
Mechanisms of the Beneficial Effects of Exercise on Brain-Derived Neurotrophic Factor Expression in Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
:1. Alzheimer’s Disease
2. Physical Exercise and Brain Health
3. Role of Brain-Derived Neurotrophic Factor
4. Mechanisms of BDNF Upregulation with Exercise
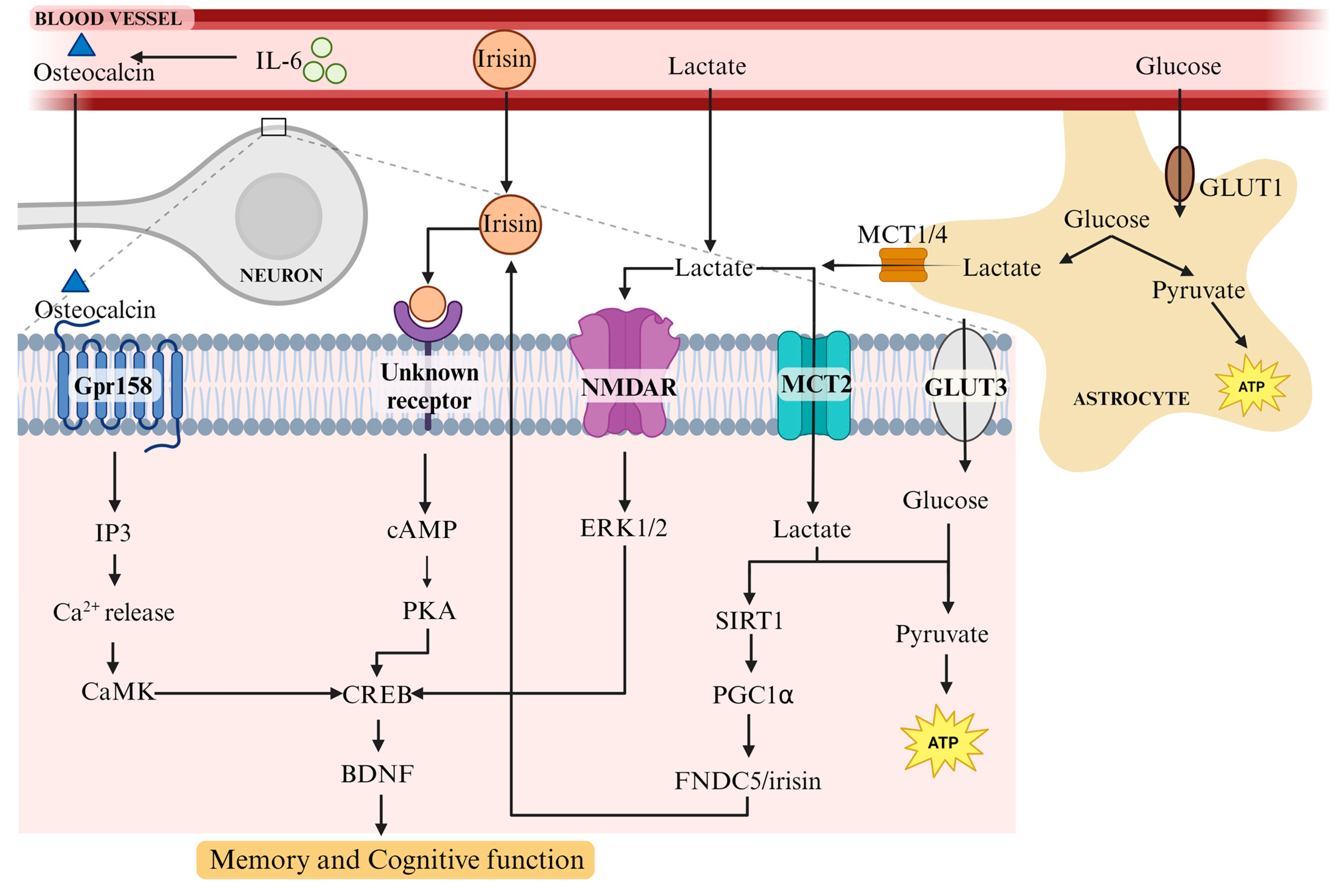
4.1. Physical Exercise Has a Multifaceted Effect on the Body
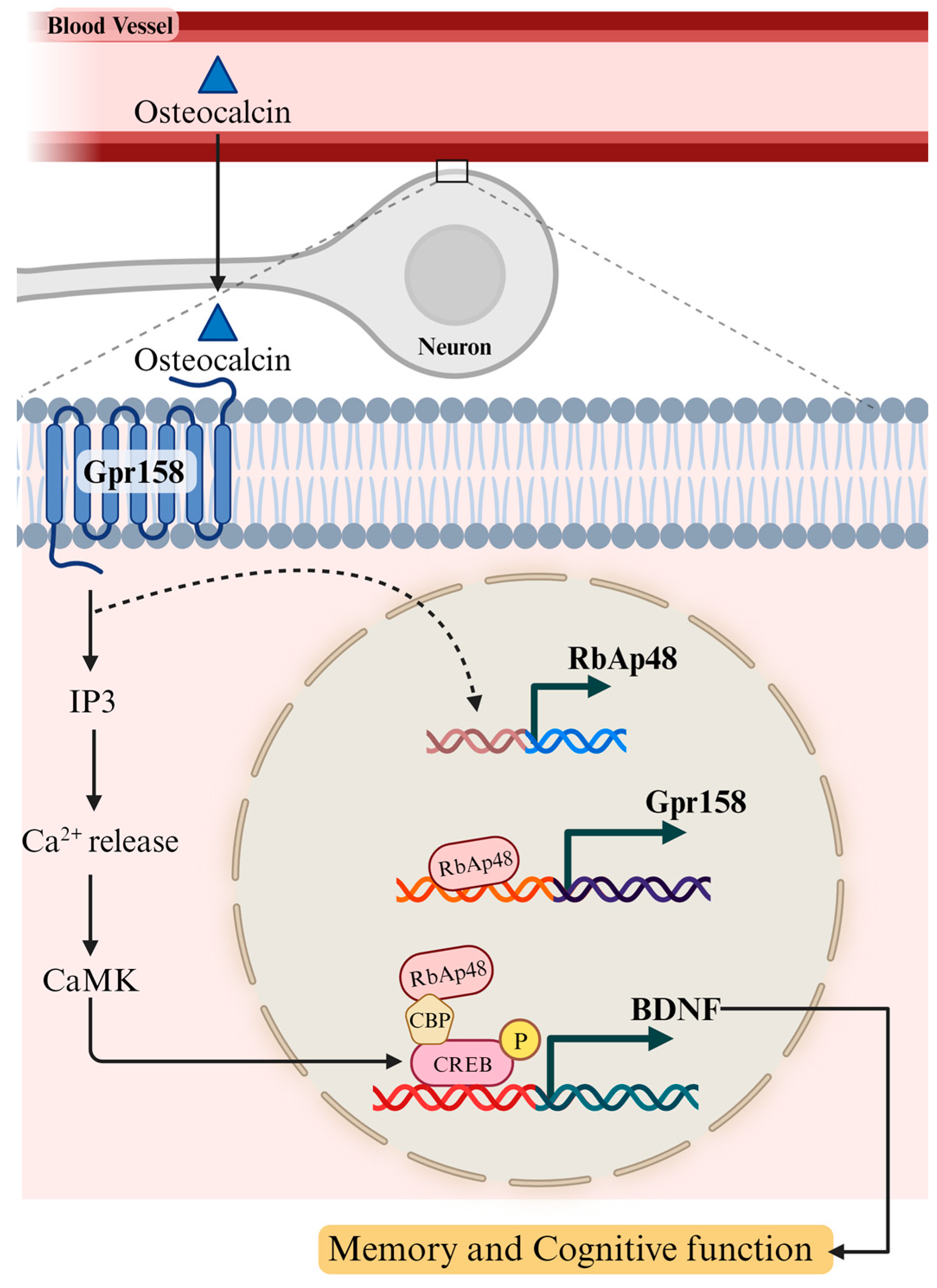
4.2. Osteocalcin, a Bone-Derived Hormone, Plays a Role in Learning and Memory via BDNF
4.3. FNDC5/Irisin Increases BDNF Levels in the Hippocampus and Supports Learning and Memory
4.4. Lactate Release from Muscle following Exercise Induces BDNF in the Hippocampus and Promotes Learning and Long-Term Memory Formation
5. Exercise-Induced BDNF Reduces APP Toxicity by Altering Its Processing
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alzheimer’s Association. 2023 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s Disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Amyloid Beta Protein Precursor and the Pathogenesis of Alzheimer’s Disease. Cell 1989, 58, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Sisodia, S.S.; Price, D.L. Role of the Beta-Amyloid Protein in Alzheimer’s Disease. FASEB J. 1995, 9, 366–370. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular Mechanisms of Neurodegeneration in Alzheimer’s Disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Mori, C.; Spooner, E.T.; Wisniewsk, K.E.; Wisniewski, T.M.; Yamaguch, H.; Saido, T.C.; Tolan, D.R.; Selkoe, D.J.; Lemere, C.A. Intraneuronal Abeta42 Accumulation in Down Syndrome Brain. Amyloid 2002, 9, 88–102. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Abeta42 Accumulation in Human Brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Bayer, T.A.; Wirths, O.; Majtényi, K.; Hartmann, T.; Multhaup, G.; Beyreuther, K.; Czech, C. Key Factors in Alzheimer’s Disease: Beta-Amyloid Precursor Protein Processing, Metabolism and Intraneuronal Transport. Brain Pathol. 2001, 11, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selective Cytotoxicity of Intracellular Amyloid β Peptide1–42 through P53 and Bax in Cultured Primary Human Neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar] [CrossRef]
- Chui, D.H.; Dobo, E.; Makifuchi, T.; Akiyama, H.; Kawakatsu, S.; Petit, A.; Checler, F.; Araki, W.; Takahashi, K.; Tabira, T. Apoptotic Neurons in Alzheimer’s Disease Frequently Show Intracellular Abeta42 Labeling. J. Alzheimers Dis. 2001, 3, 231–239. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-Associated Protein Tau. A Component of Alzheimer Paired Helical Filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I.; Smith, A.J.; George, L.; Tung, Y.C.; Zaidi, T. Identification and Localization of a Tau Peptide to Paired Helical Filaments of Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1989, 86, 5646–5650. [Google Scholar] [CrossRef]
- Lee, V.M.; Balin, B.J.; Otvos, L.; Trojanowski, J.Q. A68: A Major Subunit of Paired Helical Filaments and Derivatized Forms of Normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef]
- Alonso, A.C.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Role of Abnormally Phosphorylated Tau in the Breakdown of Microtubules in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1994, 91, 5562–5566. [Google Scholar] [CrossRef]
- Li, B.; Chohan, M.O.; Grundke-Iqbal, I.; Iqbal, K. Disruption of Microtubule Network by Alzheimer Abnormally Hyperphosphorylated Tau. Acta Neuropathol. 2007, 113, 501–511. [Google Scholar] [CrossRef]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.-P.; Hamdane, M.; Buée, L. Alzheimer’s Disease-like Tau Neuropathology Leads to Memory Deficits and Loss of Functional Synapses in a Novel Mutated Tau Transgenic Mouse without Any Motor Deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef]
- Cotman, C.W.; Berchtold, N.C.; Christie, L.-A. Exercise Builds Brain Health: Key Roles of Growth Factor Cascades and Inflammation. Trends Neurosci. 2007, 30, 464–472. [Google Scholar] [CrossRef]
- Voss, M.W.; Nagamatsu, L.S.; Liu-Ambrose, T.; Kramer, A.F. Exercise, Brain, and Cognition across the Life Span. J. Appl. Physiol. 2011, 111, 1505–1513. [Google Scholar] [CrossRef]
