pacheco, P.; Lopes, M.J.; et al. Non-AIDS-related comorbidities in people living with HIV-1 aged 50 years and older: The AGING POSITIVE study. Int. J. Infect. Dis. 2019, 79, 94–100. [Google Scholar] [CrossRef]
- Ji, Y.; Lu, H. Malignancies in HIV-Infected and AIDS Patients. Adv. Exp. Med. Biol. 2017, 1018, 167–179. [Google Scholar] [CrossRef]
- Smit, M.; Brinkman, K.; Geerlings, S.; Smit, C.; Thyagarajan, K.; Sighem, A.; de Wolf, F.; Hallett, T.B.; cohort, A.o. Future challenges for clinical care of an ageing population infected with HIV: A modelling study. Lancet Infect. Dis. 2015, 15, 810–818. [Google Scholar] [CrossRef]
- Brown, T.T.; Guaraldi, G. Multimorbidity and Burden of Disease. Interdiscip. Top. Gerontol. Geriatr. 2017, 42, 59–73. [Google Scholar] [CrossRef]
- Lerner, A.M.; Eisinger, R.W.; Fauci, A.S. Comorbidities in Persons With HIV: The Lingering Challenge. JAMA 2020, 323, 19–20. [Google Scholar] [CrossRef]
- High, K.P.; Brennan-Ing, M.; Clifford, D.B.; Cohen, M.H.; Currier, J.; Deeks, S.G.; Deren, S.; Effros, R.B.; Gebo, K.; Goronzy, J.J.; et al. HIV and aging: State of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J. Acquir. Immune Defic. Syndr. 2012, 60 (Suppl. S1), S1–S18. [Google Scholar] [CrossRef]
- Pahwa, S.; Deeks, S.; Zou, S.; Tomitch, N.; Miller-Novak, L.; Caler, E.; Justice, A.; Sacktor, N.; Gabuzda, D.; Hunt, P.W.; et al. NIH Workshop on HIV-Associated Comorbidities, Coinfections, and Complications: Summary and Recommendation for Future Research. J. Acquir. Immune Defic. Syndr. 2021, 86, 11–18. [Google Scholar] [CrossRef]
- Douek, D.C.; Brenchley, J.M.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Okamoto, Y.; Casazza, J.P.; Kuruppu, J.; Kunstman, K.; Wolinsky, S.; et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 2002, 417, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Lore, K.; Smed-Sorensen, A.; Vasudevan, J.; Mascola, J.R.; Koup, R.A. Myeloid and plasmacytoid dendritic cells transfer HIV-1 preferentially to antigen-specific CD4+ T cells. J. Exp. Med. 2005, 201, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Isaacs, S.N.; Williams, D.A.; Frank, I.; Schols, D.; De Clercq, E.; Kolson, D.L.; Collman, R.G. Role of CXCR4 in cell-cell fusion and infection of monocyte-derived macrophages by primary human immunodeficiency virus type 1 (HIV-1) strains: Two distinct mechanisms of HIV-1 dual tropism. J. Virol. 1999, 73, 7117–7125. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, J.B.; Ellis, J.E.; Hair, G.A.; Kirshenbaum, A.S.; Metcalfe, D.D.; Yi, H.; Cardona, A.C.; Lindsay, M.K.; Ansari, A.A. Human tissue mast cells are an inducible reservoir of persistent HIV infection. Blood 2007, 109, 5293–5300. [Google Scholar] [CrossRef]
- Bowers, N.L.; Helton, E.S.; Huijbregts, R.P.; Goepfert, P.A.; Heath, S.L.; Hel, Z. Immune suppression by neutrophils in HIV-1 infection: Role of PD-L1/PD-1 pathway. PLoS Pathog. 2014, 10, e1003993. [Google Scholar] [CrossRef]
- Youssefian, T.; Drouin, A.; Masse, J.M.; Guichard, J.; Cramer, E.M. Host defense role of platelets: Engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood 2002, 99, 4021–4029. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef]
- Real, F.; Capron, C.; Sennepin, A.; Arrigucci, R.; Zhu, A.; Sannier, G.; Zheng, J.; Xu, L.; Masse, J.M.; Greffe, S.; et al. Platelets from HIV-infected individuals on antiretroviral drug therapy with poor CD4(+) T cell recovery can harbor replication-competent HIV despite viral suppression. Sci. Transl. Med. 2020, 12, eaat6263. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef]
- Tintinger, G.R.; Theron, A.J.; Steel, H.C.; Cholo, M.C.; Nel, J.G.; Feldman, C.; Anderson, R. Submission for Special Issue: The Role of Platelet Activation in the Pathophysiology of HIV, Tuberculosis, and Pneumococcal Disease. Bedaquiline Suppresses ADP-Mediated Activation of Human Platelets In Vitro via Interference with Phosphatidylinositol 3-Kinase. Front. Immunol. 2020, 11, 621148. [Google Scholar] [CrossRef]
- Pretorius, E. Platelets in HIV: A Guardian of Host Defence or Transient Reservoir of the Virus? Front. Immunol. 2021, 12, 649465. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Papakonstantinou, V.; Detopoulou, P.; Fragopoulou, E.; Chini, M.; Lazanas, M.C.; Antonopoulou, S. The Role of Platelet-Activating Factor in Chronic Inflammation, Immune Activation, and Comorbidities Associated with HIV Infection. AIDS Rev. 2015, 17, 191–201. [Google Scholar] [PubMed]
- Mesquita, E.C.; Hottz, E.D.; Amancio, R.T.; Carneiro, A.B.; Palhinha, L.; Coelho, L.E.; Grinsztejn, B.; Zimmerman, G.A.; Rondina, M.T.; Weyrich, A.S.; et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci. Rep. 2018, 8, 14999. [Google Scholar] [CrossRef] [PubMed]
- Zucker-Franklin, D.; Seremetis, S.; Zheng, Z.Y. Internalization of human immunodeficiency virus type I and other retroviruses by megakaryocytes and platelets. Blood 1990, 75, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S.R.; Singh, M.V.; Dewhurst, S.; Schifitto, G.; Maggirwar, S.B. Platelets function as an acute viral reservoir during HIV-1 infection by harboring virus and T-cell complex formation. Blood Adv. 2020, 4, 4512–4521. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; McKnight, A.; Potempa, S.; Simmons, G.; Gray, P.W.; Power, C.A.; Wells, T.; Weiss, R.A.; Talbot, S.J. CD4-independent infection by HIV-2 (ROD/B): Use of the 7-transmembrane receptors CXCR-4, CCR-3, and V28 for entry. Virology 1997, 231, 130–134. [Google Scholar] [CrossRef]
- Clemetson, K.J.; Clemetson, J.M.; Proudfoot, A.E.; Power, C.A.; Baggiolini, M.; Wells, T.N. Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood 2000, 96, 4046–4054. [Google Scholar] [CrossRef]
- Lee, B.; Ratajczak, J.; Doms, R.W.; Gewirtz, A.M.; Ratajczak, M.Z. Coreceptor/chemokine receptor expression on human hematopoietic cells: Biological implications for human immunodeficiency virus-type 1 infection. Blood 1999, 93, 1145–1156. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; van Kooyk, Y. DC-SIGN: A novel HIV receptor on DCs that mediates HIV-1 transmission. Curr. Top Microbiol. Immunol. 2003, 276, 31–54. [Google Scholar] [CrossRef]
- Chaipan, C.; Soilleux, E.J.; Simpson, P.; Hofmann, H.; Gramberg, T.; Marzi, A.; Geier, M.; Stewart, E.A.; Eisemann, J.; Steinkasserer, A.; et al. DC-SIGN and CLEC-2 mediate human immunodeficiency virus type 1 capture by platelets. J. Virol. 2006, 80, 8951–8960. [Google Scholar] [CrossRef]
