
Neuroinflammation in Alzheimer’s Disease: A Potential Role of Nose-Picking in Pathogen Entry via the Olfactory System?

 ,
,  , , and
, , and 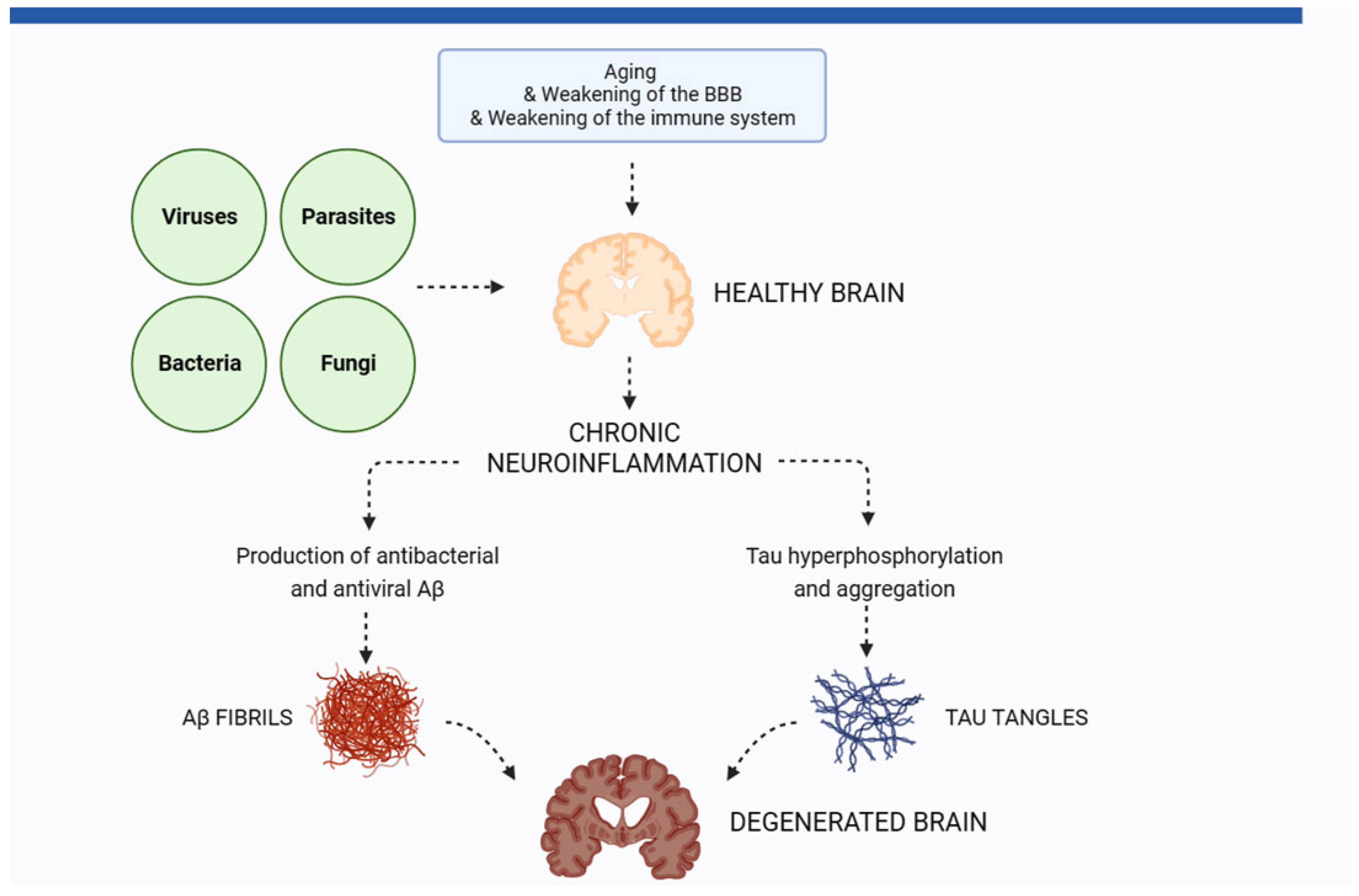{kind=link}
{kind=link}
Abstract
:1. The Role of Neuroinflammation in Alzheimer’s Disease
2. Pathogens in the Brain of AD Patients
2.1. Viruses
2.1.1. Herpes Simplex Virus 1
2.1.2. Human Herpesvirus 6 (HHV6) and Other Herpesviruses
2.1.3. SARS-CoV-2
2.2. Bacteria
2.2.1. Chlamydophila pneumoniae (Clamydia)
2.2.2. Porphyromonas gingivalis
2.2.3. Staphylococcus aureus
2.3. Fungi
3. The Olfactory System and Alzheimer’s Disease
4. A Potential Role of the Oral Microbiota in AD?
5. Conversion of a Symbiotic Nasal Microbiota to a More Pathogenic Phenotype—A Potential Role for AD Pathogenesis?
6. Animal Experiments Link Infection through the Olfactory System to AD Pathology
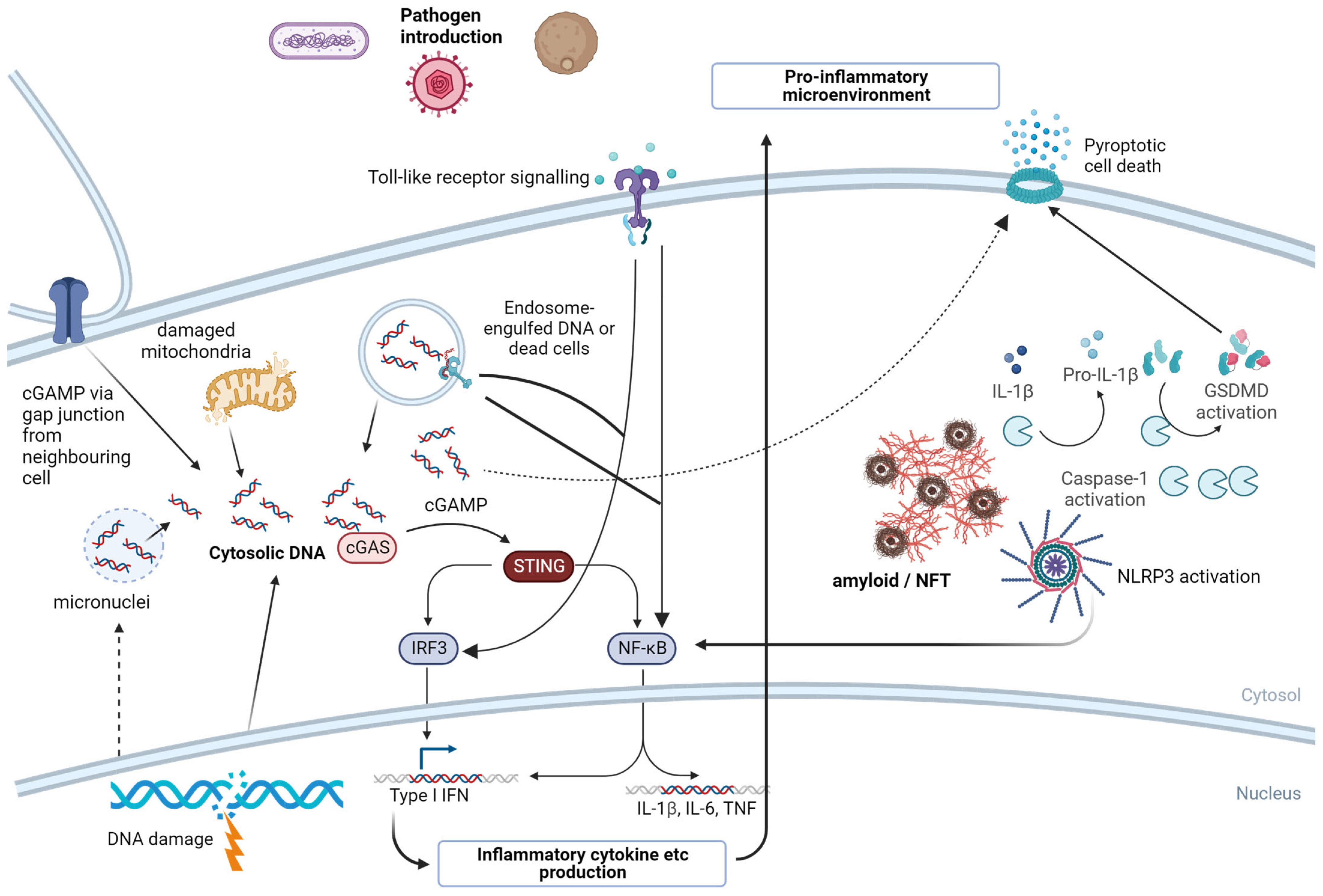
7. Pattern Recognition Receptors—A Molecular Link from Invading Pathogens to Neuroinflammation
7.1. NLRP3
7.2. cGAS/STING
7.3. Toll-like Receptors (TLRs)
8. Rhinotillexomania (Nose-Picking)—A Widespread, Unhealthy Habit?
9. Conclusions and Implications for the Prevention of AD
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shin, J.H. Dementia Epidemiology Fact Sheet 2022. Ann. Rehabil. Med. 2022, 46, 53–59. [Google Scholar] [CrossRef]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Gotz, J. Tau aggregation and its interplay with amyloid-beta. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Phuna, Z.X.; Madhavan, P. A closer look at the mycobiome in Alzheimer’s disease: Fungal species, pathogenesis and transmission. Eur. J. Neurosci. 2022, 55, 1291–1321. [Google Scholar] [CrossRef]
- Millington, C.; Sonego, S.; Karunaweera, N.; Rangel, A.; Aldrich-Wright, J.R.; Campbell, I.L.; Gyengesi, E.; Münch, G. Chronic neuroinflammation in Alzheimer’s disease: New perspectives on animal models and promising candidate drugs. BioMed Res. Int. 2014, 2014, 309129. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ye, R.D. Microglial Abeta receptors in Alzheimer’s disease. Cell Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef]
- Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Gartner, U.; Münch, G. beta-Amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon -gamma and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur. J. Neurosci. 2003, 17, 813–821. [Google Scholar] [CrossRef]
- Vojtechova, I.; Machacek, T.; Kristofikova, Z.; Stuchlik, A.; Petrasek, T. Infectious origin of Alzheimer’s disease: Amyloid beta as a component of brain antimicrobial immunity. PLoS Pathog. 2022, 18, e1010929. [Google Scholar] [CrossRef]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef]
- Bourgade, K.; Dupuis, G.; Frost, E.H.; Fulop, T. Anti-Viral Properties of Amyloid-beta Peptides. J. Alzheimers Dis. 2016, 54, 859–878. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Singh, R.; Ganeshpurkar, A.; Pokle, A.V.; Singh, R.B.; Singh, S.K.; Kumar, A. Cellular and molecular influencers of neuroinflammation in Alzheimer’s disease: Recent concepts & roles. Neurochem. Int. 2021, 151, 105212. [Google Scholar] [CrossRef]
- Banati, R.B.; Gehrmann, J.; Czech, C.; Monning, U.; Jones, L.L.; Konig, G.; Beyreuther, K.; Kreutzberg, G.W. Early and rapid de novo synthesis of Alzheimer beta A4-amyloid precursor protein (APP) in activated microglia. Glia 1993, 9, 99–210. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; Konig, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, Y. Tau and neuroinflammation in Alzheimer’s disease: Interplay mechanisms and clinical translation. J. Neuroinflamm. 2023, 20, 165. [Google Scholar] [CrossRef]
- Torres-Sanchez, E.D.; Ortiz, G.G.; Reyes-Uribe, E.; Torres-Jasso, J.H.; Salazar-Flores, J. Effect of pesticides on phosphorylation of tau protein, and its influence on Alzheimer’s disease. World J. Clin. Cases 2023, 11, 5628–5642. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional roles of reactive astrocytes in neuroinflammation and neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Song, L.; Wells, E.A.; Robinson, A.S. Critical Molecular and Cellular Contributors to Tau Pathology. Biomedicines 2021, 9, 190. [Google Scholar] [CrossRef]
- Thangaleela, S.; Sivamaruthi, B.S.; Kesika, P.; Bharathi, M.; Chaiyasut, C. Nasal Microbiota, Olfactory Health, Neurological Disorders and Aging-A Review. Microorganisms 2022, 10, 1405. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Marrero, M.; de Frias-Berzosa, B.; Hernandez-Montero, E.; Alvarez-Roger, A.; Cruz-Toro, P. Rhinotillexomania: A Manifestation of Psychiatric Illness. Acta Otorrinolaringol. Esp. 2021, 72, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Balin, B.J.; Gerard, H.C.; Arking, E.J.; Appelt, D.M.; Branigan, P.J.; Abrams, J.T.; Whittum-Hudson, J.A.; Hudson, A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med. Microbiol. Immunol. 1998, 187, 23–42. [Google Scholar] [CrossRef]
- Robinson, S.R.; Dobson, C.; Lyons, J. Challenges and directions for the pathogen hypothesis of Alzheimer’s disease. Neurobiol. Aging 2004, 25, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Nemergut, M.; Batkova, T.; Vigasova, D.; Bartos, M.; Hlozankova, M.; Schenkmayerova, A.; Liskova, B.; Sheardova, K.; Vyhnalek, M.; Hort, J.; et al. Increased occurrence of Treponema spp. and double-species infections in patients with Alzheimer’s disease. Sci. Total Environ. 2022, 844, 157114. [Google Scholar] [CrossRef] [PubMed]