- Erickson, K.I.; Leckie, R.L.; Weinstein, A.M. Physical Activity, Fitness, and Gray Matter Volume. Neurobiol. Aging 2014, 35 (Suppl. 2), S20–S28. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, D.E.; Farrar, R.P. Enhancement of Spatial Learning in F344 Rats by Physical Activity and Related Learning-Associated Alterations in Hippocampal and Cortical Cholinergic Functioning. Behav. Brain Res. 1991, 46, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Griffin, É.W.; Bechara, R.G.; Birch, A.M.; Kelly, Á.M. Exercise Enhances Hippocampal-Dependent Learning in the Rat: Evidence for a BDNF-Related Mechanism. Hippocampus 2009, 19, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.E.; Parachikova, A.I.; Cotman, C.W. Three Weeks of Running Wheel Exposure Improves Cognitive Performance in the Aged Tg2576 Mouse. Behav. Brain Res. 2007, 184, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic Exercise for Alzheimer’s Disease: A Randomized Controlled Pilot Trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef]
- Johansson, M.E.; Cameron, I.G.M.; Van der Kolk, N.M.; de Vries, N.M.; Klimars, E.; Toni, I.; Bloem, B.R.; Helmich, R.C. Aerobic Exercise Alters Brain Function and Structure in Parkinson’s Disease: A Randomized Controlled Trial. Ann. Neurol. 2022, 91, 203–216. [Google Scholar] [CrossRef]
- Tarumi, T.; Patel, N.R.; Tomoto, T.; Pasha, E.; Khan, A.M.; Kostroske, K.; Riley, J.; Tinajero, C.D.; Wang, C.; Hynan, L.S.; et al. Aerobic Exercise Training and Neurocognitive Function in Cognitively Normal Older Adults: A One-Year Randomized Controlled Trial. J. Intern. Med. 2022, 292, 788–803. [Google Scholar] [CrossRef]
- Sanders, L.M.J.; Hortobágyi, T.; Karssemeijer, E.G.A.; Van der Zee, E.A.; Scherder, E.J.A.; van Heuvelen, M.J.G. Effects of Low- and High-Intensity Physical Exercise on Physical and Cognitive Function in Older Persons with Dementia: A Randomized Controlled Trial. Alzheimers Res. Ther. 2020, 12, 28. [Google Scholar] [CrossRef]
- Lenze, E.J.; Voegtle, M.; Miller, J.P.; Ances, B.M.; Balota, D.A.; Barch, D.; Depp, C.A.; Diniz, B.S.; Eyler, L.T.; Foster, E.R.; et al. Effects of Mindfulness Training and Exercise on Cognitive Function in Older Adults: A Randomized Clinical Trial. JAMA 2022, 328, 2218–2229. [Google Scholar] [CrossRef]
- Lamb, S.E.; Sheehan, B.; Atherton, N.; Nichols, V.; Collins, H.; Mistry, D.; Dosanjh, S.; Slowther, A.M.; Khan, I.; Petrou, S.; et al. Dementia And Physical Activity (DAPA) Trial of Moderate to High Intensity Exercise Training for People with Dementia: Randomised Controlled Trial. BMJ 2018, 361, k1675. [Google Scholar] [CrossRef]
- Gates, N.; Fiatarone Singh, M.A.; Sachdev, P.S.; Valenzuela, M. The Effect of Exercise Training on Cognitive Function in Older Adults with Mild Cognitive Impairment: A Meta-Analysis of Randomized Controlled Trials. Am. J. Geriatr. Psychiatry 2013, 21, 1086–1097. [Google Scholar] [CrossRef]
- Panza, G.A.; Taylor, B.A.; MacDonald, H.V.; Johnson, B.T.; Zaleski, A.L.; Livingston, J.; Thompson, P.D.; Pescatello, L.S. Can Exercise Improve Cognitive Symptoms of Alzheimer’s Disease? J. Am. Geriatr. Soc. 2018, 66, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Öhman, H.; Savikko, N.; Strandberg, T.E.; Kautiainen, H.; Raivio, M.M.; Laakkonen, M.-L.; Tilvis, R.; Pitkälä, K.H. Effects of Exercise on Cognition: The Finnish Alzheimer Disease Exercise Trial: A Randomized, Controlled Trial. J. Am. Geriatr. Soc. 2016, 64, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Venegas-Sanabria, L.C.; Cavero-Redondo, I.; Martínez-Vizcaino, V.; Cano-Gutierrez, C.A.; Álvarez-Bueno, C. Effect of Multicomponent Exercise in Cognitive Impairment: A Systematic Review and Meta-Analysis. BMC Geriatr. 2022, 22, 617. [Google Scholar] [CrossRef] [PubMed]
- Szuhany, K.L.; Bugatti, M.; Otto, M.W. A Meta-Analytic Review of the Effects of Exercise on Brain-Derived Neurotrophic Factor. J. Psychiatr. Res. 2015, 60, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Lin, M.; Gao, J.; Xu, S.; Huang, L.; Zhu, J.; Huang, J.; Tao, J.; Chen, L. The Impact of Physical Activity on Blood Inflammatory Cytokines and Neuroprotective Factors in Individuals with Mild Cognitive Impairment: A Systematic Review and Meta-Analysis of Randomized-Controlled Trials. Aging Clin. Exp. Res. 2022, 34, 1471–1484. [Google Scholar] [CrossRef]