- Badolia, R.; Inamdar, V.; Manne, B.K.; Dangelmaier, C.; Eble, J.A.; Kunapuli, S.P. G(q) pathway regulates proximal C-type lectin-like receptor-2 (CLEC-2) signaling in platelets. J. Biol. Chem. 2017, 292, 14516–14531. [Google Scholar] [CrossRef] [PubMed]
- Chabert, A.; Hamzeh-Cognasse, H.; Pozzetto, B.; Cognasse, F.; Schattner, M.; Gomez, R.M.; Garraud, O. Human platelets and their capacity of binding viruses: Meaning and challenges? BMC Immunol. 2015, 16, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Nardi, M.A.; Li, Z. HIV-1 Tat-induced platelet activation and release of CD154 contribute to HIV-1-associated autoimmune thrombocytopenia. J. Thromb. Haemost. 2011, 9, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Madzime, M.; Rossouw, T.M.; Theron, A.J.; Anderson, R.; Steel, H.C. Interactions of HIV and Antiretroviral Therapy With Neutrophils and Platelets. Front. Immunol. 2021, 12, 634386. [Google Scholar] [CrossRef]
- Green, S.A.; Smith, M.; Hasley, R.B.; Stephany, D.; Harned, A.; Nagashima, K.; Abdullah, S.; Pittaluga, S.; Imamichi, T.; Qin, J.; et al. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: Coupling coagulation/inflammation and T cells. AIDS 2015, 29, 1297–1308. [Google Scholar] [CrossRef]
- Dib, P.R.B.; Quirino-Teixeira, A.C.; Merij, L.B.; Pinheiro, M.B.M.; Rozini, S.V.; Andrade, F.B.; Hottz, E.D. Innate immune receptors in platelets and platelet-leukocyte interactions. J. Leukoc. Biol. 2020, 108, 1157–1182. [Google Scholar] [CrossRef]
- Auerbach, D.J.; Lin, Y.; Miao, H.; Cimbro, R.; Difiore, M.J.; Gianolini, M.E.; Furci, L.; Biswas, P.; Fauci, A.S.; Lusso, P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 9569–9574. [Google Scholar] [CrossRef]
- Rozmyslowicz, T.; Majka, M.; Kijowski, J.; Murphy, S.L.; Conover, D.O.; Poncz, M.; Ratajczak, J.; Gaulton, G.N.; Ratajczak, M.Z. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 2003, 17, 33–42. [Google Scholar] [CrossRef]
- Ameglio, F.; Capobianchi, M.R.; Castilletti, C.; Cordiali Fei, P.; Fais, S.; Trento, E.; Dianzani, F. Recombinant gp120 induces IL-10 in resting peripheral blood mononuclear cells; correlation with the induction of other cytokines. Clin. Exp. Immunol. 1994, 95, 455–458. [Google Scholar] [CrossRef]
- Ankel, H.; Capobianchi, M.R.; Castilletti, C.; Dianzani, F. Interferon induction by HIV glycoprotein 120: Role of the V3 loop. Virology 1994, 205, 34–43. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Barresi, C.; Borghi, P.; Gessani, S.; Fantuzzi, L.; Ameglio, F.; Belardelli, F.; Papadia, S.; Dianzani, F. Human immunodeficiency virus type 1 gp120 stimulates cytomegalovirus replication in monocytes: Possible role of endogenous interleukin-8. J. Virol. 1997, 71, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.L.; Meltzer, M.S. Induction of IFN-alpha by HIV-1 in monocyte-enriched PBMC requires gp120-CD4 interaction but not virus replication. J. Immunol. 1993, 151, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Patella, V.; Florio, G.; Petraroli, A.; Marone, G. HIV-1 gp120 induces IL-4 and IL-13 release from human Fc epsilon RI+ cells through interaction with the VH3 region of IgE. J. Immunol. 2000, 164, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Schols, D.; De Clercq, E. Human immunodeficiency virus type 1 gp120 induces anergy in human peripheral blood lymphocytes by inducing interleukin-10 production. J. Virol. 1996, 70, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Solomon Tsegaye, T.; Gnirss, K.; Rahe-Meyer, N.; Kiene, M.; Kramer-Kuhl, A.; Behrens, G.; Munch, J.; Pohlmann, S. Platelet activation suppresses HIV-1 infection of T cells. Retrovirology 2013, 10, 48. [Google Scholar] [CrossRef]
- Rossouw, T.M.; Feldman, C. Editorial: The Role of Platelet Activation in the Pathophysiology of HIV, Tuberculosis and Pneumococcal Disease. Front. Immunol. 2021, 12, 737016. [Google Scholar] [CrossRef]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. Biomed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef]
- Kubes, P.; Ward, P.A. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000, 10, 127–135. [Google Scholar] [CrossRef]
- Speth, C.; Loffler, J.; Krappmann, S.; Lass-Florl, C.; Rambach, G. Platelets as immune cells in infectious diseases. Future Microbiol. 2013, 8, 1431–1451. [Google Scholar] [CrossRef]
- Liang, H.; Duan, Z.; Li, D.; Li, D.; Wang, Z.; Ren, L.; Shen, T.; Shao, Y. Higher levels of circulating monocyte-platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell Mol. Immunol. 2015, 12, 435–443. [Google Scholar] [CrossRef]
- Nkambule, B.B.; Davison, G.; Ipp, H. Platelet leukocyte aggregates and markers of platelet aggregation, immune activation and disease progression in HIV infected treatment naive asymptomatic individuals. J. Thromb. Thrombolysis 2015, 40, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Pircher, J.; Engelmann, B.; Massberg, S.; Schulz, C. Platelet-Neutrophil Crosstalk in Atherothrombosis. Thromb. Haemost. 2019, 119, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.F.; Campbell, R.A.; Schwertz, H.; Cody, M.J.; Franks, Z.; Tolley, N.D.; Kahr, W.H.; Lindemann, S.; Seizer, P.; Yost, C.C.; et al. Novel anti-bacterial activities of beta-defensin 1 in human platelets: Suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011, 7, e1002355. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Livada, A.C.; Morrell, C.N. Platelet and Megakaryocyte Roles in Innate and Adaptive Immunity. Circ. Res. 2022, 130, 288–308. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daeron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Yu, Q.; Yu, R.; Qin, X. The good and evil of complement activation in HIV-1 infection. Cell Mol. Immunol. 2010, 7, 334–340. [Google Scholar] [CrossRef]
- Jenabian, M.A.; Patel, M.; Kema, I.; Vyboh, K.; Kanagaratham, C.; Radzioch, D.; Thebault, P.; Lapointe, R.; Gilmore, N.; Ancuta, P.; et al. Soluble CD40-ligand (sCD40L, sCD154) plays an immunosuppressive role via regulatory T cell expansion in HIV infection. Clin. Exp. Immunol. 2014, 178, 102–111. [Google Scholar] [CrossRef]
