
Functional Consequences of Pathogenic Variants of the GJB2 Gene (Cx26) Localized in Different Cx26 Domains

 , , , and
, , , and 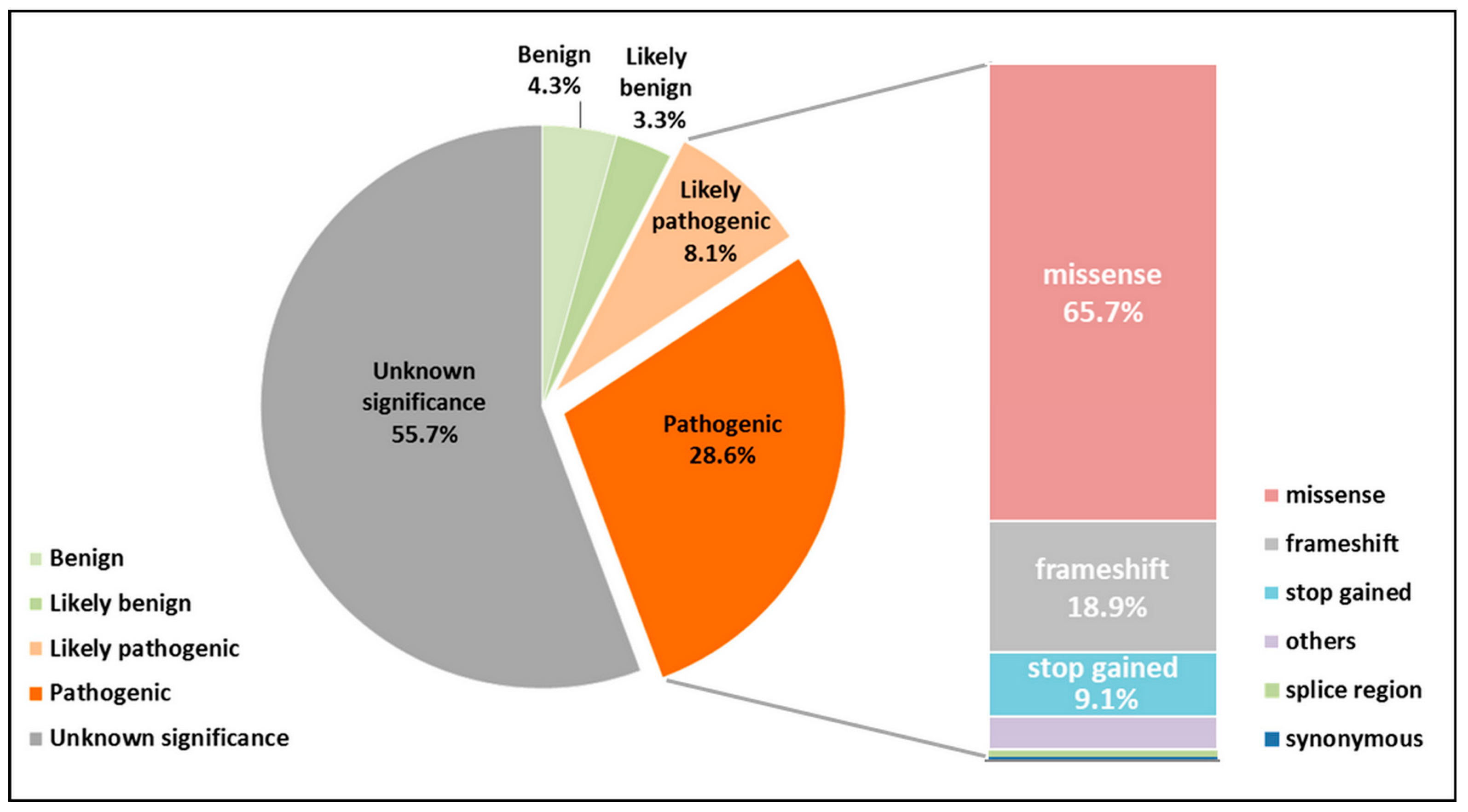{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure, Life Cycle, and Functions of Cx26
3. Current Hypotheses on the Mechanisms of Cx26-Associated Hearing Impairment
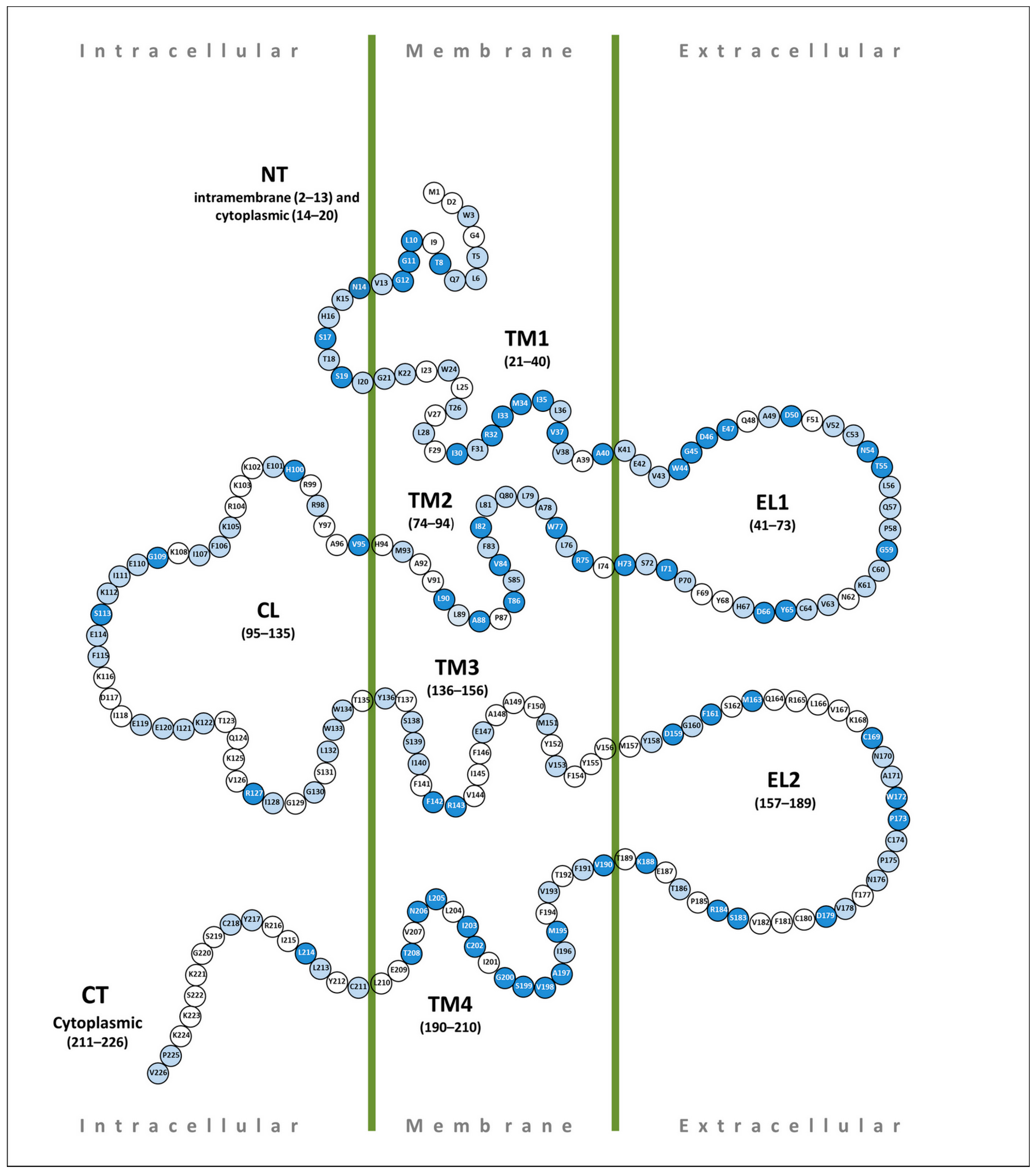
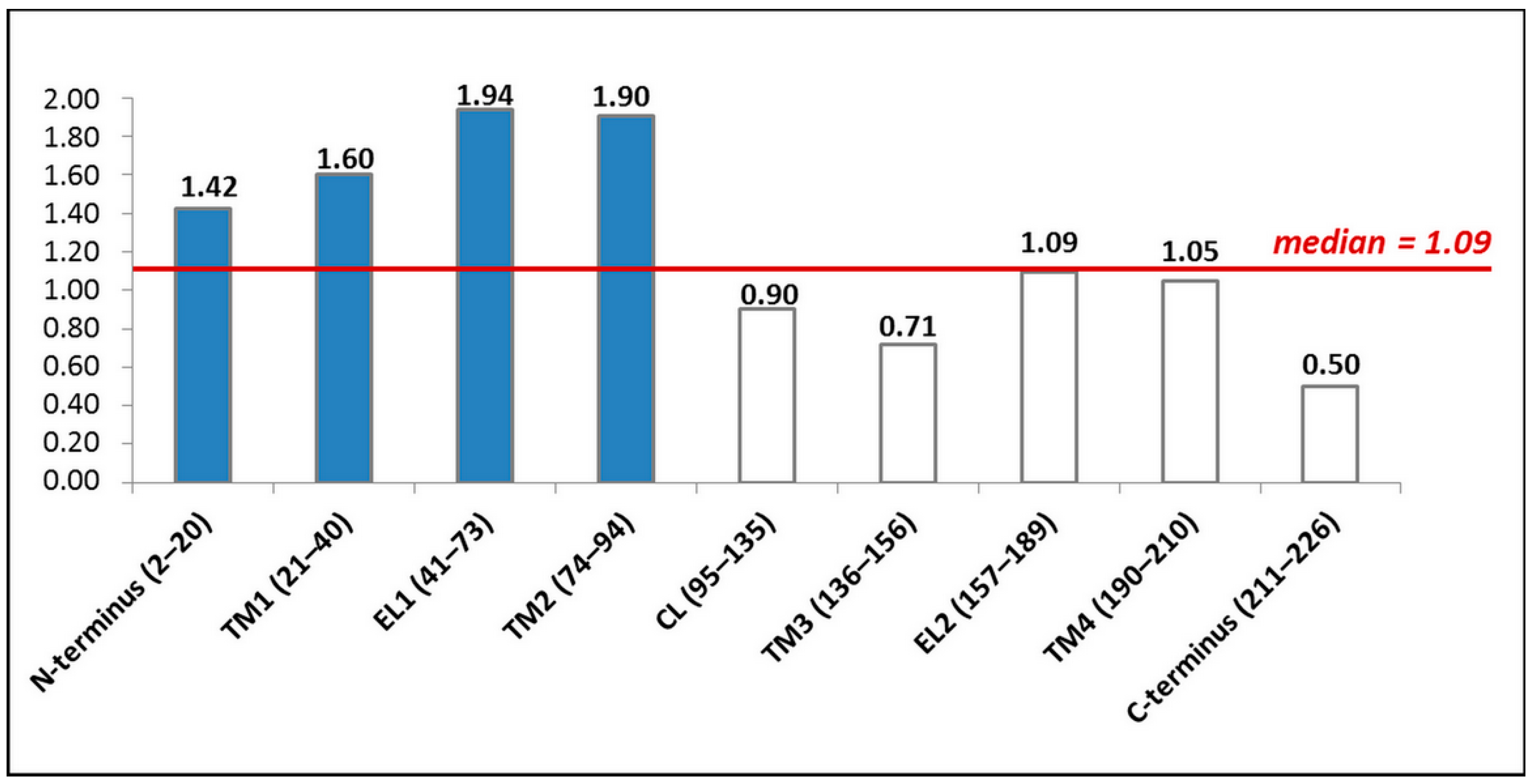
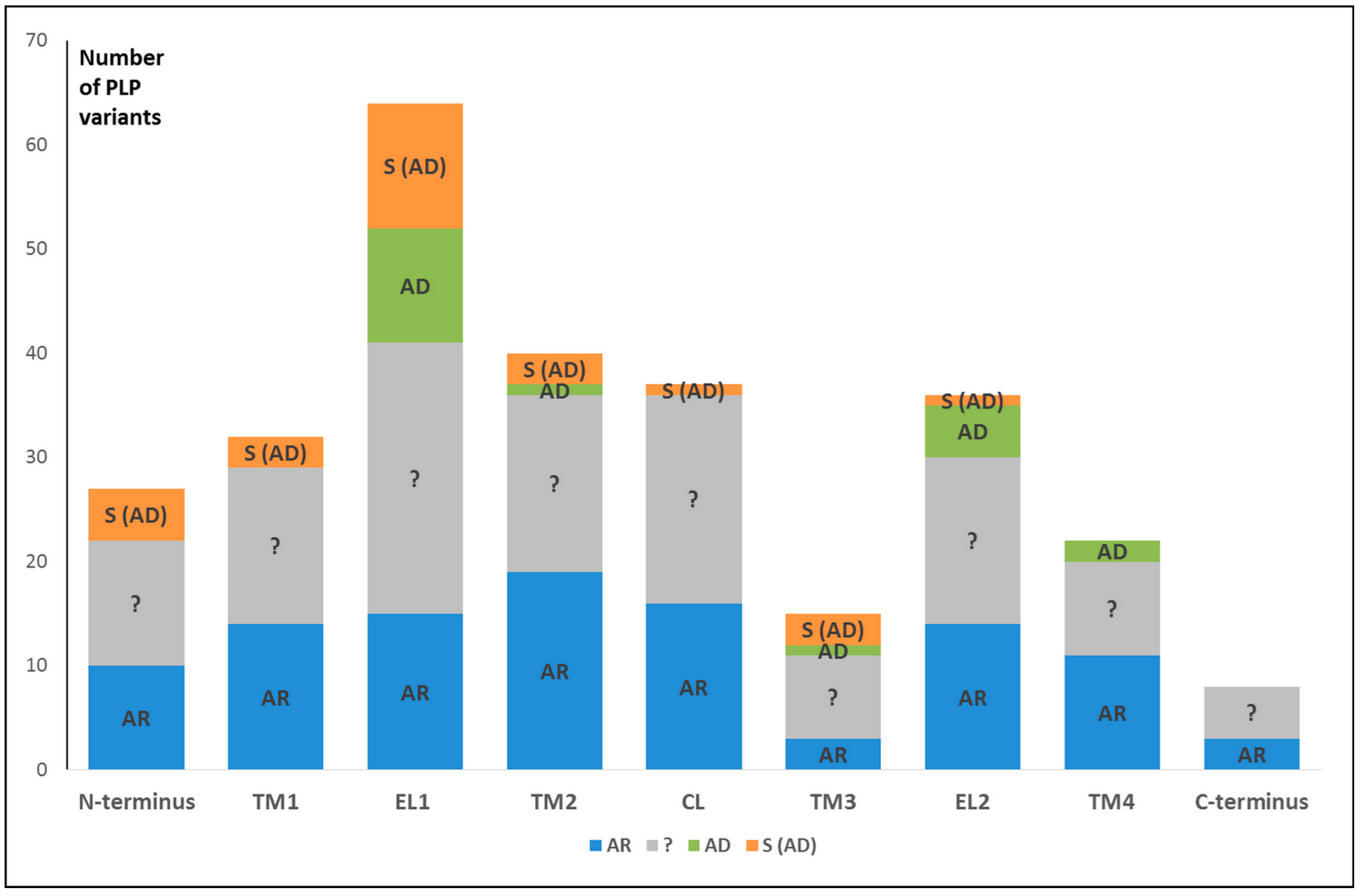
4. Pathogenic Variations in the GJB2 Gene Sequence
4.1. N-Terminus (NT)
4.1.1. Recessive Variants
4.1.2. Dominant Variants
4.2. Transmembrane Domain 1 (TM1)
4.3. Extracellular Loop 1 (EL1)
4.3.1. Recessive Variants
4.3.2. Dominant Variants
4.4. Transmembrane Domain 2 (TM2)
4.4.1. Dominant Variants
4.4.2. Recessive Variants
4.5. Cytoplasmic Loop (CL)
4.6. Transmembrane Domain 3 (TM3)
4.7. Extracellular Loop 2 (EL2)
4.7.1. Dominant Variants
4.7.2. Recessive Variants
4.8. Transmembrane Domain 4 (TM4)
4.9. C-Terminus (CT)
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M. Holstein-Rathlou NH. Gap junctions. Compr. Physiol. 2012, 2, 1981–2035. [Google Scholar] [PubMed]
- Goldberg, G.S.; Valiunas, V.; Brink, P.R. Selective permeability of gap junction channels. Biochim. Biophys. Acta 2004, 1662, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Valiunas, V.; Polosina, Y.Y.; Miller, H.; Potapova, I.A.; Valiuniene, L.; Doronin, S.; Mathias, R.T.; Robinson, R.B.; Rosen, M.R.; Cohen, I.S.; et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 2005, 568, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zong, L.; Mei, L.; Zhao, H.B. Connexin26 gap junction mediates miRNA intercellular genetic communication in the cochlea and is required for inner ear development. Sci. Rep. 2015, 5, 15647. [Google Scholar] [CrossRef] [PubMed]
- Mammano, F. Inner Ear Connexin Channels: Roles in Development and Maintenance of Cochlear Function. Cold Spring Harb. Perspect. Med. 2019, 9, a033233. [Google Scholar] [CrossRef]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef]
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef]
- Beyer, E.C.; Berthoud, V.M. Gap junction structure: Unraveled, but not fully revealed. F1000Research 2017, 6, 568. [Google Scholar] [CrossRef]
- Lukowicz-Bedford, R.M.; Farnsworth, D.R.; Miller, A.C. Connexinplexity: The spatial and temporal expression of connexin genes during vertebrate organogenesis. G3 (Bethesda) 2022, 12, jkac062. [Google Scholar] [CrossRef]
- Lagree, V.; Brunschwig, K.; Lopez, P.; Gilula, N.B.; Richard, G.; Falk, M.M. Specific amino-acid residues in the N-terminus and TM3 implicated in channel function and oligomerization compatibility of connexin43. J. Cell. Sci. 2003, 116, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.D.; Mohankumar, A.; Minogue, P.J.; Beyer, E.C.; Berthoud, V.M.; Koval, M. Cytoplasmic amino acids within the membrane interface region influence connexin oligomerization. J. Membr. Biol. 2012, 245, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.; Goodenough, D.A.; Paul, D.L. Connexins: Functions without junctions. Curr. Opin. Cell Biol. 2004, 16, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.C.; Retamal, M.A.; Basilio, D.; Bukauskas, F.F.; Bennett, M.V. Connexin-based gap junction hemichannels: Gating mechanisms. Biochim. Biophys. Acta 2005, 1711, 215–224. [Google Scholar] [CrossRef]
- Zhao, H.B.; Yu, N.; Fleming, C.R. Gap junctional hemichannel-mediated ATP release and hearing controls in the inner ear. Proc. Natl. Acad. Sci. USA 2005, 102, 18724–18729. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211. [Google Scholar] [CrossRef]
- Verselis, V.K. Connexin hemichannels and cochlear function. Neurosci. Lett. 2019, 695, 40–45. [Google Scholar] [CrossRef]
- Natha, C.M.; Vemulapalli, V.; Fiori, M.C.; Chang, C.T.; Altenberg, G.A. Connexin hemichannel inhibitors with a focus on aminoglycosides. Biochim. Biophys. Acta Mol. Basis. Dis. 2021, 1867, 166115. [Google Scholar] [CrossRef]
- Lampe, P.D.; Laird, D.W. Recent advances in connexin gap junction biology. Fac. Rev. 2022, 11, 14. [Google Scholar] [CrossRef]
- Gerido, D.A.; White, T.W. Connexin disorders of the ear, skin, and lens. Biochim. Biophys. Acta 2004, 1662, 159–170. [Google Scholar] [CrossRef]
- Richard, G. Connexin disorders of the skin. Clin. Dermatol. 2005, 23, 23–32. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Scott, C.A.; Kelsell, D.P. Key functions for gap junctions in skin and hearing. Biochem. J. 2011, 438, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Nicholson, B.J. The role of connexins in ear and skin physiology—Functional insights from disease-associated mutations. Biochim. Biophys. Acta 2013, 1828, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.J.; Simek, J.; Laird, D.W. Mechanisms linking connexin mutations to human diseases. Cell Tissue Res. 2015, 360, 701–721. [Google Scholar] [CrossRef]