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of Brain-Derived Neurotrophic Factor into the Lateral Ventricle of the Adult Rat Leads to New Neurons in the Parenchyma of the Striatum, Septum, Thalamus, and Hypothalamus. J. Neurosci. 2001, 21, 6706–6717. [Google Scholar] [CrossRef]
- Causing, C.G.; Gloster, A.; Aloyz, R.; Bamji, S.X.; Chang, E.; Fawcett, J.; Kuchel, G.; Miller, F.D. Synaptic Innervation Density Is Regulated by Neuron-Derived BDNF. Neuron 1997, 18, 257–267. [Google Scholar] [CrossRef]
- Horch, H.W.; Krüttgen, A.; Portbury, S.D.; Katz, L.C. Destabilization of Cortical Dendrites and Spines by BDNF. Neuron 1999, 23, 353–364. [Google Scholar] [CrossRef]
- Lu, B. BDNF and Activity-Dependent Synaptic Modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef]
- Scharfman, H.E. Hyperexcitability in Combined Entorhinal/Hippocampal Slices of Adult Rat After Exposure to Brain-Derived Neurotrophic Factor. J. Neurophysiol. 1997, 78, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Verpelli, C.; Piccoli, G.; Zibetti, C.; Zanchi, A.; Gardoni, F.; Huang, K.; Brambilla, D.; Di Luca, M.; Battaglioli, E.; Sala, C. Synaptic Activity Controls Dendritic Spine Morphology by Modulating eEF2-Dependent BDNF Synthesis. J. Neurosci. 2010, 30, 5830–5842. [Google Scholar] [CrossRef] [PubMed]
- Neeper, S.A.; Góauctemez-Pinilla, F.; Choi, J.; Cotman, C. Exercise and Brain Neurotrophins. Nature 1995, 373, 109. [Google Scholar] [CrossRef] [PubMed]
- Neeper, S.A.; Gómez-Pinilla, F.; Choi, J.; Cotman, C.W. Physical Activity Increases mRNA for Brain-Derived Neurotrophic Factor and Nerve Growth Factor in Rat Brain. Brain Res. 1996, 726, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pinilla, F.; Ying, Z.; Opazo, P.; Roy, R.R.; Edgerton, V.R. Differential Regulation by Exercise of BDNF and NT-3 in Rat Spinal Cord and Skeletal Muscle. Eur. J. Neurosci. 2001, 13, 1078–1084. [Google Scholar] [CrossRef]
- de Melo Coelho, F.G.; Gobbi, S.; Andreatto, C.A.A.; Corazza, D.I.; Pedroso, R.V.; Santos-Galduróz, R.F. Physical Exercise Modulates Peripheral Levels of Brain-Derived Neurotrophic Factor (BDNF): A Systematic Review of Experimental Studies in the Elderly. Arch. Gerontol. Geriatr. 2013, 56, 10–15. [Google Scholar] [CrossRef]
- Laske, C.; Banschbach, S.; Stransky, E.; Bosch, S.; Straten, G.; Machann, J.; Fritsche, A.; Hipp, A.; Niess, A.; Eschweiler, G.W. Exercise-Induced Normalization of Decreased BDNF Serum Concentration in Elderly Women with Remitted Major Depression. Int. J. Neuropsychopharmacol. 2010, 13, 595–602. [Google Scholar] [CrossRef]
- Nicolini, C.; Michalski, B.; Toepp, S.L.; Turco, C.V.; D’Hoine, T.; Harasym, D.; Gibala, M.J.; Fahnestock, M.; Nelson, A.J. A Single Bout of High-Intensity Interval Exercise Increases Corticospinal Excitability, Brain-Derived Neurotrophic Factor, and Uncarboxylated Osteolcalcin in Sedentary, Healthy Males. Neuroscience 2020, 437, 242–255. [Google Scholar] [CrossRef]
- Tsai, C.-L.; Chen, F.-C.; Pan, C.-Y.; Wang, C.-H.; Huang, T.-H.; Chen, T.-C. Impact of Acute Aerobic Exercise and Cardiorespiratory Fitness on Visuospatial Attention Performance and Serum BDNF Levels. Psychoneuroendocrinology 2014, 41, 121–131. [Google Scholar] [CrossRef]
- Håkansson, K.; Ledreux, A.; Daffner, K.; Terjestam, Y.; Bergman, P.; Carlsson, R.; Kivipelto, M.; Winblad, B.; Granholm, A.-C.; Mohammed, A.K.H. BDNF Responses in Healthy Older Persons to 35 Minutes of Physical Exercise, Cognitive Training, and Mindfulness: Associations with Working Memory Function. J. Alzheimers Dis. 2017, 55, 645–657. [Google Scholar] [CrossRef]
- Fujimura, H.; Altar, C.A.; Chen, R.; Nakamura, T.; Nakahashi, T.; Kambayashi, J.; Sun, B.; Tandon, N.N. Brain-Derived Neurotrophic Factor Is Stored in Human Platelets and Released by Agonist Stimulation. Thromb. Haemost. 2002, 87, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.J.; Tschakovsky, M.E. Exercise and Circulating BDNF: Mechanisms of Release and Implications for the Design of Exercise Interventions. Appl. Physiol. Nutr. Metab. 2018, 43, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of Brain-Derived Neurotrophic Factor across the Blood–Brain Barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a Release of Brain-Derived Neurotrophic Factor from the Brain during Exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Pardridge, W.M.; Kang, Y.S.; Buciak, J.L. Transport of Human Recombinant Brain-Derived Neurotrophic Factor (BDNF) through the Rat Blood-Brain Barrier in Vivo Using Vector-Mediated Peptide Drug Delivery. Pharm. Res. 1994, 11, 738–746. [Google Scholar] [CrossRef]