- Chapman, L.M.; Aggrey, A.A.; Field, D.J.; Srivastava, K.; Ture, S.; Yui, K.; Topham, D.J.; Baldwin, W.M., 3rd; Morrell, C.N. Platelets present antigen in the context of MHC class I. J. Immunol. 2012, 189, 916–923. [Google Scholar] [CrossRef]
- Flaujac, C.; Boukour, S.; Cramer-Borde, E. Platelets and viruses: An ambivalent relationship. Cell Mol. Life Sci. 2010, 67, 545–556. [Google Scholar] [CrossRef]
- Simon, A.Y.; Sutherland, M.R.; Pryzdial, E.L. Dengue virus binding and replication by platelets. Blood 2015, 126, 378–385. [Google Scholar] [CrossRef]
- Kullaya, V.I.; de Mast, Q.; van der Ven, A.; elMoussaoui, H.; Kibiki, G.; Simonetti, E.; de Jonge, M.I.; Ferwerda, G. Platelets Modulate Innate Immune Response Against Human Respiratory Syncytial Virus In Vitro. Viral Immunol. 2017, 30, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Seyoum, M.; Enawgaw, B.; Melku, M. Human blood platelets and viruses: Defense mechanism and role in the removal of viral pathogens. Thromb. J. 2018, 16, 16. [Google Scholar] [CrossRef] [PubMed]
- Zahn, A.; Jennings, N.; Ouwehand, W.H.; Allain, J.P. Hepatitis C virus interacts with human platelet glycoprotein VI. J. Gen. Virol. 2006, 87, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Baumer, Y.; Weatherby, T.M.; Mitchell, B.I.; SahBandar, I.N.; Premeaux, T.A.; Michelle, L.D.; Gutierrez-Huerta, C.A.; Powell-Wiley, T.M.; Brown, T.R.; Boisvert, W.A.; et al. Hiding in plain sight—platelets, the silent carriers of HIV-1. Platelets 2021, 32, 1038–1042. [Google Scholar] [CrossRef]
- Banerjee, M.; Huang, Y.; Joshi, S.; Popa, G.J.; Mendenhall, M.D.; Wang, Q.J.; Garvy, B.A.; Myint, T.; Whiteheart, S.W. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arter. Thromb. Vasc. Biol. 2020, 40, 1635–1650. [Google Scholar] [CrossRef]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV Proteins and Endothelial Dysfunction: Implications in Cardiovascular Disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef]
- Gresele, P.; Falcinelli, E.; Sebastiano, M.; Baldelli, F. Endothelial and platelet function alterations in HIV-infected patients. Thromb. Res. 2012, 129, 301–308. [Google Scholar] [CrossRef]
- Burnouf, T.; Walker, T.L. The multifaceted role of platelets in mediating brain function. Blood 2022, 140, 815–827. [Google Scholar] [CrossRef]
- Phillips, A.N.; Carr, A.; Neuhaus, J.; Visnegarwala, F.; Prineas, R.; Burman, W.J.; Williams, I.; Drummond, F.; Duprez, D.; Belloso, W.H.; et al. Interruption of antiretroviral therapy and risk of cardiovascular disease in persons with HIV-1 infection: Exploratory analyses from the SMART trial. Antivir. Ther. 2008, 13, 177–187. [Google Scholar] [CrossRef]
- Damien, P.; Cognasse, F.; Lucht, F.; Suy, F.; Pozzetto, B.; Garraud, O.; Hamzeh-Cognasse, H. Highly active antiretroviral therapy alters inflammation linked to platelet cytokines in HIV-1-infected patients. J. Infect. Dis. 2013, 208, 868–870. [Google Scholar] [CrossRef]
- Landro, L.; Ueland, T.; Otterdal, K.; Froland, S.S.; Aukrust, P. Persistently raised plasma levels of platelet-derived inflammatory mediators in HIV-infected patients during highly active anti-retroviral therapy. J. Thromb. Haemost. 2011, 9, 1075–1077. [Google Scholar] [CrossRef]
- Mayne, E.; Funderburg, N.T.; Sieg, S.F.; Asaad, R.; Kalinowska, M.; Rodriguez, B.; Schmaier, A.H.; Stevens, W.; Lederman, M.M. Increased platelet and microparticle activation in HIV infection: Upregulation of P-selectin and tissue factor expression. J. Acquir. Immune Defic. Syndr. 2012, 59, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.T.; Mayne, E.; Sieg, S.F.; Asaad, R.; Jiang, W.; Kalinowska, M.; Luciano, A.A.; Stevens, W.; Rodriguez, B.; Brenchley, J.M.; et al. Increased tissue factor expression on circulating monocytes in chronic HIV infection: Relationship to in vivo coagulation and immune activation. Blood 2010, 115, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arter. Thromb. Vasc. Biol. 2004, 24, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Henn, V.; Slupsky, J.R.; Grafe, M.; Anagnostopoulos, I.; Forster, R.; Muller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Bakogiannis, C.; Tousoulis, D.; Antonopoulos, A.S.; Stefanadis, C. The CD40/CD40 ligand system: Linking inflammation with atherothrombosis. J. Am Coll. Cardiol. 2009, 54, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.S.; Andre, P.; Yan, Y.; Phillips, D.R. The platelet CD40L/GP IIb-IIIa axis in atherothrombotic disease. Curr. Opin. Hematol. 2003, 10, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Pamukcu, B.; Lip, G.Y.; Snezhitskiy, V.; Shantsila, E. The CD40-CD40L system in cardiovascular disease. Ann. Med. 2011, 43, 331–340. [Google Scholar] [CrossRef]
- Andre, P.; Prasad, K.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Lievens, D.; Eijgelaar, W.J.; Biessen, E.A.; Daemen, M.J.; Lutgens, E. The multi-functionality of CD40L and its receptor CD40 in atherosclerosis. Thromb. Haemost. 2009, 102, 206–214. [Google Scholar] [CrossRef]
- Raadsen, M.; Du Toit, J.; Langerak, T.; van Bussel, B.; van Gorp, E.; Goeijenbier, M. Thrombocytopenia in Virus Infections. J. Clin. Med. 2021, 10, 877. [Google Scholar] [CrossRef] [PubMed]
- Christersson, C.; Johnell, M.; Siegbahn, A. Tissue factor and IL8 production by P-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of Lyn and is inhibited by IL10. J. Thromb. Haemost. 2008, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Celi, A.; Pellegrini, G.; Lorenzet, R.; De Blasi, A.; Ready, N.; Furie, B.C.; Furie, B. P-selectin induces the expression of tissue factor on monocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 8767–8771. [Google Scholar] [CrossRef] [PubMed]
- Furie, B.; Furie, B.C. P-selectin induction of tissue factor biosynthesis and expression. Haemostasis 1996, 26, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. BioChem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Graham, S.M.; Nance, R.M.; Chen, J.; Wurfel, M.M.; Hunt, P.W.; Heckbert, S.R.; Budoff, M.J.; Moore, R.D.; Jacobson, J.M.; Martin, J.N.; et al. Plasma Interleukin-6 (IL-6), Angiopoietin-2, and C-Reactive Protein Levels Predict Subsequent Type 1 Myocardial Infarction in Persons with Treated HIV Infection. J. Acquir. Immune Defic. Syndr. 2023, 93, 282–291. [Google Scholar] [CrossRef]