- Retamal, M.A.; Reyes, E.P.; García, I.E.; Pinto, B.; Martínez, A.D.; González, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef]
- García, I.E.; Prado, P.; Pupo, A.; Jara, O.; Rojas-Gómez, D.; Mujica, P.; Flores-Muñoz, C.; González-Casanova, J.; Soto-Riveros, C.; Pinto, B.I.; et al. Connexinopathies: A structural and functional glimpse. BMC Cell Biol. 2016, 17, 17. [Google Scholar] [CrossRef]
- Iyyathurai, J.; Decuypere, J.P.; Leybaert, L.; D’hondt, C.; Bultynck, G. Connexins: Substrates and regulators of autophagy. BMC Cell Biol. 2016, 17, 20. [Google Scholar] [CrossRef]
- Srinivas, M.; Verselis, V.K.; White, T.W. Human diseases associated with connexin mutations. Biochim. Biophys. Acta Biomembr. 2018, 1860, 192–201. [Google Scholar] [CrossRef]
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vega, L.; O’Shaughnessy, E.M.; Albuloushi, A.; Martin, P.E. Connexins and the Epithelial Tissue Barrier: A Focus on Connexin 26. Biology 2021, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Xu, C.; Wang, S.; Zhang, Y.; Li, W. The Role of Connexin Hemichannels in Inflammatory Diseases. Biology 2022, 11, 237. [Google Scholar] [CrossRef]
- Unger, V.M.; Kumar, N.M.; Gilula, N.B.; Yeager, M. Three-dimensional structure of a recombinant gap junction membrane channel. Science 1999, 283, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tani, K.; Hiroaki, Y.; Fujiyoshi, Y.; Sosinsky, G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA 2007, 104, 10034–10039. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Zonta, F.; Mammano, F. A fully atomistic model of the Cx32 connexon. PLoS ONE 2008, 3, e2614. [Google Scholar] [CrossRef]
- Maeda, S.; Tsukihara, T. Structure of the gap junction channel and its implications for its biological functions. Cell. Mol. Life Sci. 2011, 68, 1115–1129. [Google Scholar] [CrossRef]
- Purnick, P.E.; Benjamin, D.C.; Verselis, V.K.; Bargiello, T.A.; Dowd, T.L. Structure of the amino terminus of a gap junction protein. Arch. Biochem. Biophys. 2000, 381, 181–190. [Google Scholar] [CrossRef]
- Kwon, T.; Harris, A.L.; Rossi, A.; Bargiello, T.A. Molecular dynamics simulations of the Cx26 hemichannel: Evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 2011, 138, 475–493. [Google Scholar] [CrossRef]
- Beyer, E.C.; Lipkind, G.M.; Kyle, J.W.; Berthoud, V.M. Structural organization of intercellular channels II. Amino terminal domain of the connexins: Sequence, functional roles, and structure. Biochim. Biophys. Acta 2012, 1818, 1823–1830. [Google Scholar] [CrossRef]
- Zonta, F.; Polles, G.; Zanotti, G.; Mammano, F. Permeation pathway of homomeric connexin 26 and connexin 30 channels investigated by molecular dynamics. J. Biomol. Struct. Dyn. 2012, 29, 985–998. [Google Scholar] [CrossRef]
- Zonta, F.; Polles, G.; Sanasi, M.F.; Bortolozzi, M.; Mammano, F. The 3.5 ångström X-ray structure of the human connexin26 gap junction channel is unlikely that of a fully open channel. Cell Commun. Signal. 2013, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Batir, Y.; Bargiello, T.A.; Dowd, T.L. Structural studies of N-terminal mutants of Connexin 26 and Connexin 32 using (1)H NMR spectroscopy. Arch. Biochem. Biophys. 2016, 608, 8–19. [Google Scholar] [CrossRef]
- Albano, J.M.R.; Mussini, N.; Toriano, R.; Facelli, J.C.; Ferraro, M.B.; Pickholz, M. Calcium interactions with Cx26 hemmichannel: Spatial association between MD simulations biding sites and variant pathogenicity. Comput. Biol. Chem. 2018, 77, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.D.; Acuña, R.; Figueroa, V.; Maripillan, J.; Nicholson, B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox. Signal. 2009, 11, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.B. Hypothesis of K+-Recycling Defect Is Not a Primary Deafness Mechanism for Cx26 (GJB2) Deficiency. Front. Mol. Neurosci. 2017, 10, 162. [Google Scholar] [CrossRef]
- Aasen, T.; Johnstone, S.; Vidal-Brime, L.; Lynn, K.S.; Koval, M. Connexins: Synthesis, Post-Translational Modifications, and Trafficking in Health and Disease. Int. J. Mol. Sci. 2018, 19, 1296. [Google Scholar] [CrossRef]
- Delvaeye, T.; Vandenabeele, P.; Bultynck, G.; Leybaert, L.; Krysko, D.V. Therapeutic Targeting of Connexin Channels: New Views and Challenges. Trends Mol. Med. 2018, 24, 1036–1053. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, W.; Li, Y.; Lin, X. Structure and Function of Cochlear Gap Junctions and Implications for the Translation of Cochlear Gene Therapies. Front. Cell. Neurosci. 2019, 13, 529. [Google Scholar] [CrossRef]
- Chen, P.; Wu, W.; Zhang, J.; Chen, J.; Li, Y.; Sun, L.; Hou, S.; Yang, J. Pathological mechanisms of connexin26-related hearing loss: Potassium recycling, ATP-calcium signaling, or energy supply? Front. Mol. Neurosci. 2022, 15, 976388. [Google Scholar] [CrossRef]
- Zhu, Y. Gap Junction-Dependent and -Independent Functions of Connexin43 in Biology. Biology 2022, 11, 283. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Boström, M.; Kinnefors, A.; Rask-Andersen, H. Unique expression of connexins in the human cochlea. Hear. Res. 2009, 250, 55–62. [Google Scholar] [CrossRef]