- Phuna, Z.X.; Madhavan, P. A reappraisal on amyloid cascade hypothesis: The role of chronic infection in Alzheimer’s disease. Int. J. Neurosci. 2022, 133, 1071–1089. [Google Scholar] [CrossRef]
- Alloo, J.; Leleu, I.; Grangette, C.; Pied, S. Parasite infections, neuroinflammation, and potential contributions of gut microbiota. Front. Immunol. 2022, 13, 1024998. [Google Scholar] [CrossRef]
- Vigasova, D.; Nemergut, M.; Liskova, B.; Damborsky, J. Multi-pathogen infections and Alzheimer’s disease. Microb. Cell Fact. 2021, 20, 25. [Google Scholar] [CrossRef]
- Aurelian, L. The “viruses of love” and cancer. Am. J. Med. Technol. 1974, 40, 496–512. [Google Scholar] [PubMed]
- Tsankova, G.; Todorova, T.; Ermenlieva, N.; Nedelcheva, G.; Stoykova, Z.; Kostadinova, T. Seroprevalence of herpes simplex virus type 1 and type 2—Data from a hospital-based study in Varna, northeastern Bulgaria, 2019–2021. New Microbiol. 2023, 46, 308–310. [Google Scholar] [PubMed]
- AlMukdad, S.; Harfouche, M.; Farooqui, U.S.; Aldos, L.; Abu-Raddad, L.J. Epidemiology of herpes simplex virus type 1 and genital herpes in Australia and New Zealand: Systematic review, meta-analyses and meta-regressions. Epidemiol. Infect. 2023, 151, e33. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef]
- Gorfinkel, I.S.; Aoki, F.; McNeil, S.; Dionne, M.; Shafran, S.D.; Zickler, P.; Halperin, S.; Langley, J.; Bellamy, A.; Schulte, J.; et al. Seroprevalence of HSV-1 and HSV-2 antibodies in Canadian women screened for enrolment in a herpes simplex virus vaccine trial. Int. J. STD AIDS 2013, 24, 345–349. [Google Scholar] [CrossRef]
- Baringer, J.R. The biology of herpes simplex virus infection in humans. Surv. Ophthalmol. 1976, 21, 171–174. [Google Scholar] [CrossRef]
- McDonald, J.A.; Cherubin, S.; Goldberg, M.; Wei, Y.; Chung, W.K.; Schwartz, L.A.; Knight, J.A.; Schooling, C.M.; Santella, R.M.; Bradbury, A.R.; et al. Common Childhood Viruses and Pubertal Timing: The LEGACY Girls Study. Am. J. Epidemiol. 2021, 190, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, W.; Ibrahim, H.; Harfouche, M.; Abu Hijleh, F.; Abu-Raddad, L. Herpes simplex virus type 1 in Europe: Systematic review, meta-analyses and meta-regressions. BMJ Glob. Health 2020, 5, e002388. [Google Scholar] [CrossRef]
- Terlizzi, V.; Improta, F.; Di Fraia, T.; Sanguigno, E.; D’Amico, A.; Buono, S.; Raia, V.; Boccia, G. Primary herpes virus infection and ischemic stroke in childhood: A new association? J. Clin. Neurosci. 2014, 21, 1656–1658. [Google Scholar] [CrossRef]
- Shirtcliff, E.A.; Coe, C.L.; Pollak, S.D. Early childhood stress is associated with elevated antibody levels to herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 2009, 106, 2963–2967. [Google Scholar] [CrossRef] [PubMed]
- Boggian, I.; Buzzacaro, E.; Calistri, A.; Calvi, P.; Cavaggioni, A.; Mucignat-Caretta, C.; Palu, G. Asymptomatic herpes simplex type 1 virus infection of the mouse brain. J. Neurovirol. 2000, 6, 303–313. [Google Scholar] [CrossRef]
- Lekstrom-Himes, J.A.; Wang, K.; Pesnicak, L.; Krause, P.R.; Straus, S.E. The comparative biology of latent herpes simplex virus type 1 and type 2 infections: Latency-associated transcript promoter activity and expression in vitro and in infected mice. J. Neurovirol. 1998, 4, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Podlech, J.; Hengerer, F.; Fleck, M.; Walev, I.; Falke, D. Replication of herpes simplex virus type 1 and 2 in the medulla of the adrenal gland after vaginal infection of mice. Arch. Virol. 1996, 141, 1999–2008. [Google Scholar] [CrossRef]
- Kramm, C.M.; Rainov, N.G.; Sena-Esteves, M.; Barnett, F.H.; Chase, M.; Herrlinger, U.; Pechan, P.A.; Chiocca, E.A.; Breakefield, X.O. Long-term survival in a rodent model of disseminated brain tumors by combined intrathecal delivery of herpes vectors and ganciclovir treatment. Hum. Gene Ther. 1996, 7, 1989–1994. [Google Scholar] [CrossRef]
- Geller, A.I. Herpesviruses: Expression of genes in postmitotic brain cells. Curr. Opin. Genet. Dev. 1993, 3, 81–85. [Google Scholar] [CrossRef]
- Thompson, R.L.; Rogers, S.K.; Zerhusen, M.A. Herpes simplex virus neurovirulence and productive infection of neural cells is associated with a function which maps between 0.82 and 0.832 map units on the HSV genome. Virology 1989, 172, 435–450. [Google Scholar] [CrossRef] [PubMed]
- David, R. Neuroimmunology: Keeping HSV-1 dormant. Nat. Rev. Neurosci. 2010, 11, 790. [Google Scholar] [CrossRef]
- Burkhart, C.G. Herpes acquisition and transmission. J. Drugs Dermatol. 2005, 4, 378–383. [Google Scholar]
- Martin, C.; Aguila, B.; Araya, P.; Vio, K.; Valdivia, S.; Zambrano, A.; Concha, M.I.; Otth, C. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J. Alzheimer’s Dis. JAD 2014, 39, 849–859. [Google Scholar] [CrossRef]
- Walker, D.G.; O’Kusky, J.R.; McGeer, P.L. In situ hybridization analysis for herpes simplex virus nucleic acids in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1989, 3, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, J.; Sutton, R.N.; Gannicliffe, A.; Farragher, B.; Itzhaki, R.F. Detection of HSV1 DNA by in situ hybridisation in human brain after immunosuppression. J. Neurol. Neurosurg. Psychiatry 1986, 49, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Craske, J.; Itzhaki, R.F. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 1991, 33, 224–227. [Google Scholar] [CrossRef]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 2020, 105, 1027–1035.e1022. [Google Scholar] [CrossRef] [PubMed]