- Murphy, M.F.; Metcalfe, P.; Waters, A.H.; Carne, C.A.; Weller, I.V.; Linch, D.C.; Smith, A. Incidence and mechanism of neutropenia and thrombocytopenia in patients with human immunodeficiency virus infection. Br. J. Haematol. 1987, 66, 337–340. [Google Scholar] [CrossRef]
- Kaslow, R.A.; Phair, J.P.; Friedman, H.B.; Lyter, D.; Solomon, R.E.; Dudley, J.; Polk, B.F.; Blackwelder, W. Infection with the human immunodeficiency virus: Clinical manifestations and their relationship to immune deficiency. A report from the Multicenter AIDS Cohort Study. Ann. Intern. Med. 1987, 107, 474–480. [Google Scholar] [CrossRef]
- Rossi, G.; Gorla, R.; Stellini, R.; Franceschini, F.; Bettinzioli, M.; Cadeo, G.; Sueri, L.; Cattaneo, R.; Marinone, G. Prevalence, clinical, and laboratory features of thrombocytopenia among HIV-infected individuals. AIDS Res. Hum. Retrovir. 1990, 6, 261–269. [Google Scholar] [CrossRef]
- Peltier, J.Y.; Lambin, P.; Doinel, C.; Courouce, A.M.; Rouger, P.; Lefrere, J.J. Frequency and prognostic importance of thrombocytopenia in symptom-free HIV-infected individuals: A 5-year prospective study. AIDS 1991, 5, 381–384. [Google Scholar] [CrossRef]
- Mientjes, G.H.; van Ameijden, E.J.; Mulder, J.W.; van den Hoek, J.A.; Coutinho, R.A.; von dem Borne, A.E. Prevalence of thrombocytopenia in HIV-infected and non-HIV infected drug users and homosexual men. Br. J. Haematol. 1992, 82, 615–619. [Google Scholar] [CrossRef]
- Sloand, E.M.; Klein, H.G.; Banks, S.M.; Vareldzis, B.; Merritt, S.; Pierce, P. Epidemiology of thrombocytopenia in HIV infection. Eur. J. Haematol. 1992, 48, 168–172. [Google Scholar] [CrossRef]
- Sullivan, P.S.; Hanson, D.L.; Chu, S.Y.; Jones, J.L.; Ciesielski, C.A. Surveillance for thrombocytopenia in persons infected with HIV: Results from the multistate Adult and Adolescent Spectrum of Disease Project. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 14, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Wondimeneh, Y.; Muluye, D.; Ferede, G. Prevalence and associated factors of thrombocytopenia among HAART-naive HIV-positive patients at Gondar University Hospital, northwest Ethiopia. BMC Res. Notes 2014, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.R.; Slichter, S.J. Platelet kinetics in patients with bone marrow hypoplasia: Evidence for a fixed platelet requirement. Blood 1985, 66, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Castellares, C.; Barreiro, P.; Martin-Carbonero, L.; Labarga, P.; Vispo, M.E.; Casado, R.; Galindo, L.; Garcia-Gasco, P.; Garcia-Samaniego, J.; Soriano, V. Liver cirrhosis in HIV-infected patients: Prevalence, aetiology and clinical outcome. J. Viral. Hepat. 2008, 15, 165–172. [Google Scholar] [CrossRef]
- Ragni, M.V.; Bontempo, F.A.; Myers, D.J.; Kiss, J.E.; Oral, A. Hemorrhagic sequelae of immune thrombocytopenic purpura in human immunodeficiency virus-infected hemophiliacs. Blood 1990, 75, 1267–1272. [Google Scholar] [CrossRef]
- Lv, X.; Li, P.; Yue, P.; Tang, P.; Zhou, F. Risk factors and prognosis of thrombocytopenia in people living with HIV/AIDS. Ther. Adv. Hematol. 2023, 14, 20406207231170513. [Google Scholar] [CrossRef]
- Fan, H.W.; Guo, F.P.; Li, Y.J.; Li, N.; Li, T.S. Prevalence of thrombocytopenia among Chinese adult antiretroviral-naive HIV-positive patients. Chin. Med. J. 2015, 128, 459–464. [Google Scholar] [CrossRef]
- Koike, Y.; Yoneyama, A.; Shirai, J.; Ishida, T.; Shoda, E.; Miyazaki, K.; Sunaga, S.; Horie, R.; Aoki, K.; Koike, K.; et al. Evaluation of thrombopoiesis in thrombocytopenic disorders by simultaneous measurement of reticulated platelets of whole blood and serum thrombopoietin concentrations. Thromb. Haemost. 1998, 79, 1106–1110. [Google Scholar]
- Furrer, H. Prevalence and clinical significance of splenomegaly in asymptomatic human immunodeficiency virus type 1-infected adults. Swiss HIV cohort study. Clin. Infect. Dis. 2000, 30, 943–945. [Google Scholar] [CrossRef]
- Chelucci, C.; Federico, M.; Guerriero, R.; Mattia, G.; Casella, I.; Pelosi, E.; Testa, U.; Mariani, G.; Hassan, H.J.; Peschle, C. Productive human immunodeficiency virus-1 infection of purified megakaryocytic progenitors/precursors and maturing megakaryocytes. Blood 1998, 91, 1225–1234. [Google Scholar] [CrossRef]
- Gibellini, D.; Vitone, F.; Buzzi, M.; Schiavone, P.; De Crignis, E.; Cicola, R.; Conte, R.; Ponti, C.; Re, M.C. HIV-1 negatively affects the survival/maturation of cord blood CD34(+) hematopoietic progenitor cells differentiated towards megakaryocytic lineage by HIV-1 gp120/CD4 membrane interaction. J. Cell Physiol. 2007, 210, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Zauli, G.; Catani, L.; Gibellini, D.; Re, M.C.; Vianelli, N.; Colangeli, V.; Celeghini, C.; Capitani, S.; La Placa, M. Impaired survival of bone marrow GPIIb/IIa+ megakaryocytic cells as an additional pathogenetic mechanism of HIV-1-related thrombocytopenia. Br. J. Haematol. 1996, 92, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Ballem, P.J.; Belzberg, A.; Devine, D.V.; Lyster, D.; Spruston, B.; Chambers, H.; Doubroff, P.; Mikulash, K. Kinetic studies of the mechanism of thrombocytopenia in patients with human immunodeficiency virus infection. N. Engl. J. Med. 1992, 327, 1779–1784. [Google Scholar] [CrossRef]
- Morris, L.; Distenfeld, A.; Amorosi, E.; Karpatkin, S. Autoimmune thrombocytopenic purpura in homosexual men. Ann. Intern. Med. 1982, 96 Pt 1, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Liebman, H.A. Viral-associated immune thrombocytopenic purpura. Hematol. Am. Soc. Hematol. Educ. Program 2008, 2008, 212–218. [Google Scholar] [CrossRef]
- Dominguez, A.; Gamallo, G.; Garcia, R.; Lopez-Pastor, A.; Pena, J.M.; Vazquez, J.J. Pathophysiology of HIV related thrombocytopenia: An analysis of 41 patients. J. Clin. Pathol. 1994, 47, 999–1003. [Google Scholar] [CrossRef]
- Karpatkin, S.; Nardi, M.; Lennette, E.T.; Byrne, B.; Poiesz, B. Anti-human immunodeficiency virus type 1 antibody complexes on platelets of seropositive thrombocytopenic homosexuals and narcotic addicts. Proc. Natl. Acad. Sci. USA 1988, 85, 9763–9767. [Google Scholar] [CrossRef]
- Savona, S.; Nardi, M.A.; Lennette, E.T.; Karpatkin, S. Thrombocytopenic purpura in narcotics addicts. Ann. Intern. Med. 1985, 102, 737–741. [Google Scholar] [CrossRef]