- Lautermann, J.; ten Cate, W.J.F.; Altenhoff, P.; Grümmer, R.; Traub, O.; Frank, H.G.; Janhke, K.; Winterhager, E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998, 294, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.P.; Ikeda, K.; Katori, Y.; Oshima, T.; Kikuchi, T.; Takasaka, T. Expression of connexin 31 in the developing mouse cochlea. Neuroreport 2000, 11, 2449–2453. [Google Scholar] [CrossRef] [PubMed]
- Forge, A.; Becker, D.; Casalotti, S.; Edwards, J.; Marziano, N.; Nevill, G. Gap junctions in the inner ear: Comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J. Comp. Neurol. 2003, 467, 207–231. [Google Scholar] [CrossRef]
- Cohen-Salmon, M.; Maxeiner, S.; Krüger, O.; Theis, M.; Willecke, K.; Petit, C. Expression of the connexin43- and connexin45-encoding genes in the developing and mature mouse inner ear. Cell Tissue Res. 2004, 316, 15–22. [Google Scholar] [CrossRef]
- Ahmad, S.; Chen, S.; Sun, J.; Lin, X. Connexins 26 and 30 are co-assembled to form gap junctions in the cochlea of mice. Biochem. Biophys. Res. Commun. 2003, 307, 362–368. [Google Scholar] [CrossRef]
- Sun, J.; Ahmad, S.; Chen, S.; Tang, W.; Zhang, Y.; Chen, P.; Lin, X. Cochlear gap junctions coassembled from Cx26 and 30 show faster intercellular Ca2+ signaling than homomeric counterparts. Am. J. Physiol. Cell Physiol. 2005, 288, C613–C623. [Google Scholar] [CrossRef]
- Musil, L.S.; Goodenough, D.A. Multisubunit assembly of an integral plasma membrane channel protein, gap junction connexin43, occurs after exit from the ER. Cell 1993, 74, 1065–1077. [Google Scholar] [CrossRef]
- Maza, J.; Das Sarma, J.; Koval, M. Defining a minimal motif required to prevent connexin oligomerization in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 21115–21121. [Google Scholar] [CrossRef]
- Das, S.; Smith, T.D.; Sarma, J.D.; Ritzenthaler, J.D.; Maza, J.; Kaplan, B.E.; Cunningham, L.A.; Suaud, L.; Hubbard, M.J.; Rubenstein, R.C.; et al. ERp29 restricts Connexin43 oligomerization in the endoplasmic reticulum. Mol. Biol. Cell 2009, 20, 2593–2604. [Google Scholar] [CrossRef] [PubMed]
- Jara, O.; Acuña, R.; García, I.E.; Maripillán, J.; Figueroa, V.; Sáez, J.C.; Araya-Secchi, R.; Lagos, C.F.; Pérez-Acle, T.; Berthoud, V.M.; et al. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell 2012, 23, 3299–3311. [Google Scholar] [CrossRef]
- Martin, P.E.; Blundell, G.; Ahmad, S.; Errington, R.J.; Evans, W.H. Multiple pathways in the trafficking and assembly of connexin 26, 32 and 43 into gap junction intercellular communication channels. J. Cell Sci. 2001, 114, 3845–3855. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Jordan, K.; Simek, J.; Shao, Q.; Jedeszko, C.; Walton, P.; Laird, D.W. Mechanisms of Cx43 and Cx26 transport to the plasma membrane and gap junction regeneration. J. Cell Sci. 2005, 118, 4451–4462. [Google Scholar] [CrossRef] [PubMed]
- Defourny, J.; Thelen, N.; Thiry, M. Actin-independent trafficking of cochlear connexin 26 to non-lipid raft gap junction plaques. Hear. Res. 2019, 374, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Z.; Jin, Y.; Chen, S.; Xu, K.; Xie, L.; Qiu, Y.; Wang, X.H.; Sun, Y.; Kong, W.J. F-Actin Dysplasia Involved in Organ of Corti Deformity in GJB2 Knockdown Mouse Model. Front. Mol. Neurosci. 2022, 14, 808553. [Google Scholar] [CrossRef]
- Qu, C.; Gardner, P.; Schrijver, I. The role of the cytoskeleton in the formation of gap junctions by Connexin 30. Exp. Cell Res. 2009, 315, 1683–1692. [Google Scholar] [CrossRef]
- Thévenin, A.F.; Kowal, T.J.; Fong, J.T.; Kells, R.M.; Fisher, C.G.; Falk, M.M. Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology (Bethesda) 2013, 28, 93–116. [Google Scholar] [CrossRef]
- Hervé, J.C.; Derangeon, M.; Sarrouilhe, D.; Bourmeyster, N. Influence of the scaffolding protein Zonula Occludens (ZOs) on membrane channels. Biochim. Biophys. Acta. 2014, 1838, 595–604. [Google Scholar] [CrossRef]
- Defourny, J.; Thiry, M. Recent insights into gap junction biogenesis in the cochlea. Dev. Dyn. 2023, 252, 239–246. [Google Scholar] [CrossRef]
- Locke, D.; Bian, S.; Li, H.; Harris, A.L. Post-translational modifications of connexin26 revealed by mass spectrometry. Biochem. J. 2009, 424, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Batissoco, A.C.; Salazar-Silva, R.; Oiticica, J.; Bento, R.F.; Mingroni-Netto, R.C.; Haddad, L.A. A Cell Junctional Protein Network Associated with Connexin-26. Int. J. Mol. Sci. 2018, 19, 2535. [Google Scholar] [CrossRef] [PubMed]
- Wingard, J.C.; Zhao, H.B. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss—A Common Hereditary Deafness. Front. Cell. Neurosci. 2015, 9, 202. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- Qiu, Y.; Zheng, J.; Chen, S.; Sun, Y. Connexin Mutations and Hereditary Diseases. Int. J. Mol. Sci. 2022, 23, 4255. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Wang, Y.; An, L.; Zeng, B.; Wang, Y.; Frishman, D.; Liu, M.; Chen, Y.; Tang, W.; Xu, H. Molecular Mechanisms and Clinical Phenotypes of GJB2 Missense Variants. Biology 2023, 12, 505. [Google Scholar] [CrossRef]