- Schwiegelshohn, B.; Presley, J.F.; Gorecki, M.; Vogel, T.; Carpentier, Y.A.; Maxfield, F.R.; Deckelbaum, R.J. Effects of apoprotein E on intracellular metabolism of model triglyceride-rich particles are distinct from effects on cell particle uptake. J. Biol. Chem. 1995, 270, 1761–1769. [Google Scholar] [CrossRef]
- Poirier, J. Apolipoprotein E in the brain and its role in Alzheimer’s disease. J. Psychiatry Neurosci. JPN 1996, 21, 128–134. [Google Scholar]
- Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E: The Alzheimer’s disease connection. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 1485–1494. [Google Scholar] [CrossRef]
- Sandbrink, R.; Hartmann, T.; Masters, C.L.; Beyreuther, K. Genes contributing to Alzheimer’s disease. Mol. Psychiatry 1996, 1, 27–40. [Google Scholar]
- Dobson, C.B.; Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease. Neurobiol. Aging 1999, 20, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Protto, V.; Marcocci, M.E.; Miteva, M.T.; Piacentini, R.; Li Puma, D.D.; Grassi, C.; Palamara, A.T.; De Chiara, G. Role of HSV-1 in Alzheimer’s disease pathogenesis: A challenge for novel preventive/therapeutic strategies. Curr. Opin. Pharmacol. 2022, 63, 102200. [Google Scholar] [CrossRef] [PubMed]
- De Vlieger, L.; Vandenbroucke, R.E.; Van Hoecke, L. Recent insights into viral infections as a trigger and accelerator in alzheimer’s disease. Drug Discov. Today 2022, 27, 103340. [Google Scholar] [CrossRef]
- Yong, S.J.; Yong, M.H.; Teoh, S.L.; Soga, T.; Parhar, I.; Chew, J.; Lim, W.L. The Hippocampal Vulnerability to Herpes Simplex Virus Type I Infection: Relevance to Alzheimer’s Disease and Memory Impairment. Front. Cell. Neurosci. 2021, 15, 695738. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Overwhelming Evidence for a Major Role for Herpes Simplex Virus Type 1 (HSV1) in Alzheimer’s Disease (AD); Underwhelming Evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ke, Q.; Wei, W.; Cui, L.; Wang, Y. Apolipoprotein E and viral infection: Risks and Mechanisms. Mol. Ther. Nucleic Acids 2023, 33, 529–542. [Google Scholar] [CrossRef]
- Zhang, H.; Shao, L.; Lin, Z.; Long, Q.X.; Yuan, H.; Cai, L.; Jiang, G.; Guo, X.; Yang, R.; Zhang, Z.; et al. APOE interacts with ACE2 inhibiting SARS-CoV-2 cellular entry and inflammation in COVID-19 patients. Signal Transduct. Target. Ther. 2022, 7, 261. [Google Scholar] [CrossRef]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Possible mechanisms and signposts. FASEB J. 2017, 31, 3216–3226. [Google Scholar] [CrossRef]
- Tzeng, N.S.; Chung, C.H.; Lin, F.H.; Chiang, C.P.; Yeh, C.B.; Huang, S.Y.; Lu, R.B.; Chang, H.A.; Kao, Y.C.; Yeh, H.W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections-a Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Romanescu, C.; Schreiner, T.G.; Mukovozov, I. The Role of Human Herpesvirus 6 Infection in Alzheimer’s Disease Pathogenicity-A Theoretical Mosaic. J. Clin. Med. 2022, 11, 3061. [Google Scholar] [CrossRef]
- Huang, C.; Liu, W.; Ren, X.; Lv, Y.; Wang, L.; Huang, J.; Zhu, F.; Wu, D.; Zhou, L.; Huang, X.; et al. Association between human herpesvirus 6 (HHV-6) and cognitive function in the elderly population in Shenzhen, China. Aging Clin. Exp. Res. 2022, 34, 2407–2415. [Google Scholar] [CrossRef]
- Santpere, G.; Telford, M.; Andres-Benito, P.; Navarro, A.; Ferrer, I. The Presence of Human Herpesvirus 6 in the Brain in Health and Disease. Biomolecules 2020, 10, 1520. [Google Scholar] [CrossRef]
- Romeo, M.A.; Gilardini Montani, M.S.; Gaeta, A.; D’Orazi, G.; Faggioni, A.; Cirone, M. HHV-6A infection dysregulates autophagy/UPR interplay increasing beta amyloid production and tau phosphorylation in astrocytoma cells as well as in primary neurons, possible molecular mechanisms linking viral infection to Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165647. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Itzhaki, R.F.; Kaplan, D.L. Potential Involvement of Varicella Zoster Virus in Alzheimer’s Disease via Reactivation of Quiescent Herpes Simplex Virus Type 1. J. Alzheimers Dis. 2022, 88, 1189–1200. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lathe, R. Herpes Viruses and Senile Dementia: First Population Evidence for a Causal Link. J. Alzheimers Dis. 2018, 64, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Preda, M.; Manolescu, L.S.C.; Chivu, R.D. Advances in Alpha Herpes Viruses Vaccines for Human. Vaccines 2023, 11, 1094. [Google Scholar] [CrossRef]
- Awasthi, S.; Friedman, H.M. An mRNA vaccine to prevent genital herpes. Transl. Res. 2022, 242, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Aschner, C.B.; Herold, B.C. Alphaherpesvirus Vaccines. Curr. Issues Mol. Biol. 2021, 41, 469–508. [Google Scholar] [CrossRef]
- Sandgren, K.J.; Truong, N.R.; Smith, J.B.; Bertram, K.; Cunningham, A.L. Vaccines for Herpes Simplex: Recent Progress Driven by Viral and Adjuvant Immunology. Methods Mol. Biol. 2020, 2060, 31–56. [Google Scholar] [CrossRef]