- Klein, A.B.; Williamson, R.; Santini, M.A.; Clemmensen, C.; Ettrup, A.; Rios, M.; Knudsen, G.M.; Aznar, S. Blood BDNF Concentrations Reflect Brain-Tissue BDNF Levels across Species. Int. J. Neuropsychopharmacol. 2011, 14, 347–353. [Google Scholar] [CrossRef]
- Sartorius, A.; Hellweg, R.; Litzke, J.; Vogt, M.; Dormann, C.; Vollmayr, B.; Danker-Hopfe, H.; Gass, P. Correlations and Discrepancies between Serum and Brain Tissue Levels of Neurotrophins after Electroconvulsive Treatment in Rats. Pharmacopsychiatry 2009, 42, 270–276. [Google Scholar] [CrossRef]
- Karege, F.; Schwald, M.; Cisse, M. Postnatal Developmental Profile of Brain-Derived Neurotrophic Factor in Rat Brain and Platelets. Neurosci. Lett. 2002, 328, 261–264. [Google Scholar] [CrossRef]
- Want, A.; Morgan, J.E.; Barde, Y.-A. Brain-Derived Neurotrophic Factor Measurements in Mouse Serum and Plasma Using a Sensitive and Specific Enzyme-Linked Immunosorbent Assay. Sci. Rep. 2023, 13, 7740. [Google Scholar] [CrossRef]
- Heisz, J.J.; Clark, I.B.; Bonin, K.; Paolucci, E.M.; Michalski, B.; Becker, S.; Fahnestock, M. The Effects of Physical Exercise and Cognitive Training on Memory and Neurotrophic Factors. J. Cogn. Neurosci. 2017, 29, 1895–1907. [Google Scholar] [CrossRef]
- Arazi, H.; Babaei, P.; Moghimi, M.; Asadi, A. Acute Effects of Strength and Endurance Exercise on Serum BDNF and IGF-1 Levels in Older Men. BMC Geriatr. 2021, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.J.; Green, D.J.; Ellis, K.A.; Cerin, E.; Heinonen, I.; Naylor, L.H.; Larsen, R.; Wennberg, P.; Boraxbekk, C.-J.; Lewis, J.; et al. Distinct Effects of Acute Exercise and Breaks in Sitting on Working Memory and Executive Function in Older Adults: A Three-Arm, Randomised Cross-over Trial to Evaluate the Effects of Exercise with and without Breaks in Sitting on Cognition. Br. J. Sports Med. 2020, 54, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Coelho, F.M.; Pereira, D.S.; Lustosa, L.P.; Silva, J.P.; Dias, J.M.D.; Dias, R.C.D.; Queiroz, B.Z.; Teixeira, A.L.; Teixeira, M.M.; Pereira, L.S.M. Physical Therapy Intervention (PTI) Increases Plasma Brain-Derived Neurotrophic Factor (BDNF) Levels in Non-Frail and Pre-Frail Elderly Women. Arch. Gerontol. Geriatr. 2012, 54, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Ruscheweyh, R.; Willemer, C.; Krüger, K.; Duning, T.; Warnecke, T.; Sommer, J.; Völker, K.; Ho, H.V.; Mooren, F.; Knecht, S.; et al. Physical Activity and Memory Functions: An Interventional Study. Neurobiol. Aging 2011, 32, 1304–1319. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, A.; Fenesi, B.; Paolucci, E.; Heisz, J.J. The Effects of Aerobic Exercise Intensity on Memory in Older Adults. Appl. Physiol. Nutr. Metab. 2020, 45, 591–600. [Google Scholar] [CrossRef]
- Erickson, K.I.; Voss, M.W.; Prakash, R.S.; Basak, C.; Szabo, A.; Chaddock, L.; Kim, J.S.; Heo, S.; Alves, H.; White, S.M.; et al. Exercise Training Increases Size of Hippocampus and Improves Memory. Proc. Natl. Acad. Sci. USA 2011, 108, 3017–3022. [Google Scholar] [CrossRef]
- Fahnestock, M. BDNF: The Link between Beta-Amyloid and Memory Loss. Future Neurol. 2011, 6, 627–639. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and Synaptic Plasticity, Cognitive Function, and Dysfunction. In Neurotrophic Factors; Lewin, G.R., Carter, B.D., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin, Heidelberg, 2014; pp. 223–250. ISBN 978-3-642-45106-5. [Google Scholar]
- Binder, D.K.; Scharfman, H.E. Brain-Derived Neurotrophic Factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in Neuronal Development and Function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Lu, Y.; Christian, K.; Lu, B. BDNF: A Key Regulator for Protein-Synthesis Dependent LTP and Long-Term Memory? Neurobiol. Learn. Mem. 2008, 89, 312–323. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor Form of Brain-Derived Neurotrophic Factor and Mature Brain-Derived Neurotrophic Factor Are Decreased in the Pre-Clinical Stages of Alzheimer’s Disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Siuda, J.; Patalong-Ogiewa, M.; Żmuda, W.; Targosz-Gajniak, M.; Niewiadomska, E.; Matuszek, I.; Jędrzejowska-Szypułka, H.; Lewin-Kowalik, J.; Rudzińska-Bar, M. Cognitive Impairment and BDNF Serum Levels. Neurol. Neurochir. Pol. 2017, 51, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Michalski, B.; Fahnestock, M. Pro-Brain-Derived Neurotrophic Factor Is Decreased in Parietal Cortex in Alzheimer’s Disease. Mol. Brain Res. 2003, 111, 148–154. [Google Scholar] [CrossRef]