- Xiang, J.; Sun, X.; Song, N.; Ramaswamy, S.; Abou Tayoun, A.N.; Peng, Z. Comprehensive interpretation of single-nucleotide substitutions in GJB2 reveals the genetic and phenotypic landscape of GJB2-related hearing loss. Hum. Genet. 2023, 142, 33–43. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Sabag, A.D.; Ophir, E.; Avraham, K.B.; Ben-Tal, N. The structural context of disease-causing mutations in gap junctions. J. Biol. Chem. 2006, 281, 28958–28963. [Google Scholar] [CrossRef]
- Yilmaz, A. Bioinformatic Analysis of GJB2 Gene Missense Mutations. Cell Biochem. Biophys. 2015, 71, 1623–1642. [Google Scholar] [CrossRef]
- Bai, D. Structural analysis of key gap junction domains--Lessons from genome data and disease-linked mutants. Semin. Cell Dev. Biol. 2016, 50, 74–82. [Google Scholar] [CrossRef]
- Kim, H.R.; Oh, S.K.; Lee, E.S.; Choi, S.Y.; Roh, S.E.; Kim, S.J.; Tsukihara, T.; Lee, K.Y.; Jeon, C.J.; Kim, U.K. The pathological effects of connexin 26 variants related to hearing loss by in silico and in vitro analysis. Hum. Genet. 2016, 135, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Meşe, G.; Londin, E.; Mui, R.; Brink, P.R.; White, T.W. Altered gating properties of functional Cx26 mutants associated with recessive non-syndromic hearing loss. Hum. Genet. 2004, 115, 191–199. [Google Scholar] [CrossRef]
- Meşe, G.; Valiunas, V.; Brink, P.R.; White, T.W. Connexin26 deafness associated mutations show altered permeability to large cationic molecules. Am. J. Physiol. Cell Physiol. 2008, 295, C966–C974. [Google Scholar] [CrossRef] [PubMed]
- Dalamon, V.; Fiori, M.C.; Figueroa, V.A.; Oliva, C.A.; Del Rio, R.; Gonzalez, W.; Canan, J.; Elgoyhen, A.B.; Altenberg, G.A.; Retamal, M.A. Gap-junctional channel and hemichannel activity of two recently identified connexin 26 mutants associated with deafness. Pflugers Arch. 2016, 468, 909–918. [Google Scholar] [CrossRef]
- D’Andrea, P.; Veronesi, V.; Bicego, M.; Melchionda, S.; Zelante, L.; Di Iorio, E.; Bruzzone, R.; Gasparini, P. Hearing loss: Frequency and functional studies of the most common connexin26 alleles. Biochem. Biophys. Res. Commun. 2002, 296, 685–691. [Google Scholar] [CrossRef] [PubMed]
- García, I.E.; Maripillán, J.; Jara, O.; Ceriani, R.; Palacios-Muñoz, A.; Ramachandran, J.; Olivero, P.; Perez-Acle, T.; González, C.; Sáez, J.C.; et al. Keratitis-ichthyosis-deafness syndrome-associated Cx26 mutants produce nonfunctional gap junctions but hyperactive hemichannels when co-expressed with wild type Cx43. J. Investig. Dermatol. 2015, 135, 1338–1347. [Google Scholar] [CrossRef]
- Haack, B.; Schmalisch, K.; Palmada, M.; Böhmer, C.; Kohlschmidt, N.; Keilmann, A.; Zechner, U.; Limberger, A.; Beckert, S.; Zenner, H.P.; et al. Deficient membrane integration of the novel p.N14D-GJB2 mutant associated with non-syndromic hearing impairment. Hum. Mutat. 2006, 27, 1158–1159. [Google Scholar] [CrossRef]
- Terrinoni, A.; Codispoti, A.; Serra, V.; Bruno, E.; Didona, B.; Paradisi, M.; Nisticò, S.; Campione, E.; Napolitano, B.; Diluvio, L.; et al. Connexin 26 (GJB2) mutations as a cause of the KID syndrome with hearing loss. Biochem. Biophys. Res. Commun. 2010, 395, 25–30. [Google Scholar] [CrossRef]
- Terrinoni, A.; Codispoti, A.; Serra, V.; Didona, B.; Bruno, E.; Nisticò, R.; Giustizieri, M.; Alessandrini, M.; Campione, E.; Melino, G. Connexin 26 (GJB2) mutations, causing KID Syndrome, are associated with cell death due to calcium gating deregulation. Biochem. Biophys. Res. Commun. 2010, 394, 909–914. [Google Scholar] [CrossRef]
- Bailey, R.A.; Beahm, D.L.; Skerrett, I.M. The Complex and Critical Role of Glycine 12 (G12) in Beta-Connexins of Human Skin. Int. J. Mol. Sci. 2021, 22, 2615. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.R.; Derosa, A.M.; White, T.W. Connexin mutations causing skin disease and deafness increase hemichannel activity and cell death when expressed in Xenopus oocytes. J. Investig. Dermatol. 2009, 129, 870–878. [Google Scholar] [CrossRef] [PubMed]
- García, I.E.; Villanelo, F.; Contreras, G.F.; Pupo, A.; Pinto, B.I.; Contreras, J.E.; Pérez-Acle, T.; Alvarez, O.; Latorre, R.; Martínez, A.D.; et al. The syndromic deafness mutation G12R impairs fast and slow gating in Cx26 hemichannels. J. Gen. Physiol. 2018, 150, 697–711. [Google Scholar] [CrossRef]
- Albuloushi, A.; Lovgren, M.L.; Steel, A.; Yeoh, Y.; Waters, A.; Zamiri, M.; Martin, P.E. A heterozygous mutation in GJB2 (Cx26F142L) associated with deafness and recurrent skin rashes results in connexin assembly deficiencies. Exp. Dermatol. 2020, 29, 970–979. [Google Scholar] [CrossRef]