- Walsh, C.M.; Nardi, M.A.; Karpatkin, S. On the mechanism of thrombocytopenic purpura in sexually active homosexual men. N. Engl. J. Med. 1984, 311, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S.; Nardi, M. Autoimmune anti-HIV-1gp120 antibody with antiidiotype-like activity in sera and immune complexes of HIV-1-related immunologic thrombocytopenia. J. Clin. Investig. 1992, 89, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Oksenhendler, E.; Duedari, N.; Bierling, P. Cross-reactive antibodies between HIV-gp120 and platelet gpIIIa (CD61) in HIV-related immune thrombocytopenic purpura. Clin. Exp. Immunol. 1996, 103, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Chia, W.K.; Blanchette, V.; Mody, M.; Wright, J.F.; Freedman, J. Characterization of HIV-1-specific antibodies and HIV-1-crossreactive antibodies to platelets in HIV-1-infected haemophiliac patients. Br. J. Haematol. 1998, 103, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Conejero, R.; Rivera, J.; Rosillo, M.C.; Cano, A.; Rodriguez, T.; Vicente, V. Association of autoantibodies against platelet glycoproteins Ib/IX and IIb/IIIa, and platelet-reactive anti-HIV antibodies in thrombocytopenic narcotic addicts. Br. J. Haematol. 1996, 93, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Nardi, M.; Tomlinson, S.; Greco, M.A.; Karpatkin, S. Complement-independent, peroxide-induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell 2001, 106, 551–561. [Google Scholar] [CrossRef]
- Karpatkin, S.; Nardi, M.A.; Hymes, K.B. Sequestration of antiplatelet GPIIIa antibody in rheumatoid factor immune complexes of human immunodeficiency virus 1 thrombocytopenic patients. Proc. Natl. Acad. Sci. USA 1995, 92, 2263–2267. [Google Scholar] [CrossRef]
- Nardi, M.A.; Liu, L.X.; Karpatkin, S. GPIIIa-(49-66) is a major pathophysiologically relevant antigenic determinant for antiplatelet GPIIIa of HIV-1-related immunologic thrombocytopenia. Proc. Natl. Acad. Sci. USA 1997, 94, 7589–7594. [Google Scholar] [CrossRef]
- Nardi, M.; Karpatkin, S. Antiidiotype antibody against platelet anti-GPIIIa contributes to the regulation of thrombocytopenia in HIV-1-ITP patients. J. Exp. Med. 2000, 191, 2093–2100. [Google Scholar] [CrossRef]
- Quadri, M.I.; Lee, C.A.; Goodall, A.H.; Lim, S.G.; Kernoff, P.B. Antibodies to platelet glycoproteins in haemophiliacs infected with HIV. Clin. Lab Haematol. 1992, 14, 109–120. [Google Scholar] [CrossRef]
- Najean, Y.; Rain, J.D. The mechanism of thrombocytopenia in patients with HIV infection. J. Lab Clin. Med. 1994, 123, 415–420. [Google Scholar]
- Servais, J.; Nkoghe, D.; Schmit, J.C.; Arendt, V.; Robert, I.; Staub, T.; Moutschen, M.; Schneider, F.; Hemmer, R. HIV-associated hematologic disorders are correlated with plasma viral load and improve under highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2001, 28, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Getawa, S.; Aynalem, M.; Bayleyegn, B.; Adane, T. The global prevalence of thrombocytopenia among HIV-infected adults: A systematic review and meta-analysis. Int. J. Infect. Dis. 2021, 105, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Damtie, S.; Workineh, L.; Kiros, T.; Eyayu, T.; Tiruneh, T. Hematological Abnormalities of Adult HIV-Infected Patients Before and After Initiation of Highly Active Antiretroviral Treatment at Debre Tabor Comprehensive Specialized Hospital, Northcentral Ethiopia: A Cross-Sectional Study. HIV AIDS 2021, 13, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Woldeamanuel, G.G.; Wondimu, D.H. Prevalence of thrombocytopenia before and after initiation of HAART among HIV infected patients at black lion specialized hospital, Addis Ababa, Ethiopia: A cross sectional study. BMC Hematol. 2018, 18, 9. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Seligmann, M. HIV-related thrombocytopenia. Immunodefic. Rev. 1990, 2, 221–231. [Google Scholar]
- Ambler, K.L.; Vickars, L.M.; Leger, C.S.; Foltz, L.M.; Montaner, J.S.; Harris, M.; Dias Lima, V.; Leitch, H.A. Clinical Features, Treatment, and Outcome of HIV-Associated Immune Thrombocytopenia in the HAART Era. Adv. Hematol. 2012, 2012, 910954. [Google Scholar] [CrossRef]
- Tan, Y.; Che, L.; Bi, H.; Fan, S.; Zhou, Z.; Min, H. Clinical features and treatment effect of HIV-associated immune thrombocytopenia-single center Ten-Years data summary. Platelets 2023, 34, 2200836. [Google Scholar] [CrossRef]
- Lord, R.V.; Coleman, M.J.; Milliken, S.T. Splenectomy for HIV-related immune thrombocytopenia: Comparison with results of splenectomy for non-HIV immune thrombocytopenic purpura. Arch. Surg. 1998, 133, 205–210. [Google Scholar] [CrossRef]
- Alonso, M.; Gossot, D.; Bourstyn, E.; Galera, M.J.; Oksenhendler, E.; Celerier, M.; Clot, P. Splenectomy in human immunodeficiency virus-related thrombocytopenia. Br. J. Surg. 1993, 80, 330–333. [Google Scholar] [CrossRef]
- Ferguson, C.M. Splenectomy for immune thrombocytopenia related to human immunodeficiency virus. Surg. Gynecol. Obs. 1988, 167, 300–302. [Google Scholar]
- Aboolian, A.; Ricci, M.; Shapiro, K.; Connors, A.; LaRaja, R.D. Surgical treatment of HIV-related immune thrombocytopenia. Int. Surg. 1999, 84, 81–85. [Google Scholar] [PubMed]
- Kim, H.C.; Raska, K., Jr.; Trooskin, S.; Saidi, P. Immune thrombocytopenia in hemophiliacs infected with human immunodeficiency virus and their response to splenectomy. Arch. Intern. Med. 1989, 149, 1685–1688. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, P.; Wood, L.; Dent, D.M. Results of treatment in immune thrombocytopenia. Q. J. Med. 1986, 58, 153–165. [Google Scholar]
- Chaturvedi, S.; Arnold, D.M.; McCrae, K.R. Splenectomy for immune thrombocytopenia: Down but not out. Blood 2018, 131, 1172–1182. [Google Scholar] [CrossRef]
- Aslam, M.I.; Cardile, A.P.; Crawford, G.E. Use of peptide thrombopoietin receptor agonist romiplostim (Nplate) in a case of primary HIV-associated thrombocytopenia. J. Int. Assoc. Provid. AIDS Care 2014, 13, 22–23. [Google Scholar] [CrossRef]
- Soliman, M.; Engel, E.; Rico, J.; Rodriguez, C. Successful Use of Eltrombopag in a Pediatric Patient With Human Immunodeficiency Virus (HIV)-Associated Thrombocytopenia. J. Pediatr. Pharmacol. Ther. 2019, 24, 242–246. [Google Scholar] [CrossRef]
- Kowalczyk, M.; Rubinstein, P.G.; Aboulafia, D.M. Initial Experience with the Use of Thrombopoetin Receptor Agonists in Patients with Refractory HIV-Associated Immune Thrombocytopenic Purpura: A Case Series. J. Int. Assoc. Provid. AIDS Care 2015, 14, 211–216. [Google Scholar] [CrossRef]