- Pruunsild, P.; Sepp, M.; Orav, E.; Koppel, I.; Timmusk, T. Identification of Cis-Elements and Transcription Factors Regulating Neuronal Activity-Dependent Transcription of Human BDNF Gene. J. Neurosci. 2011, 31, 3295–3308. [Google Scholar] [CrossRef]
- Esvald, E.-E.; Tuvikene, J.; Sirp, A.; Patil, S.; Bramham, C.R.; Timmusk, T. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J. Neurosci. 2020, 40, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Moya-Alvarado, G.; Tiburcio-Felix, R.; Ibáñez, M.R.; Aguirre-Soto, A.A.; Guerra, M.V.; Wu, C.; Mobley, W.C.; Perlson, E.; Bronfman, F.C. BDNF/TrkB Signaling Endosomes in Axons Coordinate CREB/mTOR Activation and Protein Synthesis in the Cell Body to Induce Dendritic Growth in Cortical Neurons. eLife 2023, 12, e77455. [Google Scholar] [CrossRef] [PubMed]
- Narasimhamurthy, R.K.; Andrade, D.; Mumbrekar, K.D. Modulation of CREB and Its Associated Upstream Signaling Pathways in Pesticide-Induced Neurotoxicity. Mol. Cell. Biochem. 2022, 477, 2581–2593. [Google Scholar] [CrossRef]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid β-Peptide Inhibition of the PKA/CREB Pathway and Long-Term Potentiation: Reversibility by Drugs That Enhance cAMP Signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef]
- Garzon, D.J.; Fahnestock, M. Oligomeric Amyloid Decreases Basal Levels of Brain-Derived Neurotrophic Factor (BDNF) mRNA via Specific Downregulation of BDNF Transcripts IV and V in Differentiated Human Neuroblastoma Cells. J. Neurosci. 2007, 27, 2628–2635. [Google Scholar] [CrossRef]
- Rosa, E.; Fahnestock, M. CREB Expression Mediates Amyloid β-Induced Basal BDNF Downregulation. Neurobiol. Aging 2015, 36, 2406–2413. [Google Scholar] [CrossRef]
- Belrose, J.C.; Masoudi, R.; Michalski, B.; Fahnestock, M. Increased Pro-Nerve Growth Factor and Decreased Brain-Derived Neurotrophic Factor in Non-Alzheimer’s Disease Tauopathies. Neurobiol. Aging 2014, 35, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Rosa, E.; Mahendram, S.; Ke, Y.D.; Ittner, L.M.; Ginsberg, S.D.; Fahnestock, M. Tau Downregulates BDNF Expression in Animal and Cellular Models of Alzheimer’s Disease. Neurobiol. Aging 2016, 48, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yin, Y.; Liu, H.; Fang, L.; Tao, X.; Wei, L.; Zuo, Y.; Yin, Y.; Ke, D.; Wang, J.-Z. Tau Inhibits PKA by Nuclear Proteasome-Dependent PKAR2α Elevation with Suppressed CREB/GluA1 Phosphorylation. Aging Cell 2020, 19, e13055. [Google Scholar] [CrossRef]
- Pedersen, B.K. Physical Activity and Muscle–Brain Crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392. [Google Scholar] [CrossRef]
- Panati, K.; Suneetha, Y.; Narala, V.R. Irisin/FNDC5--An Updated Review. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 689–697. [Google Scholar] [PubMed]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-Linked FNDC5/Irisin Rescues Synaptic Plasticity and Memory Defects in Alzheimer’s Models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef]
- Yang, J.; Ruchti, E.; Petit, J.-M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate Promotes Plasticity Gene Expression by Potentiating NMDA Signaling in Neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef]
- El Hayek, L.; Khalifeh, M.; Zibara, V.; Abi Assaad, R.; Emmanuel, N.; Karnib, N.; El-Ghandour, R.; Nasrallah, P.; Bilen, M.; Ibrahim, P.; et al. Lactate Mediates the Effects of Exercise on Learning and Memory through SIRT1-Dependent Activation of Hippocampal Brain-Derived Neurotrophic Factor (BDNF). J. Neurosci. 2019, 39, 2369–2382. [Google Scholar] [CrossRef]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.; Greig, N.; Mattison, J.; et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef]
- Agudelo, L.Z.; Femenía, T.; Orhan, F.; Porsmyr-Palmertz, M.; Goiny, M.; Martinez-Redondo, V.; Correia, J.C.; Izadi, M.; Bhat, M.; Schuppe-Koistinen, I.; et al. Skeletal Muscle PGC-1α1 Modulates Kynurenine Metabolism and Mediates Resilience to Stress-Induced Depression. Cell 2014, 159, 33–45. [Google Scholar] [CrossRef]