- Arita, K.; Akiyama, M.; Aizawa, T.; Umetsu, Y.; Segawa, I.; Goto, M.; Sawamura, D.; Demura, M.; Kawano, K.; Shimizu, H. A novel N14Y mutation in Connexin26 in keratitis-ichthyosis-deafness syndrome: Analyses of altered gap junctional communication and molecular structure of N terminus of mutated Connexin26. Am. J. Pathol. 2006, 169, 416–423. [Google Scholar] [CrossRef] [PubMed]
- de Zwart-Storm, E.A.; Rosa, R.F.; Martin, P.E.; Foelster-Holst, R.; Frank, J.; Bau, A.E.; Zen, P.R.; Graziadio, C.; Paskulin, G.A.; Kamps, M.A.; et al. Molecular analysis of connexin26 asparagine14 mutations associated with syndromic skin phenotypes. Exp. Dermatol. 2011, 20, 408–412. [Google Scholar] [CrossRef]
- Sanchez, H.A.; Slavi, N.; Srinivas, M.; Verselis, V.K. Syndromic deafness mutations at Asn 14 differentially alter the open stability of Cx26 hemichannels. J. Gen. Physiol. 2016, 148, 25–42. [Google Scholar] [CrossRef]
- Press, E.R.; Shao, Q.; Kelly, J.J.; Chin, K.; Alaga, A.; Laird, D.W. Induction of cell death and gain-of-function properties of connexin26 mutants predict severity of skin disorders and hearing loss. J. Biol. Chem. 2017, 292, 9721–9732. [Google Scholar] [CrossRef]
- Valdez Capuccino, J.M.; Chatterjee, P.; García, I.E.; Botello-Smith, W.M.; Zhang, H.; Harris, A.L.; Luo, Y.; Contreras, J.E. The connexin26 human mutation N14K disrupts cytosolic intersubunit interactions and promotes channel opening. J. Gen. Physiol. 2019, 151, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Tóth, T.; Kupka, S.; Haack, B.; Riemann, K.; Braun, S.; Fazakas, F.; Zenner, H.P.; Muszbek, L.; Blin, N.; Pfister, M.; et al. GJB2 mutations in patients with non-syndromic hearing loss from Northeastern Hungary. Hum. Mutat. 2004, 23, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Rouan, F.; Willoughby, C.E.; Brown, N.; Chung, P.; Ryynänen, M.; Jabs, E.W.; Bale, S.J.; DiGiovanna, J.J.; Uitto, J.; et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis-ichthyosis-deafness syndrome. Am. J. Hum. Genet. 2002, 70, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Mazereeuw-Hautier, J.; Chiaverini, C.; Jonca, N.; Bieth, E.; Dreyfus, I.; Maza, A.; Cardot-Leccia, N.; Perrin, C.; Lacour, J.P. Lethal form of keratitis-ichthyosis-deafness syndrome caused by the GJB2 mutation p.Ser17Phe. Acta Derm. Venereol. 2014, 94, 591–592. [Google Scholar] [CrossRef]
- Abbott, A.C.; García, I.E.; Villanelo, F.; Flores-Muñoz, C.; Ceriani, R.; Maripillán, J.; Novoa-Molina, J.; Figueroa-Cares, C.; Pérez-Acle, T.; Sáez, J.C.; et al. Expression of KID syndromic mutation Cx26S17F produces hyperactive hemichannels in supporting cells of the organ of Corti. Front. Cell Dev. Biol. 2023, 10, 1071202. [Google Scholar] [CrossRef]
- Aypek, H.; Bay, V.; Meşe, G. Altered cellular localization and hemichannel activities of KID syndrome associated connexin26 I30N and D50Y mutations. BMC Cell Biol. 2016, 17, 5. [Google Scholar] [CrossRef]
- Mani, R.S.; Ganapathy, A.; Jalvi, R.; Srikumari Srisailapathy, C.R.; Malhotra, V.; Chadha, S.; Agarwal, A.; Ramesh, A.; Rangasayee, R.R.; Anand, A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009, 17, 502–509. [Google Scholar] [CrossRef]
- Xiao, Z.; Yang, Z.; Liu, X.; Xie, D. Impaired membrane targeting and aberrant cellular localization of human Cx26 mutants associated with inherited recessive hearing loss. Acta Otolaryngol. 2011, 131, 59–66. [Google Scholar] [CrossRef]
- Beach, R.; Abitbol, J.M.; Allman, B.L.; Esseltine, J.L.; Shao, Q.; Laird, D.W. GJB2 Mutations Linked to Hearing Loss Exhibit Differential Trafficking and Functional Defects as Revealed in Cochlear-Relevant Cells. Front. Cell Dev. Biol. 2020, 8, 215. [Google Scholar] [CrossRef]
- Shen, J.; Oza, A.M.; Del Castillo, I.; Duzkale, H.; Matsunaga, T.; Pandya, A.; Kang, H.P.; Mar-Heyming, R.; Guha, S.; Moyer, K.; et al. Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genet. Med. 2019, 21, 2442–2452. [Google Scholar] [CrossRef]
- Kelsell, D.P.; Dunlop, J.; Stevens, H.P.; Lench, N.J.; Liang, J.N.; Parry, G.; Mueller, R.F.; Leigh, I.M. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 1997, 387, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Kelley, P.M.; Harris, D.J.; Comer, B.C.; Askew, J.W.; Fowler, T.; Smith, S.D.; Kimberling, W.J. Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. Am. J. Hum. Genet. 1998, 62, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.; Skórka, A.; Mueller-Malesińska, M.; Kostrzewa, G.; Kisiel, B.; Waligóra, J.; Krajewski, P.; Ołdak, M.; Korniszewski, L.; Skarzyński, H.; et al. M34T and V37I mutations in GJB2 associated hearing impairment: Evidence for pathogenicity and reduced penetrance. Am. J. Med. Genet. A 2007, 143A, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Skerrett, I.M.; Di, W.L.; Kasperek, E.M.; Kelsell, D.P.; Nicholson, B.J. Aberrant gating, but a normal expression pattern, underlies the recessive phenotype of the deafness mutant Connexin26M34T. FASEB J. 2004, 18, 860–862. [Google Scholar] [CrossRef]