- Moake, J.L. Thrombotic microangiopathies. N. Engl. J. Med. 2002, 347, 589–600. [Google Scholar] [CrossRef]
- Jokela, J.; Flynn, T.; Henry, K. Thrombotic thrombocytopenic purpura in a human immunodeficiency virus (HIV)-seropositive homosexual man. Am. J. Hematol. 1987, 25, 341–343. [Google Scholar] [CrossRef]
- Boccia, R.V.; Gelmann, E.P.; Baker, C.C.; Marti, G.; Longo, D.L. A hemolytic-uremic syndrome with the acquired immunodeficiency syndrome. Ann. Intern. Med. 1984, 101, 716–717. [Google Scholar] [CrossRef] [PubMed]
- Leaf, A.N.; Laubenstein, L.J.; Raphael, B.; Hochster, H.; Baez, L.; Karpatkin, S. Thrombotic thrombocytopenic purpura associated with human immunodeficiency virus type 1 (HIV-1) infection. Ann. Intern. Med. 1988, 109, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Gervasoni, C.; Ridolfo, A.L.; Vaccarezza, M.; Parravicini, C.; Vago, L.; Adorni, F.; Cappelletti, A.; d’Arminio Monforte, A.; Galli, M. Thrombotic microangiopathy in patients with acquired immunodeficiency syndrome before and during the era of introduction of highly active antiretroviral therapy. Clin. Infect. Dis. 2002, 35, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Fusco, G.; Fusco, J.; Balu, R.; Gangjee, S.; Brennan, C.; Feinberg, J.; Collaborations in HIV Outcomes Research/US Cohort. HIV-associated thrombotic microangiopathy in the era of highly active antiretroviral therapy: An observational study. Clin. Infect. Dis. 2004, 39, S267–S275. [Google Scholar] [CrossRef]
- Hart, D.; Sayer, R.; Miller, R.; Edwards, S.; Kelly, A.; Baglin, T.; Hunt, B.; Benjamin, S.; Patel, R.; Machin, S.; et al. Human immunodeficiency virus associated thrombotic thrombocytopenic purpura—Favourable outcome with plasma exchange and prompt initiation of highly active antiretroviral therapy. Br. J. Haematol. 2011, 153, 515–519. [Google Scholar] [CrossRef]
- Bade, N.A.; Giffi, V.S.; Baer, M.R.; Zimrin, A.B.; Law, J.Y. Thrombotic microangiopathy in the setting of human immunodeficiency virus infection: High incidence of severe thrombocytopenia. J. Clin. Apher. 2018, 33, 342–348. [Google Scholar] [CrossRef]
- Louw, S.; Gounden, R.; Mayne, E.S. Thrombotic thrombocytopenic purpura (TTP)-like syndrome in the HIV era. Thromb. J. 2018, 16, 35. [Google Scholar] [CrossRef]
- Hymes, K.B.; Karpatkin, S. Human immunodeficiency virus infection and thrombotic microangiopathy. Semin. Hematol. 1997, 34, 117–125. [Google Scholar]
- Furlan, M.; Lammle, B. Haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura—New insights into underlying biochemical mechanisms. Nephrol. Dial. Transpl. 2000, 15, 1112–1114. [Google Scholar] [CrossRef]
- Bianchi, V.; Robles, R.; Alberio, L.; Furlan, M.; Lammle, B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: A severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood 2002, 100, 710–713. [Google Scholar] [CrossRef]
- Moake, J.L.; Rudy, C.K.; Troll, J.H.; Weinstein, M.J.; Colannino, N.M.; Azocar, J.; Seder, R.H.; Hong, S.L.; Deykin, D. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N. Engl. J. Med. 1982, 307, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.F.; Scully, M.; Cohen, H.; Roedling, S.; Starke, R.; Edwards, S.G.; Machin, S.J. Thrombotic thrombocytopaenic purpura in HIV-infected patients. Int. J. STD AIDS 2005, 16, 538–542. [Google Scholar] [CrossRef]
- Gunther, K.; Garizio, D.; Nesara, P. ADAMTS13 activity and the presence of acquired inhibitors in human immunodeficiency virus-related thrombotic thrombocytopenic purpura. Transfusion 2007, 47, 1710–1716. [Google Scholar] [CrossRef]
- Park, Y.A.; Hay, S.N.; Brecher, M.E. ADAMTS13 activity levels in patients with human immunodeficiency virus-associated thrombotic microangiopathy and profound CD4 deficiency. J. Clin. Apher. 2009, 24, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Malak, S.; Wolf, M.; Millot, G.A.; Mariotte, E.; Veyradier, A.; Meynard, J.L.; Korach, J.M.; Malot, S.; Bussel, A.; Azoulay, E.; et al. Human immunodeficiency virus-associated thrombotic microangiopathies: Clinical characteristics and outcome according to ADAMTS13 activity. Scand. J. Immunol. 2008, 68, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Jokiranta, T.S. HUS and atypical HUS. Blood 2017, 129, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Masoet, A.; Bassa, F.; Chothia, M.Y. HIV-associated thrombotic thrombocytopaenic purpura: A retrospective cohort study during the anti-retroviral therapy era. J. Clin. Apher. 2019, 34, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Freist, M.; Garrouste, C.; Szlavik, N.; Coppo, P.; Lautrette, A.; Heng, A.E. Efficacy of eculizumab in an adult patient with HIV-associated hemolytic uremic syndrome: A case report. Medicine 2017, 96, e9358. [Google Scholar] [CrossRef]
- Gonzalez, D.E.T.; Misol, D.D.; Ordonez, N.A.; Zamora, F.A.S.; Millan Prada, H.A.; Zuniga, R.E.; Trivino, A.M. Atypical hemolytic uremic syndrome in a patient with HIV treated with eculizumab: A case report. IDCases 2023, 31, e01692. [Google Scholar] [CrossRef]
- Popescu, N.I.; Lupu, C.; Lupu, F. Disseminated intravascular coagulation and its immune mechanisms. Blood 2022, 139, 1973–1986. [Google Scholar] [CrossRef]
- Mayne, E.S.; Mayne, A.L.H.; Louw, S.J. Pathogenic factors associated with development of disseminated intravascular coagulopathy (DIC) in a tertiary academic hospital in South Africa. PLoS ONE 2018, 13, e0195793. [Google Scholar] [CrossRef] [PubMed]
- Kyrle, P.A.; Eichinger, S. Deep vein thrombosis. Lancet 2005, 365, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Peng, F.; Li, M.; Yi, Q.; Tang, W.; Wu, S. Elevated Risk of Venous Thromboembolism in People Living with HIV. Viruses 2022, 14, 590. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, C.; Jourdi, G.; Adjambri, E.; Walborn, A.; Patel, P.; Fareed, J.; Elalamy, I.; Hoppensteadt, D.; Gerotziafas, G.T. Disseminated Intravascular Coagulation: An Update on Pathogenesis, Diagnosis, and Therapeutic Strategies. Clin. Appl. Thromb. Hemost. 2018, 24, 8S–28S. [Google Scholar] [CrossRef] [PubMed]
- Chernysh, I.N.; Nagaswami, C.; Kosolapova, S.; Peshkova, A.D.; Cuker, A.; Cines, D.B.; Cambor, C.L.; Litvinov, R.I.; Weisel, J.W. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci. Rep. 2020, 10, 5112. [Google Scholar] [CrossRef]