- Hamrick, M.W. A Role for Myokines in Muscle-Bone Interactions. Exerc. Sport. Sci. Rev. 2011, 39, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Berg, U.; Bang, P. Exercise and Circulating Insulin-like Growth Factor I. Horm. Res. 2004, 62 (Suppl. 1), 50–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Russo-Neustadt, A.A. Running Exercise-Induced up-Regulation of Hippocampal Brain-Derived Neurotrophic Factor Is CREB-Dependent. Hippocampus 2009, 19, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.S.; Pedersen, B.K.; Xu, G.; Lehmann, R.; Weigert, C.; Plomgaard, P. Exercise-Induced Secretion of FGF21 and Follistatin Are Blocked by Pancreatic Clamp and Impaired in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 2816–2825. [Google Scholar] [CrossRef]
- Sa-Nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Satjaritanun, P.; Wang, X.; Liang, G.; Li, X.; Jiang, C.; Pratchayasakul, W.; Chattipakorn, N.; et al. FGF21 Improves Cognition by Restored Synaptic Plasticity, Dendritic Spine Density, Brain Mitochondrial Function and Cell Apoptosis in Obese-Insulin Resistant Male Rats. Horm. Behav. 2016, 85, 86–95. [Google Scholar] [CrossRef]
- Bradburn, S.; McPhee, J.S.; Bagley, L.; Sipila, S.; Stenroth, L.; Narici, M.V.; Pääsuke, M.; Gapeyeva, H.; Osborne, G.; Sassano, L.; et al. Association between Osteocalcin and Cognitive Performance in Healthy Older Adults. Age Ageing 2016, 45, 844–849. [Google Scholar] [CrossRef]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise Promotes the Expression of Brain Derived Neurotrophic Factor (BDNF) through the Action of the Ketone Body β-Hydroxybutyrate. eLife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Moser, S.C.; van der Eerden, B.C.J. Osteocalcin—A Versatile Bone-Derived Hormone. Front. Endocrinol. 2019, 9, 794. [Google Scholar] [CrossRef]
- Aktitiz, S.; Atakan, M.M.; Turnagöl, H.H.; Koşar, Ş.N. Interleukin-6, Undercarboxylated Osteocalcin, and Brain-Derived Neurotrophic Factor Responses to Single and Repeated Sessions of High-Intensity Interval Exercise. Peptides 2022, 157, 170864. [Google Scholar] [CrossRef]
- Levinger, I.; Jerums, G.; Stepto, N.K.; Parker, L.; Serpiello, F.R.; McConell, G.K.; Anderson, M.; Hare, D.L.; Byrnes, E.; Ebeling, P.R.; et al. The Effect of Acute Exercise on Undercarboxylated Osteocalcin and Insulin Sensitivity in Obese Men. J. Bone Miner. Res. 2014, 29, 2571–2576. [Google Scholar] [CrossRef]
- Chowdhury, S.; Schulz, L.; Palmisano, B.; Singh, P.; Berger, J.M.; Yadav, V.K.; Mera, P.; Ellingsgaard, H.; Hidalgo, J.; Brüning, J.; et al. Muscle-Derived Interleukin 6 Increases Exercise Capacity by Signaling in Osteoblasts. J. Clin. Investig. 2020, 130, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Puig, J.; Blasco, G.; Daunis-i-Estadella, J.; Moreno, M.; Molina, X.; Alberich-Bayarri, A.; Xifra, G.; Pedraza, S.; Ricart, W.; Fernández-Aranda, F.; et al. Lower Serum Osteocalcin Concentrations Are Associated with Brain Microstructural Changes and Worse Cognitive Performance. Clin. Endocrinol. 2016, 84, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Zhang, D.; Ma, D.-N.; Hou, Y.-F.; Zhuang, Q.-Q.; Gong, Y.-L.; Sun, L.-H.; Zhao, H.-Y.; Tao, B.; Yang, Y.-Y.; et al. Osteocalcin Ameliorates Cognitive Dysfunctions in a Mouse Model of Alzheimer’s Disease by Reducing Amyloid β Burden and Upregulating Glycolysis in Neuroglia. Cell Death Discov. 2023, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.; Lee, H.; Srinivas, P.; Gao, X.-B.; et al. Maternal and Offspring Pools of Osteocalcin Influence Brain Development and Functions. Cell 2013, 155, 228–241. [Google Scholar] [CrossRef]
- Nakamura, M.; Imaoka, M.; Takeda, M. Interaction of Bone and Brain: Osteocalcin and Cognition. Int. J. Neurosci. 2021, 131, 1115–1123. [Google Scholar] [CrossRef]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young Blood Reverses Age-Related Impairments in Cognitive Function and Synaptic Plasticity in Mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef]
- Khrimian, L.; Obri, A.; Ramos-Brossier, M.; Rousseaud, A.; Moriceau, S.; Nicot, A.-S.; Mera, P.; Kosmidis, S.; Karnavas, T.; Saudou, F.; et al. Gpr158 Mediates Osteocalcin’s Regulation of Cognition. J. Exp. Med. 2017, 214, 2859–2873. [Google Scholar] [CrossRef]