- Schnier, C.; Janbek, J.; Lathe, R.; Haas, J. Reduced dementia incidence after varicella zoster vaccination in Wales 2013–2020. Alzheimers Dement 2022, 8, e12293. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.S. Infection control of herpes simplex virus infections in obstetrics and gynecology. J. Reprod. Med. 1986, 31, 395–398. [Google Scholar]
- Lewis, M.A. Herpes simplex virus: An occupational hazard in dentistry. Int. Dent. J. 2004, 54, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019: A Review. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Bostanciklioglu, M. Temporal Correlation Between Neurological and Gastrointestinal Symptoms of SARS-CoV-2. Inflamm. Bowel Dis. 2020, 26, e89–e91. [Google Scholar] [CrossRef]
- Younger, D.S. COVID-19 (novel SARS-CoV-2) neurological illness. Handb. Clin. Neurol. 2023, 195, 159–179. [Google Scholar] [CrossRef]
- Teodoro, T.; Chen, J.; Gelauff, J.; Edwards, M.J. Functional neurological disorder in people with long COVID: A systematic review. Eur. J. Neurol. 2023, 30, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Taruffi, L.; Muccioli, L.; Mitolo, M.; Ferri, L.; Descovich, C.; Mazzoni, S.; Michelucci, R.; Lodi, R.; Liguori, R.; Cortelli, P.; et al. Neurological Manifestations of Long COVID: A Single-Center One-Year Experience. Neuropsychiatr. Dis. Treat. 2023, 19, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, P.; Shen, J.; Jiang, Y.; Wu, L.; Nie, X.; Liu, J.; Chen, W. Neurological Manifestations of hospitalized patients with mild to moderate infection with SARS-CoV-2 Omicron variant in Shanghai, China. J. Infect. Public. Health 2023, 16, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Montazerlotfelahi, H.; Norouzi, M.; Askarimoghaddam, F.; Hashemnejad, M.A.; Bastan Sarabi, N.; Qorbani, M.; Dehghani, M.; Ashrafi, M.; Mostafavi, K.; Ketabforoush, A.; et al. Neurological Involvements in COVID-19: A hospital-based study. Iran. J. Child. Neurol. 2023, 17, 69–80. [Google Scholar] [CrossRef]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Anghelescu, B.A.; Bruno, V.; Martino, D.; Roach, P. Effects of the COVID-19 Pandemic on Parkinson’s Disease: A Single-Centered Qualitative Study. Can. J. Neurol. Sci. 2022, 49, 171–183. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Z. COVID-19 and the risk of Alzheimer’s disease, amyotrophic lateral sclerosis, and multiple sclerosis. Ann. Clin. Transl. Neurol. 2022, 9, 1953–1961. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, J.; Lin, J.; Shang, H. COVID-19 and risk of neurodegenerative disorders: A Mendelian randomization study. Transl. Psychiatry 2022, 12, 283. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Das, S.; Ghosh, R.; Dubey, M.J.; Chakraborty, A.P.; Roy, D.; Das, G.; Dutta, A.; Santra, A.; Sengupta, S.; et al. The Effects of SARS-CoV-2 Infection on the Cognitive Functioning of Patients with Pre-Existing Dementia. J. Alzheimers Dis. Rep. 2023, 7, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Suster, D.; Sawant, D.; Mishra, A.; Michaille, J.J.; Tili, E. The amplification of CNS damage in Alzheimer’s disease due to SARS-CoV2 infection. Ann. Diagn. Pathol. 2022, 61, 152057. [Google Scholar] [CrossRef]
- Joo, S.H.; Hahn, C.T.; Lee, C.U. The Impact of the COVID-19 Pandemic and Social Distancing on Cognition of Alzheimer’s Disease Patients. Psychiatry Investig. 2022, 19, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wang, Y.; Zheng, J. COVID-19 and Alzheimer’s disease: How one crisis worsens the other. Transl. Neurodegener. 2021, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, S.; Scarioni, M.; Di Lorenzo, F.; Hort, J.; Georges, J.; Tomic, S.; Nobili, F.; Frederiksen, K.S.; The Management Group of the EAN Dementia; Cognitive Disorders Scientific Panel. Dementia and COVID-19, a Bidirectional Liaison: Risk Factors, Biomarkers, and Optimal Health Care. J. Alzheimers Dis. 2021, 82, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023, 111, 1086–1093.e1082. [Google Scholar] [CrossRef]
- Bubak, A.N.; Merle, L.; Niemeyer, C.S.; Baxter, B.D.; Gentile Polese, A.; Ramakrishnan, V.; Gomez, J.; Madrigal, L.; Villegas-Lanau, A.; Lopera, F.; et al. Signatures for viral infection and inflammation in the proximal olfactory system in familial Alzheimer’s disease. Neurobiol. Aging 2023, 123, 75–82. [Google Scholar] [CrossRef]
- Gautam, J.; Krawiec, C. Chlamydia Pneumonia. In StatPearls; Treasure Island (FL) Ineligible Companies: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ma, Y.; Sun, J.; Che, G.; Cheng, H. Systematic Infection of Chlamydia Pneumoniae. Clin. Lab. 2022, 68, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Gerard, H.C.; Dreses-Werringloer, U.; Wildt, K.S.; Deka, S.; Oszust, C.; Balin, B.J.; Frey, W.H., 2nd; Bordayo, E.Z.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol. Med. Microbiol. 2006, 48, 355–366. [Google Scholar] [CrossRef]
- Ring, R.H.; Lyons, J.M. Failure to detect Chlamydia pneumoniae in the late-onset Alzheimer’s brain. J. Clin. Microbiol. 2000, 38, 2591–2594. [Google Scholar] [CrossRef]
- Nochlin, D.; Shaw, C.M.; Campbell, L.A.; Kuo, C.C. Failure to detect Chlamydia pneumoniae in brain tissues of Alzheimer’s disease. Neurology 1999, 53, 1888. [Google Scholar] [CrossRef]
- Maheshwari, P.; Eslick, G.D. Bacterial infection and Alzheimer’s disease: A meta-analysis. J. Alzheimers Dis. 2015, 43, 957–966. [Google Scholar] [CrossRef]