- Bicego, M.; Beltramello, M.; Melchionda, S.; Carella, M.; Piazza, V.; Zelante, L.; Bukauskas, F.F.; Arslan, E.; Cama, E.; Pantano, S.; et al. Pathogenetic role of the deafness-related M34T mutation of Cx26. Hum. Mol. Genet. 2006, 15, 2569–2587. [Google Scholar] [CrossRef]
- Martin, P.E.; Coleman, S.L.; Casalotti, S.O.; Forge, A.; Evans, W.H. Properties of connexin26 gap junctional proteins derived from mutations associated with non-syndromal heriditary deafness. Hum. Mol. Genet. 1999, 8, 2369–2376. [Google Scholar] [CrossRef]
- Thönnissen, E.; Rabionet, R.; Arbonès, M.L.; Estivill, X.; Willecke, K.; Ott, T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum. Genet. 2002, 111, 190–197. [Google Scholar] [CrossRef]
- Palmada, M.; Schmalisch, K.; Böhmer, C.; Schug, N.; Pfister, M.; Lang, F.; Blin, N. Loss of function mutations of the GJB2 gene detected in patients with DFNB1-associated hearing impairment. Neurobiol. Dis. 2006, 22, 112–118. [Google Scholar] [CrossRef]
- Oshima, A.; Doi, T.; Mitsuoka, K.; Maeda, S.; Fujiyoshi, Y. Roles of Met-34, Cys-64, and Arg-75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J. Biol. Chem. 2003, 278, 1807–1816. [Google Scholar] [CrossRef]
- Zonta, F.; Buratto, D.; Cassini, C.; Bortolozzi, M.; Mammano, F. Molecular dynamics simulations highlight structural and functional alterations in deafness-related M34T mutation of connexin 26. Front. Physiol. 2014, 5, 85. [Google Scholar] [CrossRef]
- Bruzzone, R.; Veronesi, V.; Gomès, D.; Bicego, M.; Duval, N.; Marlin, S.; Petit, C.; D’Andrea, P.; White, T.W. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003, 533, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, J.; Lee, M.G.; Choi, J.Y.; Lee, K.A. Non-syndromic hearing loss caused by the dominant cis mutation R75Q with the recessive mutation V37I of the GJB2 (Connexin 26) gene. Exp. Mol. Med. 2015, 47, e169. [Google Scholar] [CrossRef]
- Montgomery, J.R.; White, T.W.; Martin, B.L.; Turner, M.L.; Holland, S.M. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J. Am. Acad. Dermatol. 2004, 51, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, W.; Ahmad, S.; Sipp, J.A.; Chen, P.; Lin, X. Gap junction-mediated intercellular biochemical coupling in cochlear supporting cells is required for normal cochlear functions. Proc. Natl. Acad. Sci. USA 2005, 102, 15201–15206. [Google Scholar] [CrossRef]
- Gerido, D.A.; DeRosa, A.M.; Richard, G.; White, T.W. Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafness. Am. J. Physiol. Cell Physiol. 2007, 293, C337–C345. [Google Scholar] [CrossRef]
- Sánchez, H.A.; Mese, G.; Srinivas, M.; White, T.W.; Verselis, V.K. Differentially altered Ca2+ regulation and Ca2+ permeability in Cx26 hemichannels formed by the A40V and G45E mutations that cause keratitis ichthyosis deafness syndrome. J. Gen. Physiol. 2010, 136, 47–62. [Google Scholar] [CrossRef]
- Stong, B.C.; Chang, Q.; Ahmad, S.; Lin, X. A novel mechanism for connexin 26 mutation linked deafness: Cell death caused by leaky gap junction hemichannels. Laryngoscope 2006, 116, 2205–2210. [Google Scholar] [CrossRef]
- Melchionda, S.; Bicego, M.; Marciano, E.; Franzè, A.; Morgutti, M.; Bortone, G.; Zelante, L.; Carella, M.; D’Andrea, P. Functional characterization of a novel Cx26 (T55N) mutation associated to non-syndromic hearing loss. Biochem. Biophys. Res. Commun. 2005, 337, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.R.; Alesutan, I.; Föller, M.; Sopjani, M.; Bress, A.; Baur, M.; Salama, R.H.; Bakr, M.S.; Mohamed, M.A.; Blin, N.; et al. Functional analysis of a novel I71N mutation in the GJB2 gene among Southern Egyptians causing autosomal recessive hearing loss. Cell. Physiol. Biochem. 2010, 26, 959–966. [Google Scholar] [CrossRef]
- Bruzzone, R.; Gomès, D.; Denoyelle, E.; Duval, N.; Perea, J.; Veronesi, V.; Weil, D.; Petit, C.; Gabellec, M.M.; D’Andrea, P.; et al. Functional analysis of a dominant mutation of human connexin26 associated with nonsyndromic deafness. Cell Commun. Adhes. 2001, 8, 425–431. [Google Scholar] [CrossRef]
- Rouan, F.; White, T.W.; Brown, N.; Taylor, A.M.; Lucke, T.W.; Paul, D.L.; Munro, C.S.; Uitto, J.; Hodgins, M.B.; Richard, G. Trans-dominant inhibition of connexin-43 by mutant connexin-26: Implications for dominant connexin disorders affecting epidermal differentiation. J. Cell Sci. 2001, 114, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Marziano, N.K.; Casalotti, S.O.; Portelli, A.E.; Becker, D.L.; Forge, A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum. Mol. Genet. 2003, 12, 805–812. [Google Scholar] [CrossRef]
- Yum, S.W.; Zhang, J.; Scherer, S.S. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol. Dis. 2010, 38, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Scherer, S.S.; Yum, S.W. Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26. Mol. Cell. Neurosci. 2011, 47, 71–78. [Google Scholar] [CrossRef]