- Mikoshiba, K. IP3 Receptor/Ca2+ Channel: From Discovery to New Signaling Concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Nakashima, T.; Takemoto-Kimura, S.; Aoki, K.; Morishita, Y.; Asahara, H.; Ohya, K.; Yamaguchi, A.; Takai, T.; et al. Regulation of Osteoclast Differentiation and Function by the CaMK-CREB Pathway. Nat. Med. 2006, 12, 1410–1416. [Google Scholar] [CrossRef]
- Kosmidis, S.; Polyzos, A.; Harvey, L.; Youssef, M.; Denny, C.A.; Dranovsky, A.; Kandel, E.R. RbAp48 Protein Is a Critical Component of GPR158/OCN Signaling and Ameliorates Age-Related Memory Loss. Cell Rep. 2018, 25, 959–973.e6. [Google Scholar] [CrossRef]
- Zhang, Q.; Vo, N.; Goodman, R.H. Histone Binding Protein RbAp48 Interacts with a Complex of CREB Binding Protein and Phosphorylated CREB. Mol. Cell. Biol. 2000, 20, 4970–4978. [Google Scholar] [CrossRef] [PubMed]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP Histone Acetyltransferase Activity Is a Critical Component of Memory Consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone Deacetylase Inhibitors Enhance Memory and Synaptic Plasticity via CREB:CBP-Dependent Transcriptional Activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise Induces Hippocampal BDNF through a PGC-1α/FNDC5 Pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef]
- Hashemi, M.-S.; Ghaedi, K.; Salamian, A.; Karbalaie, K.; Emadi-Baygi, M.; Tanhaei, S.; Nasr-Esfahani, M.H.; Baharvand, H. Fndc5 Knockdown Significantly Decreased Neural Differentiation Rate of Mouse Embryonic Stem Cells. Neuroscience 2013, 231, 296–304. [Google Scholar] [CrossRef]
- Islam, M.R.; Valaris, S.; Young, M.F.; Haley, E.B.; Luo, R.; Bond, S.F.; Mazuera, S.; Kitchen, R.R.; Caldarone, B.J.; Bettio, L.E.B.; et al. Exercise Hormone Irisin Is a Critical Regulator of Cognitive Function. Nat. Metab. 2021, 3, 1058–1070. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-Dependent Myokine That Drives Brown-Fat-like Development of White Fat and Thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Park, J.; Kim, J.; Mikami, T. Exercise Hormone Irisin Prevents Physical Inactivity-Induced Cognitive Decline in Mice. Behav. Brain Res. 2022, 433, 114008. [Google Scholar] [CrossRef]
- Vijay, N.; Morris, M.E. Role of Monocarboxylate Transporters in Drug Delivery to the Brain. Curr. Pharm. Des. 2014, 20, 1487–1498. [Google Scholar] [CrossRef]
- Riske, L.; Thomas, R.K.; Baker, G.B.; Dursun, S.M. Lactate in the Brain: An Update on Its Relevance to Brain Energy, Neurons, Glia and Panic Disorder. Ther. Adv. Psychopharmacol. 2017, 7, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate Uptake into Astrocytes Stimulates Aerobic Glycolysis: A Mechanism Coupling Neuronal Activity to Glucose Utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Steinman, M.Q.; Gao, V.; Alberini, C.M. The Role of Lactate-Mediated Metabolic Coupling between Astrocytes and Neurons in Long-Term Memory Formation. Front. Integr. Neurosci. 2016, 10, 10. [Google Scholar] [CrossRef]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate Produced by Glycogenolysis in Astrocytes Regulates Memory Processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef]
- Liguori, C.; Stefani, A.; Sancesario, G.; Sancesario, G.M.; Marciani, M.G.; Pierantozzi, M. CSF Lactate Levels, τ Proteins, Cognitive Decline: A Dynamic Relationship in Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, 655–659. [Google Scholar] [CrossRef]
- Zebhauser, P.T.; Berthele, A.; Goldhardt, O.; Diehl-Schmid, J.; Priller, J.; Ortner, M.; Grimmer, T. Cerebrospinal Fluid Lactate Levels along the Alzheimer’s Disease Continuum and Associations with Blood-Brain Barrier Integrity, Age, Cognition, and Biomarkers. Alzheimer’s Res. Ther. 2022, 14, 61. [Google Scholar] [CrossRef]
- Schiffer, T.; Schulte, S.; Sperlich, B.; Achtzehn, S.; Fricke, H.; Strüder, H.K. Lactate Infusion at Rest Increases BDNF Blood Concentration in Humans. Neurosci. Lett. 2011, 488, 234–237. [Google Scholar] [CrossRef]
- Chen, Q.; He, S.; Hu, X.-L.; Yu, J.; Zhou, Y.; Zheng, J.; Zhang, S.; Zhang, C.; Duan, W.-H.; Xiong, Z.-Q. Differential Roles of NR2A- and NR2B-Containing NMDA Receptors in Activity-Dependent Brain-Derived Neurotrophic Factor Gene Regulation and Limbic Epileptogenesis. J. Neurosci. 2007, 27, 542–552. [Google Scholar] [CrossRef]