- Chacko, A.; Delbaz, A.; Walkden, H.; Basu, S.; Armitage, C.W.; Eindorf, T.; Trim, L.K.; Miller, E.; West, N.P.; St John, J.A.; et al. Chlamydia pneumoniae can infect the central nervous system via the olfactory and trigeminal nerves and contributes to Alzheimer’s disease risk. Sci. Rep. 2022, 12, 2759. [Google Scholar] [CrossRef] [PubMed]
- Little, C.S.; Hammond, C.J.; MacIntyre, A.; Balin, B.J.; Appelt, D.M. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol. Aging 2004, 25, 419–429. [Google Scholar] [CrossRef]
- Lim, C.; Hammond, C.J.; Hingley, S.T.; Balin, B.J. Chlamydia pneumoniae infection of monocytes in vitro stimulates innate and adaptive immune responses relevant to those in Alzheimer’s disease. J. Neuroinflamm. 2014, 11, 217. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Tuomisto, K.; Jousilahti, P.; Havulinna, A.S.; Sundvall, J.; Salomaa, V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Amano, A.; Kuboniwa, M.; Kataoka, K.; Tazaki, K.; Inoshita, E.; Nagata, H.; Tamagawa, H.; Shizukuishi, S. Binding of hemoglobin by Porphyromonas gingivalis. FEMS Microbiol. Lett. 1995, 134, 63–67. [Google Scholar] [CrossRef]
- Chu, L.; Bramanti, T.E.; Ebersole, J.L.; Holt, S.C. Hemolytic activity in the periodontopathogen Porphyromonas gingivalis: Kinetics of enzyme release and localization. Infect. Immun. 1991, 59, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Nie, R.; Wu, Z.; Ni, J.; Zeng, F.; Yu, W.; Zhang, Y.; Kadowaki, T.; Kashiwazaki, H.; Teeling, J.L.; Zhou, Y. Porphyromonas gingivalis Infection Induces Amyloid-beta Accumulation in Monocytes/Macrophages. J. Alzheimers Dis. 2019, 72, 479–494. [Google Scholar] [CrossRef]
- Leira, Y.; Iglesias-Rey, R.; Gomez-Lado, N.; Aguiar, P.; Campos, F.; D’Aiuto, F.; Castillo, J.; Blanco, J.; Sobrino, T. Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch. Oral. Biol. 2019, 99, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Raineri, E.J.M.; Altulea, D.; van Dijl, J.M. Staphylococcal trafficking and infection-from ‘nose to gut’ and back. FEMS Microbiol. Rev. 2022, 46, fuab041. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jia, C.; Wang, X.; Shen, J.; Tan, J.; Wei, Z.; Wang, S.; Sun, D.; Xie, Z.; Luo, F. The impact of methicillin resistance on clinical outcome among patients with Staphylococcus aureus osteomyelitis: A retrospective cohort study of 482 cases. Sci. Rep. 2023, 13, 7990. [Google Scholar] [CrossRef]
- Siciliano, V.; Passerotto, R.A.; Chiuchiarelli, M.; Leanza, G.M.; Ojetti, V. Difficult-to-Treat Pathogens: A Review on the Management of Multidrug-Resistant Staphylococcus epidermidis. Life 2023, 13, 1126. [Google Scholar] [CrossRef] [PubMed]
- Corsini Campioli, C.; Castillo Almeida, N.E.; O’Horo, J.C.; Wilson, W.R.; Cano, E.; DeSimone, D.C.; Baddour, L.M.; Sohail, M.R. Diagnosis, management, and outcomes of brain abscess due to gram-negative versus gram-positive bacteria. Int. J. Infect. Dis. 2022, 115, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial-Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of pathogens in multiple sclerosis. Int. Rev. Immunol. 2014, 33, 266–283. [Google Scholar] [CrossRef]
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, P.; Condic, M.; Herrmann, M.; Oberstein, T.J.; Scharin-Mehlmann, M.; Gilbert, D.F.; Friedrich, O.; Gromer, T.; Kornhuber, J.; Lang, R.; et al. Amyloidogenic amyloid-beta-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 2016, 6, 32228. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Lee, J.E.; Kim, H.K.; Park, Y.J.; Jeon, J.H.; Park, S.A.; Lee, N.; Lee, I.H.; Kim, D.H.; Yang, S.H.; et al. Human Palatine Tonsils Are Linked to Alzheimer’s Disease through Function of Reservoir of Amyloid Beta Protein Associated with Bacterial Infection. Cells 2022, 11, 2285. [Google Scholar] [CrossRef]
- Lu, Y.; Saibro-Girardi, C.; Fitz, N.F.; McGuire, M.R.; Ostach, M.A.; Mamun-Or-Rashid, A.N.M.; Lefterov, I.; Koldamova, R. Multi-transcriptomics reveals brain cellular responses to peripheral infection in Alzheimer’s disease model mice. Cell Rep. 2023, 42, 112785. [Google Scholar] [CrossRef]
- Yadav, P.; Lee, Y.H.; Panday, H.; Kant, S.; Bajwa, N.; Parashar, R.; Jha, S.K.; Jha, N.K.; Nand, P.; Lee, S.S.; et al. Implications of Microorganisms in Alzheimer’s Disease. Curr. Issues Mol. Biol. 2022, 44, 4584–4615. [Google Scholar] [CrossRef]
- Parady, B. Innate Immune and Fungal Model of Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2018, 2, 139–152. [Google Scholar] [CrossRef]
- Pisa, D.; Alonso, R.; Rabano, A.; Rodal, I.; Carrasco, L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Pisa, D.; Aguado, B.; Carrasco, L. Identification of Fungal Species in Brain Tissue from Alzheimer’s Disease by Next-Generation Sequencing. J. Alzheimers Dis. 2017, 58, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Itzhaki, R.F.; Balin, B.J.; Miklossy, J.; Barron, A.E. Role of Microbes in the Development of Alzheimer’s Disease: State of the Art—An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front. Genet. 2018, 9, 362. [Google Scholar] [CrossRef]
- Schubert, C.R.; Carmichael, L.L.; Murphy, C.; Klein, B.E.; Klein, R.; Cruickshanks, K.J. Olfaction and the 5-year incidence of cognitive impairment in an epidemiological study of older adults. J. Am. Geriatr. Soc. 2008, 56, 1517–1521. [Google Scholar] [CrossRef] [PubMed]