- Rodriguez-Paris, J.; Waldhaus, J.; Gordhandas, J.A.; Pique, L.; Schrijver, I. Comparative functional characterization of novel non-syndromic GJB2 gene variant p.Gly45Arg and lethal syndromic variant p.Gly45Glu. PeerJ 2016, 4, e2494. [Google Scholar] [CrossRef]
- Choi, S.Y.; Park, H.J.; Lee, K.Y.; Dinh, E.H.; Chang, Q.; Ahmad, S.; Lee, S.H.; Bok, J.; Lin, X.; Kim, U.K. Different functional consequences of two missense mutations in the GJB2 gene associated with non-syndromic hearing loss. Hum. Mutat. 2009, 30, E716–E727. [Google Scholar] [CrossRef]
- Forge, A.; Marziano, N.K.; Casalotti, S.O.; Becker, D.L.; Jagger, D. The inner ear contains heteromeric channels composed of cx26 and cx30 and deafness-related mutations in cx26 have a dominant negative effect on cx30. Cell Commun. Adhes. 2003, 10, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Telford, D.; Laird, D.W. Functional domain mapping and selective trans-dominant effects exhibited by Cx26 disease-causing mutations. J. Biol. Chem. 2004, 279, 19157–19168. [Google Scholar] [CrossRef] [PubMed]
- de Zwart-Storm, E.A.; Hamm, H.; Stoevesandt, J.; Steijlen, P.M.; Martin, P.E.; van Geel, M.; van Steensel, M.A. A novel missense mutation in GJB2 disturbs gap junction protein transport and causes focal palmoplantar keratoderma with deafness. J. Med. Genet. 2008, 45, 161–166. [Google Scholar] [CrossRef]
- Shuja, Z.; Li, L.; Gupta, S.; Meşe, G.; White, T.W. Connexin26 Mutations Causing Palmoplantar Keratoderma and Deafness Interact with Connexin43, Modifying Gap Junction and Hemichannel Properties. J. Investig. Dermatol. 2016, 136, 225–235. [Google Scholar] [CrossRef]
- van Geel, M.; van Steensel, M.A.; Küster, W.; Hennies, H.C.; Happle, R.; Steijlen, P.M.; König, A. HID and KID syndromes are associated with the same connexin 26 mutation. Br. J. Dermatol. 2002, 146, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, H.A.; Villone, K.; Srinivas, M.; Verselis, V.K. The D50N mutation and syndromic deafness: Altered Cx26 hemichannel properties caused by effects on the pore and intersubunit interactions. J. Gen. Physiol. 2013, 142, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Lopez, W.; Gonzalez, J.; Liu, Y.; Harris, A.L.; Contreras, J.E. Insights on the mechanisms of Ca(2+) regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y). J. Gen. Physiol. 2013, 142, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mhaske, P.V.; Levit, N.A.; Li, L.; Wang, H.Z.; Lee, J.R.; Shuja, Z.; Brink, P.R.; White, T.W. The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am. J. Physiol. Cell Physiol. 2013, 304, C1150–C1158. [Google Scholar] [CrossRef]
- Janecke, A.R.; Hennies, H.C.; Günther, B.; Gansl, G.; Smolle, J.; Messmer, E.M.; Utermann, G.; Rittinger, O. GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal form. Am. J. Med. Genet. A 2005, 133A, 128–131. [Google Scholar] [CrossRef]
- Lilly, E.; Bunick, C.G.; Maley, A.M.; Zhang, S.; Spraker, M.K.; Theos, A.J.; Vivar, K.L.; Seminario-Vidal, L.; Bennett, A.E.; Sidbury, R.; et al. More than keratitis, ichthyosis, and deafness: Multisystem effects of lethal GJB2 mutations. J. Am. Acad. Dermatol. 2019, 80, 617–625. [Google Scholar] [CrossRef]
- Mese, G.; Sellitto, C.; Li, L.; Wang, H.Z.; Valiunas, V.; Richard, G.; Brink, P.R.; White, T.W. The Cx26-G45E mutation displays increased hemichannel activity in a mouse model of the lethal form of keratitis-ichthyosis-deafness syndrome. Mol. Biol. Cell 2011, 22, 4776–4786. [Google Scholar] [CrossRef]
- Zhang, Y.; Hao, H. Conserved glycine at position 45 of major cochlear connexins constitutes a vital component of the Ca2+ sensor for gating of gap junction hemichannels. Biochem. Biophys. Res. Commun. 2013, 436, 424–429. [Google Scholar] [CrossRef]
- Ogawa, Y.; Takeichi, T.; Kono, M.; Hamajima, N.; Yamamoto, T.; Sugiura, K.; Akiyama, M. Revertant mutation releases confined lethal mutation, opening Pandora’s box: A novel genetic pathogenesis. PLoS Genet. 2014, 10, e1004276. [Google Scholar] [CrossRef]
- Bakirtzis, G.; Choudhry, R.; Aasen, T.; Shore, L.; Brown, K.; Bryson, S.; Forrow, S.; Tetley, L.; Finbow, M.; Greenhalgh, D.; et al. Targeted epidermal expression of mutant Connexin 26(D66H) mimics true Vohwinkel syndrome and provides a model for the pathogenesis of dominant connexin disorders. Hum. Mol. Genet. 2003, 12, 1737–1744. [Google Scholar] [CrossRef]
- de Zwart-Storm, E.A.; van Geel, M.; Veysey, E.; Burge, S.; Cooper, S.; Steijlen, P.M.; Martin, P.E.; van Steensel, M.A. A novel missense mutation in GJB2, p.Tyr65His, causes severe Vohwinkel syndrome. Br. J. Dermatol. 2011, 164, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; White, T.W.; Smith, L.E.; Bailey, R.A.; Compton, J.G.; Paul, D.L.; Bale, S.J. Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 1998, 103, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Aasen, T.; Hodgins, M.; Laird, D.W. Transport and function of cx26 mutants involved in skin and deafness disorders. Cell Commun. Adhes. 2003, 10, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Deng, Y.; Bao, X.; Reuss, L.; Altenberg, G.A. Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness. FASEB J. 2005, 19, 1516–1518. [Google Scholar] [CrossRef]