- Yao, W.; Cao, Q.; Luo, S.; He, L.; Yang, C.; Chen, J.; Qi, Q.; Hashimoto, K.; Zhang, J. Microglial ERK-NRBP1-CREB-BDNF Signaling in Sustained Antidepressant Actions of (R)-Ketamine. Mol. Psychiatry 2022, 27, 1618–1629. [Google Scholar] [CrossRef] [PubMed]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA Receptor Is Coupled to the ERK Pathway by a Direct Interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Laurin, D.; Verreault, R.; Lindsay, J.; MacPherson, K.; Rockwood, K. Physical Activity and Risk of Cognitive Impairment and Dementia in Elderly Persons. Arch. Neurol. 2001, 58, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Friedland, R.P.; Fritsch, T.; Smyth, K.A.; Koss, E.; Lerner, A.J.; Chen, C.H.; Petot, G.J.; Debanne, S.M. Patients with Alzheimer’s Disease Have Reduced Activities in Midlife Compared with Healthy Control-Group Members. Proc. Natl. Acad. Sci. USA 2001, 98, 3440–3445. [Google Scholar] [CrossRef]
- Rolland, Y.; van Kan, G.A.; Vellas, B. Physical Activity and Alzheimer’s Disease: From Prevention to Therapeutic Perspectives. J. Am. Med. Dir. Assoc. 2008, 9, 390–405. [Google Scholar] [CrossRef]
- Alty, J.; Farrow, M.; Lawler, K. Exercise and Dementia Prevention. Pract. Neurol. 2020, 20, 234–240. [Google Scholar] [CrossRef]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle Interventions to Prevent Cognitive Impairment, Dementia and Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Brown, B.M.; Peiffer, J.J.; Taddei, K.; Lui, J.K.; Laws, S.M.; Gupta, V.B.; Taddei, T.; Ward, V.K.; Rodrigues, M.A.; Burnham, S.; et al. Physical Activity and Amyloid-β Plasma and Brain Levels: Results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol. Psychiatry 2013, 18, 875–881. [Google Scholar] [CrossRef]
- Arancibia, S.; Silhol, M.; Moulière, F.; Meffre, J.; Höllinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective Effect of BDNF against Beta-Amyloid Induced Neurotoxicity in Vitro and in Vivo in Rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef]
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF Signaling Control Amyloidogenic Route and Aβ Production in Hippocampal Neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144. [Google Scholar] [CrossRef]
- Wang, Z.-H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.-Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. Neuromolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-Secretase Cleaves Amyloid Precursor Protein and Regulates the Pathogenesis in Alzheimer’s Disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Wu, W.; Kang, S.S.; Liu, X.; Wu, Z.; Peng, J.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Liu, X.; et al. BDNF Inhibits Neurodegenerative Disease-Associated Asparaginyl Endopeptidase Activity via Phosphorylation by AKT. JCI Insight 2018, 3, e99007. [Google Scholar] [CrossRef]
- Baranowski, B.J.; Hayward, G.C.; Marko, D.M.; MacPherson, R.E.K. Examination of BDNF Treatment on BACE1 Activity and Acute Exercise on Brain BDNF Signaling. Front. Cell. Neurosci. 2021, 15, 665867. [Google Scholar] [CrossRef] [PubMed]
- Baranowski, B.J.; Mohammad, A.; Finch, M.S.; Brown, A.; Dhaliwal, R.; Marko, D.M.; LeBlanc, P.J.; McCormick, C.M.; Fajardo, V.A.; MacPherson, R.E.K. Exercise Training and BDNF Injections Alter Amyloid Precursor Protein (APP) Processing Enzymes and Improve Cognition. J. Appl. Physiol. 2023, 135, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and Regulated Alpha-Secretase Cleavage of Alzheimer’s Amyloid Precursor Protein by a Disintegrin Metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef]
- Nigam, S.M.; Xu, S.; Kritikou, J.S.; Marosi, K.; Brodin, L.; Mattson, M.P. Exercise and BDNF Reduce Aβ Production by Enhancing α-Secretase Processing of APP. J. Neurochem. 2017, 142, 286–296. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaberi, S.; Fahnestock, M. Mechanisms of the Beneficial Effects of Exercise on Brain-Derived Neurotrophic Factor Expression in Alzheimer’s Disease. Biomolecules 2023, 13, 1577. https://doi.org/10.3390/biom13111577
Jaberi S, Fahnestock M. Mechanisms of the Beneficial Effects of Exercise on Brain-Derived Neurotrophic Factor Expression in Alzheimer’s Disease. Biomolecules. 2023; 13(11):1577. https://doi.org/10.3390/biom13111577
Chicago/Turabian StyleJaberi, Sama, and Margaret Fahnestock. 2023. "Mechanisms of the Beneficial Effects of Exercise on Brain-Derived Neurotrophic Factor Expression in Alzheimer’s Disease" Biomolecules 13, no. 11: 1577. https://doi.org/10.3390/biom13111577