- GoodSmith, M.S.; Wroblewski, K.E.; Schumm, L.P.; McClintock, M.K.; Pinto, J.M. Association of APOE epsilon4 Status With Long-term Declines in Odor Sensitivity, Odor Identification, and Cognition in Older US Adults. Neurology 2023, 101, e1341–e1350. [Google Scholar] [CrossRef]
- Yan, Y.; Aierken, A.; Wang, C.; Song, D.; Ni, J.; Wang, Z.; Quan, Z.; Qing, H. A potential biomarker of preclinical Alzheimer’s disease: The olfactory dysfunction and its pathogenesis-based neural circuitry impairments. Neurosci. Biobehav. Rev. 2022, 132, 857–869. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. Olfactory tau pathology in Alzheimer disease and mild cognitive impairment. Clin. Neuropathol. 2006, 25, 265–271. [Google Scholar] [PubMed]
- Jellinger, K.A.; Attems, J. Alzheimer pathology in the olfactory bulb. Neuropathol. Appl. Neurobiol. 2005, 31, 203. [Google Scholar] [CrossRef]
- Attems, J.; Lintner, F.; Jellinger, K.A. Olfactory involvement in aging and Alzheimer’s disease: An autopsy study. J. Alzheimers Dis. 2005, 7, 149–157, discussion 173–180. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Wszolek, Z.K.; Graff-Radford, N.R.; Cookson, N.; Dickson, D.W. Tau pathology in the olfactory bulb correlates with Braak stage, Lewy body pathology and apolipoprotein epsilon4. Neuropathol. Appl. Neurobiol. 2003, 29, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, T.; Cairns, N.J.; Lantos, P.L. beta-amyloid deposition and neurofibrillary tangle formation in the olfactory bulb in ageing and Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 1999, 25, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Prandota, J. Possible link between Toxoplasma gondii and the anosmia associated with neurodegenerative diseases. Am. J. Alzheimers Dis. Other Demen. 2014, 29, 205–214. [Google Scholar] [CrossRef]
- Davies, D.C.; Brooks, J.W.; Lewis, D.A. Axonal loss from the olfactory tracts in Alzheimer’s disease. Neurobiol. Aging 1993, 14, 353–357. [Google Scholar] [CrossRef]
- Mann, D.M.; Tucker, C.M.; Yates, P.O. Alzheimer’s disease: An olfactory connection? Mech. Ageing Dev. 1988, 42, 1–15. [Google Scholar] [CrossRef]
- Pagelow, D.; Chhatbar, C.; Beineke, A.; Liu, X.; Nerlich, A.; van Vorst, K.; Rohde, M.; Kalinke, U.; Forster, R.; Halle, S.; et al. The olfactory epithelium as a port of entry in neonatal neurolisteriosis. Nat. Commun. 2018, 9, 4269. [Google Scholar] [CrossRef]
- van Riel, D.; Verdijk, R.; Kuiken, T. The olfactory nerve: A shortcut for influenza and other viral diseases into the central nervous system. J. Pathol. 2015, 235, 277–287. [Google Scholar] [CrossRef]
- Chaudhuri, J.D. Blood brain barrier and infection. Med. Sci. Monit. 2000, 6, 1213–1222. [Google Scholar]
- Ueno, M.; Akiguchi, I.; Hosokawa, M.; Shinnou, M.; Sakamoto, H.; Takemura, M.; Higuchi, K. Ultrastructural and permeability features of microvessels in the olfactory bulbs of SAM mice. Acta Neuropathol. 1998, 96, 261–270. [Google Scholar] [CrossRef]
- Garg, Y.; Kapoor, D.N.; Sharma, A.K.; Bhatia, A. Drug Delivery Systems and Strategies to Overcome the Barriers of Brain. Curr. Pharm. Des. 2022, 28, 619–641. [Google Scholar] [CrossRef]
- Selvaraj, K.; Gowthamarajan, K.; Karri, V. Nose to brain transport pathways an overview: Potential of nanostructured lipid carriers in nose to brain targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, J.F.; Tranter, K.M.; Robinson, B.L.; Hanig, J.P.; Faubion, M.G.; Sarkar, S. The time course of blood brain barrier leakage and its implications on the progression of methamphetamine-induced seizures. Neurotoxicology 2018, 69, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Bourganis, V.; Kammona, O.; Alexopoulos, A.; Kiparissides, C. Recent advances in carrier mediated nose-to-brain delivery of pharmaceutics. Eur. J. Pharm. Biopharm. 2018, 128, 337–362. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Wolak, D.J.; Pizzo, M.E.; Thorne, R.G. Rapid transport within cerebral perivascular spaces underlies widespread tracer distribution in the brain after intranasal administration. J. Cereb. Blood Flow. Metab. 2015, 35, 371–381. [Google Scholar] [CrossRef]
- Bake, S.; Sohrabji, F. 17beta-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology 2004, 145, 5471–5475. [Google Scholar] [CrossRef]
- Bearer, E.L.; Breakefield, X.O.; Schuback, D.; Reese, T.S.; LaVail, J.H. Retrograde axonal transport of herpes simplex virus: Evidence for a single mechanism and a role for tegument. Proc. Natl. Acad. Sci. USA 2000, 97, 8146–8150. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Cassell, M.D.; Perlman, S. Anterograde, transneuronal transport of herpes simplex virus type 1 strain H129 in the murine visual system. J. Virol. 1996, 70, 5405–5413. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, R.; Mateen, F.J.; Rabinstein, A.A. Outcome of fulminant bacterial meningitis in adult patients. Eur. J. Neurol. 2014, 21, 447–453. [Google Scholar] [CrossRef]
- Caesar, N.M.; Myers, K.A.; Fan, X. Neisseria meningitidis serogroup B vaccine development. Microb. Pathog. 2013, 57, 33–40. [Google Scholar] [CrossRef]
- Crum, N.F. Update on Listeria monocytogenes infection. Curr. Gastroenterol. Rep. 2002, 4, 287–296. [Google Scholar] [CrossRef]
- Bonadio, W.A.; Mannenbach, M.; Krippendorf, R. Bacterial meningitis in older children. Am. J. Dis. Child. 1990, 144, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Lockey, M.W. Primary amoebic meningoencephalitis. Laryngoscope 1978, 88, 484–503. [Google Scholar] [CrossRef] [PubMed]