- Mason, R.G.; Sharp, D.; Chuang, H.Y.; Mohammad, S.F. The endothelium: Roles in thrombosis and hemostasis. Arch. Pathol. Lab. Med. 1977, 101, 61–64. [Google Scholar]
- Vandendries, E.R.; Furie, B.C.; Furie, B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb. Haemost. 2004, 92, 459–466. [Google Scholar] [CrossRef]
- Diacovo, T.G.; Catalina, M.D.; Siegelman, M.H.; von Andrian, U.H. Circulating activated platelets reconstitute lymphocyte homing and immunity in L-selectin-deficient mice. J. Exp. Med. 1998, 187, 197–204. [Google Scholar] [CrossRef]
- Li, B.; Zhang, L.; Liu, Y.; Xiao, J.; Wang, X.; Wei, Y.; Fan, L.; Duan, Y.; Li, G.; Kong, Y.; et al. Manifestations and Related Risk Factors of Thrombocyte Abnormalities in HIV-Positive Patients Before and After the Initiation of ART. Infect. Drug Resist. 2021, 14, 4809–4819. [Google Scholar] [CrossRef]
- Schafer, A.I. Thrombocytosis. N. Engl. J. Med. 2004, 350, 1211–1219. [Google Scholar] [CrossRef]
- Ortel, T.L.; Neumann, I.; Ageno, W.; Beyth, R.; Clark, N.P.; Cuker, A.; Hutten, B.A.; Jaff, M.R.; Manja, V.; Schulman, S.; et al. American Society of Hematology 2020 guidelines for management of venous thromboembolism: Treatment of deep vein thrombosis and pulmonary embolism. Blood Adv. 2020, 4, 4693–4738. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.M.; Chane, T.; Patel, M.; Chen, S.; Xue, W.; Easley, K.A. Warfarin therapy in the HIV medical home model: Low rates of therapeutic anticoagulation despite adherence and differences in dosing based on specific antiretrovirals. AIDS Patient Care STDS 2012, 26, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Hokusai, V.T.E.I.; Buller, H.R.; Decousus, H.; Grosso, M.A.; Mercuri, M.; Middeldorp, S.; Prins, M.H.; Raskob, G.E.; Schellong, S.M.; Schwocho, L.; et al. Edoxaban versus warfarin for the treatment of symptomatic venous thromboembolism. N. Engl. J. Med. 2013, 369, 1406–1415. [Google Scholar] [CrossRef]
- Investigators, E.; Bauersachs, R.; Berkowitz, S.D.; Brenner, B.; Buller, H.R.; Decousus, H.; Gallus, A.S.; Lensing, A.W.; Misselwitz, F.; Prins, M.H.; et al. Oral rivaroxaban for symptomatic venous thromboembolism. N. Engl. J. Med. 2010, 363, 2499–2510. [Google Scholar] [CrossRef]
- Agnelli, G.; Buller, H.R.; Cohen, A.; Curto, M.; Gallus, A.S.; Johnson, M.; Masiukiewicz, U.; Pak, R.; Thompson, J.; Raskob, G.E.; et al. Oral apixaban for the treatment of acute venous thromboembolism. N. Engl. J. Med. 2013, 369, 799–808. [Google Scholar] [CrossRef]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; Eriksson, H.; Baanstra, D.; Schnee, J.; Goldhaber, S.Z.; Group, R.-C.S. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N. Engl. J. Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef]
- Ganji, R.; Ala, S.; Aarabi, M.; Baghery, B.; Salehifar, E. Comparison of Dabigatran vs. Warfarin in Acute Vnous Thromboemboly: Systematic Review. Iran J. Pharm. Res. 2016, 15, 611–617. [Google Scholar]
- Egan, G.; Hughes, C.A.; Ackman, M.L. Drug interactions between antiplatelet or novel oral anticoagulant medications and antiretroviral medications. Ann. Pharmacother. 2014, 48, 734–740. [Google Scholar] [CrossRef]
- Oliveira, R.; Patel, R.K.; Taylor, C.; Czuprynska, J.; Arya, R.; Roberts, L.N. Direct oral anticoagulants for the management of venous thromboembolism in patients with HIV—A single centre experience. Br. J. Haematol. 2019, 186, e148–e151. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.C.; Kuller, L.H.; Skanderson, M.; Lowy, E.; Kraemer, K.L.; Butt, A.A.; Bidwell Goetz, M.; Leaf, D.; Oursler, K.A.; et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.C.; Regan, S.; Feske, S.; Meigs, J.B.; Grinspoon, S.K.; Triant, V.A. Comparison of ischemic stroke incidence in HIV-infected and non-HIV-infected patients in a US health care system. J. Acquir. Immune Defic. Syndr. 2012, 60, 351–358. [Google Scholar] [CrossRef]
- Rasmussen, L.D.; Engsig, F.N.; Christensen, H.; Gerstoft, J.; Kronborg, G.; Pedersen, C.; Obel, N. Risk of cerebrovascular events in persons with and without HIV: A Danish nationwide population-based cohort study. AIDS 2011, 25, 1637–1646. [Google Scholar] [CrossRef]
- Lin, H.L.; Muo, C.H.; Lin, C.Y.; Chen, H.J.; Chen, P.C. Incidence of stroke in patients with HIV infection: A population-based study in Taiwan. PLoS ONE 2019, 14, e0217147. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Duncan, M.S.; Alcorn, C.W.; So-Armah, K.; Butt, A.A.; Goetz, M.B.; Tindle, H.A.; Sico, J.J.; Tracy, R.P.; Justice, A.C.; et al. Association of Human Immunodeficiency Virus Infection and Risk of Peripheral Artery Disease. Circulation 2018, 138, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Grinspoon, S.; Carr, A. Cardiovascular risk and body-fat abnormalities in HIV-infected adults. N. Engl. J. Med. 2005, 352, 48–62. [Google Scholar] [CrossRef]
- Pao, V.; Lee, G.A.; Grunfeld, C. HIV therapy, metabolic syndrome, and cardiovascular risk. Curr. Atheroscler. Rep. 2008, 10, 61–70. [Google Scholar] [CrossRef]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1-Associated Atherosclerosis: Unraveling the Missing Link. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef]
- D’Ascenzo, F.; Cerrato, E.; Calcagno, A.; Grossomarra, W.; Ballocca, F.; Omede, P.; Montefusco, A.; Veglia, S.; Barbero, U.; Gili, S.; et al. High prevalence at computed coronary tomography of non-calcified plaques in asymptomatic HIV patients treated with HAART: A meta-analysis. Atherosclerosis 2015, 240, 197–204. [Google Scholar] [CrossRef]
- Zanni, M.V.; Abbara, S.; Lo, J.; Wai, B.; Hark, D.; Marmarelis, E.; Grinspoon, S.K. Increased coronary atherosclerotic plaque vulnerability by coronary computed tomography angiography in HIV-infected men. AIDS 2013, 27, 1263–1272. [Google Scholar] [CrossRef]
- Willoughby, S.; Holmes, A.; Loscalzo, J. Platelets and cardiovascular disease. Eur J. Cardiovasc. Nurs. 2002, 1, 273–288. [Google Scholar] [CrossRef]
- Khodadi, E. Platelet Function in Cardiovascular Disease: Activation of Molecules and Activation by Molecules. Cardiovasc. Toxicol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Force, U.S.P.S.T.; Davidson, K.W.; Barry, M.J.; Mangione, C.M.; Cabana, M.; Chelmow, D.; Coker, T.R.; Davis, E.M.; Donahue, K.E.; Jaen, C.R.; et al. Aspirin Use to Prevent Cardiovascular Disease: US Preventive Services Task Force Recommendation Statement. JAMA 2022, 327, 1577–1584. [Google Scholar] [CrossRef]
- O’Brien, M.P.; Hunt, P.W.; Kitch, D.W.; Klingman, K.; Stein, J.H.; Funderburg, N.T.; Berger, J.S.; Tebas, P.; Clagett, B.; Moisi, D.; et al. A Randomized Placebo Controlled Trial of Aspirin Effects on Immune Activation in Chronically Human Immunodeficiency Virus-Infected Adults on Virologically Suppressive Antiretroviral Therapy. Open Forum Infect. Dis. 2017, 4, ofw278. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.; Montenont, E.; Hu, L.; Nardi, M.A.; Valdes, V.; Merolla, M.; Gettenberg, G.; Cavanagh, K.; Aberg, J.A.; Bhardwaj, N.; et al. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: A pilot study. J. Acquir. Immune Defic. Syndr. 2013, 63, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, E.; Garshick, M.S.; Schwartz, T.; Ratnapala, N.; Cambria, M.; Dann, R.; O’Brien, M.; Heguy, A.; Berger, J.S. Antiplatelet Effects of Clopidogrel Vs Aspirin in Virologically Controlled HIV: A Randomized Controlled Trial. JACC Basic Transl. Sci. 2022, 7, 1086–1097. [Google Scholar] [CrossRef]
- Feinstein, M.J.; Hsue, P.Y.; Benjamin, L.A.; Bloomfield, G.S.; Currier, J.S.; Freiberg, M.S.; Grinspoon, S.K.; Levin, J.; Longenecker, C.T.; Post, W.S. Characteristics, Prevention, and Management of Cardiovascular Disease in People Living With HIV: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e98–e124. [Google Scholar] [CrossRef]
- Grinspoon, S.K.; Fitch, K.V.; Zanni, M.V.; Fichtenbaum, C.J.; Umbleja, T.; Aberg, J.A.; Overton, E.T.; Malvestutto, C.D.; Bloomfield, G.S.; Currier, J.S.; et al. Pitavastatin to Prevent Cardiovascular Disease in HIV Infection. N. Engl. J. Med. 2023, 389, 687–699. [Google Scholar] [CrossRef]
- Boccara, F.; Mary-Krause, M.; Teiger, E.; Lang, S.; Lim, P.; Wahbi, K.; Beygui, F.; Milleron, O.; Gabriel Steg, P.; Funck-Brentano, C.; et al. Acute coronary syndrome in human immunodeficiency virus-infected patients: Characteristics and 1 year prognosis. Eur. Heart J. 2011, 32, 41–50. [Google Scholar] [CrossRef]
- D’Ascenzo, F.; Cerrato, E.; Biondi-Zoccai, G.; Moretti, C.; Omede, P.; Sciuto, F.; Bollati, M.; Modena, M.G.; Gaita, F.; Sheiban, I. Acute coronary syndromes in human immunodeficiency virus patients: A meta-analysis investigating adverse event rates and the role of antiretroviral therapy. Eur. Heart J. 2012, 33, 875–880. [Google Scholar] [CrossRef]
- Hauguel-Moreau, M.; Boccara, F.; Boyd, A.; Salem, J.E.; Brugier, D.; Curjol, A.; Hulot, J.S.; Kerneis, M.; Galier, S.; Cohen, A.; et al. Platelet reactivity in human immunodeficiency virus infected patients on dual antiplatelet therapy for an acute coronary syndrome: The EVERE2ST-HIV study. Eur. Heart J. 2017, 38, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Vannappagari, V.; Nkhoma, E.T.; Atashili, J.; Laurent, S.S.; Zhao, H. Prevalence, severity, and duration of thrombocytopenia among HIV patients in the era of highly active antiretroviral therapy. Platelets 2011, 22, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Kim, I.; Kim, N.J.; Lee, S.A.; Choi, Y.A.; Bae, J.Y.; Kwon, J.H.; Choe, P.G.; Park, W.B.; Yoon, S.S.; et al. Hematological manifestations of human immunodeficiency virus infection and the effect of highly active anti-retroviral therapy on cytopenia. Korean J. Hematol. 2011, 46, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Metcalf Pate, K.A.; Mankowski, J.L. HIV and SIV Associated Thrombocytopenia: An Expanding Role for Platelets in the Pathogenesis of HIV. Drug Discov. Today Dis. Mech. 2011, 8, e25–e32. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug | Rate of Thrombocytopenia * |
|---|---|
| Nucleoside/nucleotide reverse transcriptase inhibitor (NRTI) | |
| Abacavir | Grades 3/4: 1% |
| Didanosine | <1%, post-marketing and/or case reports |
| Emtricitabine | None reported in the FDA product label |
| Lamivudine | Adults: 4%; Children: 1% |
| Stavudine | <1%, post-marketing and/or case reports |
| Tenofovir disoproxil fumarate | None reported in the FDA product label |
| Tenofovir alafenamide | None reported in the FDA product label |
| Zidovudine | Infants, children, and adolescents, grades 3/4: 1% |
| Non-nucleoside reverse transcriptase inhibitor (NNRTI) | |
| Doravirine | None reported in the FDA product label |
| Efavirenz | None reported in the FDA product label |
| Etravirine | None reported in the FDA product label |
| Nevirapine | Thrombocytopenia rates similar to placebo |
| Rilpivirine | None reported in the FDA product label |
| Protease inhibitor | |
| Atazanavir | Grades ¾: 2% |
| Darunavir | None reported in the FDA product label |
| Fosamprenavir calcium | None reported in the FDA product label |
| Lopinavir/ritonavir | Grade 3/4: children: 4% |
| Integrase inhibitor | |
| Bictegravir | None reported in the FDA product label |
| Cabotegravir | None reported in the FDA product label |
| Dolutegravir | None reported in the FDA product label |
| Raltegravir | Post-marketing reports of thrombocytopenia |
| Other pharmacologic category | |
| Cobicistat ** | None reported in the FDA product label |
| Enfuvirtide | <1%, post-marketing, and/or case reports |
| Fostemsavir | None reported in the FDA product label |
| Ibalizumab | Decreased platelet count < 50,000/mm3: 3% |
| Lenacapavir | None reported in the FDA product label |
| Maraviroc | None reported in the FDA product label |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awamura, T.; Nakasone, E.S.; Gangcuangco, L.M.; Subia, N.T.; Bali, A.-J.; Chow, D.C.; Shikuma, C.M.; Park, J. Platelet and HIV Interactions and Their Contribution to Non-AIDS Comorbidities. Biomolecules 2023, 13, 1608. https://doi.org/10.3390/biom13111608
Awamura T, Nakasone ES, Gangcuangco LM, Subia NT, Bali A-J, Chow DC, Shikuma CM, Park J. Platelet and HIV Interactions and Their Contribution to Non-AIDS Comorbidities. Biomolecules. 2023; 13(11):1608. https://doi.org/10.3390/biom13111608
Chicago/Turabian StyleAwamura, Thomas, Elizabeth S. Nakasone, Louie Mar Gangcuangco, Natalie T. Subia, Aeron-Justin Bali, Dominic C. Chow, Cecilia M. Shikuma, and Juwon Park. 2023. "Platelet and HIV Interactions and Their Contribution to Non-AIDS Comorbidities" Biomolecules 13, no. 11: 1608. https://doi.org/10.3390/biom13111608