- Bulgart, H.R.; Neczypor, E.W.; Wold, L.E.; Mackos, A.R. Microbial involvement in Alzheimer disease development and progression. Mol. Neurodegener. 2020, 15, 42. [Google Scholar] [CrossRef]
- Mattos-Graner, R.O.; Klein, M.I.; Alves, L.A. The complement system as a key modulator of the oral microbiome in health and disease. Crit. Rev. Microbiol. 2023, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Blum, J.; Silva, M.; Byrne, S.J.; Butler, C.A.; Adams, G.G.; Reynolds, E.C.; Dashper, S.G. Temporal development of the infant oral microbiome. Crit. Rev. Microbiol. 2022, 48, 730–742. [Google Scholar] [CrossRef]
- Morrison, A.G.; Sarkar, S.; Umar, S.; Lee, S.T.M.; Thomas, S.M. The Contribution of the Human Oral Microbiome to Oral Disease: A Review. Microorganisms 2023, 11, 318. [Google Scholar] [CrossRef]
- Bowland, G.B.; Weyrich, L.S. The Oral-Microbiome-Brain Axis and Neuropsychiatric Disorders: An Anthropological Perspective. Front. Psychiatry 2022, 13, 8. [Google Scholar] [CrossRef]
- Jungbauer, G.; Stähli, A.; Zhu, X.; Auber Alberi, L.; Sculean, A.; Eick, S. Periodontal microorganisms and Alzheimer disease—A causative relationship? Periodontol. 2000 2022, 89, 59–82. [Google Scholar] [CrossRef]
- Guo, H.; Li, B.; Yao, H.; Liu, D.; Chen, R.; Zhou, S.; Ji, Y.; Zeng, L.; Du, M. Profiling the oral microbiomes in patients with Alzheimer’s disease. Oral. Dis. 2023, 29, 1341–1355. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.M.; Scarmeas, N.; Celenti, R.S.; Elkind, M.S.; Wright, C.B.; Schupf, N.; Papapanou, P.N. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS ONE 2014, 9, e114959. [Google Scholar] [CrossRef]
- Sparks Stein, P.; Steffen, M.J.; Smith, C.; Jicha, G.; Ebersole, J.L.; Abner, E.; Dawson, D., 3rd. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Gancz, A.S.; Weyrich, L.S. Studying ancient human oral microbiomes could yield insights into the evolutionary history of noncommunicable diseases. F1000Res 2023, 12, 109. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Jiao, B.; Liao, X.X.; Guo, L.N.; Yuan, Z.H.; Wang, X.; Xiao, X.W.; Zhang, X.Y.; Tang, B.S.; Shen, L. Analysis of Salivary Microbiome in Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2019, 72, 633–640. [Google Scholar] [CrossRef]
- Wu, Y.F.; Lee, W.F.; Salamanca, E.; Yao, W.L.; Su, J.N.; Wang, S.Y.; Hu, C.J.; Chang, W.J. Oral Microbiota Changes in Elderly Patients, an Indicator of Alzheimer’s Disease. Int. J. Environ. Res. Public. Health 2021, 18, 4211. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Huang, C.-P.; Lan, H.; Lau, H.-G.; Chiang, C.-P.; Chen, Y.-W. Association of periodontitis and oral microbiomes with Alzheimer’s disease: A narrative systematic review. J. Dent. Sci. 2022, 17, 1762–1779. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Tian, S.; Liu, J.; Cao, R.; Yue, P.; Cai, X.; Shang, Q.; Yang, M.; Han, L.; Zhang, D.-k. Dual role of the nasal microbiota in neurological diseases—An unignorable risk factor or a potential therapy carrier. Pharmacol. Res. 2022, 179, 106189. [Google Scholar] [CrossRef]
- Harrass, S.; Yi, C.; Chen, H. Chronic Rhinosinusitis and Alzheimer’s Disease—A Possible Role for the Nasal Microbiome in Causing Neurodegeneration in the Elderly. Int. J. Mol. Sci. 2021, 22, 11207. [Google Scholar] [CrossRef] [PubMed]
- Gay, F. Bacterial transportable toxins of the nasopharyngeal microbiota in multiple sclerosis. Nose-to-brain direct. Rev. Neurol. 2019, 175, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Balin, B.J.; Hammond, C.J.; Little, C.S.; Hingley, S.T.; Al-Atrache, Z.; Appelt, D.M.; Whittum-Hudson, J.A.; Hudson, A.P. Chlamydia pneumoniae: An Etiologic Agent for Late-Onset Dementia. Front. Aging Neurosci. 2018, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T.; Jalouli, M.; Rahman, M.A.; Jeandet, P.; Behl, T.; Alexiou, A.; Albadrani, G.M.; Abdel-Daim, M.M.; Perveen, A.; et al. Neuroinflammatory Signaling in the Pathogenesis of Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 126–146. [Google Scholar] [CrossRef]
- Kania, G.; Siegert, S.; Behnke, S.; Prados-Rosales, R.; Casadevall, A.; Luscher, T.F.; Luther, S.A.; Kopf, M.; Eriksson, U.; Blyszczuk, P. Innate signaling promotes formation of regulatory nitric oxide-producing dendritic cells limiting T-cell expansion in experimental autoimmune myocarditis. Circulation 2013, 127, 2285–2294. [Google Scholar] [CrossRef]
- Tripathi, P.; Tripathi, P.; Kashyap, L.; Singh, V. The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 2007, 51, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Shichita, T. Role of alarmins in poststroke inflammation and neuronal repair. Semin. Immunopathol. 2023, 45, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. Role of DAMPs and cell death in autoimmune diseases: The example of multiple sclerosis. Genes. Immun. 2023, 24, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Kunze, R.; Fischer, S.; Marti, H.H.; Preissner, K.T. Brain alarm by self-extracellular nucleic acids: From neuroinflammation to neurodegeneration. J. Biomed. Sci. 2023, 30, 64. [Google Scholar] [CrossRef]
- Moriyama, K.; Nishida, O. Targeting Cytokines, Pathogen-Associated Molecular Patterns, and Damage-Associated Molecular Patterns in Sepsis via Blood Purification. Int. J. Mol. Sci. 2021, 22, 8882. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nunez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.A.; Magalhaes, J.G.; Tattoli, I.; Philpott, D.J.; Travassos, L.H. Nod-like proteins in inflammation and disease. J. Pathol. 2008, 214, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Z.; Zheng, Y.; Yu, Q.; Zeng, M.; Bai, L.; Yang, L.; Guo, M.; Jiang, X.; Gan, J. Inhibitors of the NLRP3 inflammasome pathway as promising therapeutic candidates for inflammatory diseases (Review). Int. J. Mol. Med. 2023, 51, 35. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Harashima, A.; Xia, T.; Konno, H.; Konno, K.; Morales, A.; Ahn, J.; Gutman, D.; Barber, G.N. STING recognition of cytoplasmic DNA instigates cellular defense. Mol. Cell 2013, 50, 5–15. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Gluck, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L.; et al. A rat model of ataxia-telangiectasia: Evidence for a neurodegenerative phenotype. Hum. Mol. Genet. 2017, 26, 109–123. [Google Scholar] [CrossRef]
- Sun, Z.; Hornung, V. cGAS-STING signaling. Curr. Biol. 2022, 32, R730–R734. [Google Scholar] [CrossRef]
- Squillace, S.; Salvemini, D. Toll-like receptor-mediated neuroinflammation: Relevance for cognitive dysfunctions. Trends Pharmacol. Sci. 2022, 43, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, H.R.; Haukedal, H.; Freude, K. Cell Type Specific Expression of Toll-Like Receptors in Human Brains and Implications in Alzheimer’s Disease. Biomed. Res. Int. 2019, 2019, 7420189. [Google Scholar] [CrossRef]
- Chen, K.; Iribarren, P.; Hu, J.; Chen, J.; Gong, W.; Cho, E.H.; Lockett, S.; Dunlop, N.M.; Wang, J.M. Activation of Toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J. Biol. Chem. 2006, 281, 3651–3659. [Google Scholar] [CrossRef]
- Qin, Y.; Liu, Y.; Hao, W.; Decker, Y.; Tomic, I.; Menger, M.D.; Liu, C.; Fassbender, K. Stimulation of TLR4 Attenuates Alzheimer’s Disease-Related Symptoms and Pathology in Tau-Transgenic Mice. J. Immunol. 2016, 197, 3281–3292. [Google Scholar] [CrossRef]
- Herrera-Rivero, M.; Santarelli, F.; Brosseron, F.; Kummer, M.P.; Heneka, M.T. Dysregulation of TLR5 and TAM Ligands in the Alzheimer’s Brain as Contributors to Disease Progression. Mol. Neurobiol. 2019, 56, 6539–6550. [Google Scholar] [CrossRef]
- Chen, Y.C.; Yip, P.K.; Huang, Y.L.; Sun, Y.; Wen, L.L.; Chu, Y.M.; Chen, T.F. Sequence variants of toll like receptor 4 and late-onset Alzheimer’s disease. PLoS ONE 2012, 7, e50771. [Google Scholar] [CrossRef]
- Yu, J.T.; Miao, D.; Cui, W.Z.; Ou, J.R.; Tian, Y.; Wu, Z.C.; Zhang, W.; Tan, L. Common variants in toll-like receptor 4 confer susceptibility to Alzheimer’s disease in a Han Chinese population. Curr. Alzheimer Res. 2012, 9, 458–466. [Google Scholar]
- Jefferson, J.W.; Thompson, T.D. Rhinotillexomania: Psychiatric disorder or habit? J. Clin. Psychiatry 1995, 56, 56–59. [Google Scholar]
- Andrade, C.; Srihari, B.S. A preliminary survey of rhinotillexomania in an adolescent sample. J. Clin. Psychiatry 2001, 62, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K. Aetiological evaluation of foreign bodies in the ear and nose. J. Laryngol. Otol. 1984, 98, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; van Kleef, M.; Vos, M.C.; Ott, A.; Verbrugh, H.A.; Fokkens, W. Nose picking and nasal carriage of Staphylococcus aureus. Infect. Control Hosp. Epidemiol. 2006, 27, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Rathore, D.; Ahmed, S.K.; Ahluwalia, H.S.; Mehta, P. Rhinotillexomania: A rare cause of medial orbital wall erosion. Ophthalmic Plast. Reconstr. Surg. 2013, 29, e134–e135. [Google Scholar] [CrossRef]
- Shah, K.; Sankrithi, P.; Shah, S.; Smith, M.A. Chronic Rhinotillexomania Leading to Multiple Infectious Sequelae in a 66-Year-Old Female. Cureus 2020, 12, e9856. [Google Scholar] [CrossRef]
- Lavell, A.H.A.; Tijdink, J.; Buis, D.T.P.; Smulders, Y.M.; Bomers, M.K.; Sikkens, J.J. Why not to pick your nose: Association between nose picking and SARS-CoV-2 incidence, a cohort study in hospital health care workers. PLoS ONE 2023, 18, e0288352. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.S.; Jorm, A.F.; Korten, A.E.; Creasey, H.; McCusker, E.; Broe, G.A.; Longley, W.; Anthony, J.C. Environmental risk factors for Alzheimer’s disease: Their relationship to age of onset and to familial or sporadic types. Psychol. Med. 1992, 22, 429–436. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Kumar, P.; Bhuyan, D.J.; Jensen, S.O.; Roberts, T.L.; Münch, G.W. Neuroinflammation in Alzheimer’s Disease: A Potential Role of Nose-Picking in Pathogen Entry via the Olfactory System? Biomolecules 2023, 13, 1568. https://doi.org/10.3390/biom13111568
Zhou X, Kumar P, Bhuyan DJ, Jensen SO, Roberts TL, Münch GW. Neuroinflammation in Alzheimer’s Disease: A Potential Role of Nose-Picking in Pathogen Entry via the Olfactory System? Biomolecules. 2023; 13(11):1568. https://doi.org/10.3390/biom13111568
Chicago/Turabian StyleZhou, Xian, Paayal Kumar, Deep J. Bhuyan, Slade O. Jensen, Tara L. Roberts, and Gerald W. Münch. 2023. "Neuroinflammation in Alzheimer’s Disease: A Potential Role of Nose-Picking in Pathogen Entry via the Olfactory System?" Biomolecules 13, no. 11: 1568. https://doi.org/10.3390/biom13111568