- Deng, Y.; Chen, Y.; Reuss, L.; Altenberg, G.A. Mutations of connexin 26 at position 75 and dominant deafness: Essential role of arginine for the generation of functional gap-junctional channels. Hear. Res. 2006, 220, 87–94. [Google Scholar] [CrossRef]
- Kudo, T.; Kure, S.; Ikeda, K.; Xia, A.P.; Katori, Y.; Suzuki, M.; Kojima, K.; Ichinohe, A.; Suzuki, Y.; Aoki, Y.; et al. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum. Mol. Genet. 2003, 12, 995–1004. [Google Scholar] [CrossRef]
- Maeda, Y.; Fukushima, K.; Kawasaki, A.; Nishizaki, K.; Smith, R.J. Cochlear expression of a dominant-negative GJB2R75W construct delivered through the round window membrane in mice. Neurosci. Res. 2007, 58, 250–254. [Google Scholar] [CrossRef]
- Inoshita, A.; Iizuka, T.; Okamura, H.O.; Minekawa, A.; Kojima, K.; Furukawa, M.; Kusunoki, T.; Ikeda, K. Postnatal development of the organ of Corti in dominant-negative GJB2 transgenic mice. Neuroscience 2008, 156, 1039–1047. [Google Scholar] [CrossRef]
- Inoshita, A.; Karasawa, K.; Funakubo, M.; Miwa, A.; Ikeda, K.; Kamiya, K. Dominant negative connexin26 mutation R75W causing severe hearing loss influences normal programmed cell death in postnatal organ of Corti. BMC Genet. 2014, 15, 1. [Google Scholar] [CrossRef]
- Feldmann, D.; Denoyelle, F.; Blons, H.; Lyonnet, S.; Loundon, N.; Rouillon, I.; Hadj-Rabia, S.; Petit, C.; Couderc, R.; Garabédian, E.N.; et al. The GJB2 mutation R75Q can cause nonsyndromic hearing loss DFNA3 or hereditary palmoplantar keratoderma with deafness. Am. J. Med. Genet. A 2005, 137, 225–227. [Google Scholar] [CrossRef]
- Piazza, V.; Beltramello, M.; Menniti, M.; Colao, E.; Malatesta, P.; Argento, R.; Chiarella, G.; Gallo, L.V.; Catalano, M.; Perrotti, N.; et al. Functional analysis of R75Q mutation in the gene coding for Connexin 26 identified in a family with nonsyndromic hearing loss. Clin. Genet. 2005, 68, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Birkenhäger, R.; Lüblinghoff, N.; Prera, E.; Schild, C.; Aschendorff, A.; Arndt, S. Autosomal dominant prelingual hearing loss with palmoplantar keratoderma syndrome: Variability in clinical expression from mutations of R75W and R75Q in the GJB2 gene. Am. J. Med. Genet. A 2010, 152A, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Chang, W.T.; Li, A.H.; Yeh, T.H.; Wu, C.Y.; Chen, M.S.; Huang, P.C. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 2003, 84, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Beltramello, M.; Piazza, V.; Bukauskas, F.F.; Pozzan, T.; Mammano, F. Impaired permeability to Ins(1,4,5)P3 in a mutant connexin underlies recessive hereditary deafness. Nat. Cell Biol. 2005, 7, 63–69. [Google Scholar] [CrossRef]
- Wei, Q.; Liu, Y.; Wang, S.; Liu, T.; Lu, Y.; Xing, G.; Cao, X. A novel compound heterozygous mutation in the GJB2 gene causing non-syndromic hearing loss in a family. Int. J. Mol. Med. 2014, 33, 310–316. [Google Scholar] [CrossRef]
- Shi, X.; Zhang, Y.; Qiu, S.; Zhuang, W.; Yuan, N.; Sun, T.; Gao, J.; Qiao, Y.; Liu, K. A Novel GJB2 compound heterozygous mutation c.257C>G (p.T86R)/c.176del16 (p.G59A fs*18) causes sensorineural hearing loss in a Chinese family. J. Clin. Lab. Anal. 2018, 32, e22444. [Google Scholar] [CrossRef]
- Kupka, S.; Braun, S.; Aberle, S.; Haack, B.; Ebauer, M.; Zeissler, U.; Zenner, H.P.; Blin, N.; Pfister, M. Frequencies of GJB2 mutations in German control individuals and patients showing sporadic non-syndromic hearing impairment. Hum. Mutat. 2002, 20, 77–78. [Google Scholar] [CrossRef]
- Dalamón, V.; Lotersztein, V.; Béhèran, A.; Lipovsek, M.; Diamante, F.; Pallares, N.; Francipane, L.; Frechtel, G.; Paoli, B.; Mansilla, E.; et al. GJB2 and GJB6 genes: Molecular study and identification of novel GJB2 mutations in the hearing-impaired Argentinean population. Audiol. Neurootol. 2010, 15, 194–202. [Google Scholar] [CrossRef]
- Brown, C.W.; Levy, M.L.; Flaitz, C.M.; Reid, B.S.; Manolidis, S.; Hebert, A.A.; Bender, M.M.; Heilstedt, H.A.; Plunkett, K.S.; Fang, P.; et al. A novel GJB2 (connexin 26) mutation, F142L, in a patient with unusual mucocutaneous findings and deafness. J. Investig. Dermatol. 2003, 121, 1221–1223. [Google Scholar] [CrossRef]
- Ibáñez, M.M.; Alcalde, M.M.; Jiménez, M.R.; Muñoz, M.D.; Díez-Delgado, F.J. An unusual mucocutaneous syndrome with sensorineural deafness due to connexin 26 mutations. Pediatr. Dermatol. 2013, 30, e138–e142. [Google Scholar] [CrossRef]
- Chen, T.; Jiang, L.; Liu, C.; Shan, H.; Chen, J.; Yang, B.; Ou, Q. Update of the spectrum of GJB2 mutations in 107 patients with nonsyndromic hearing loss in the Fujian population of China. Ann. Hum. Genet. 2014, 78, 235–242. [Google Scholar] [CrossRef]
- Meyer, C.G.; Amedofu, G.K.; Brandner, J.M.; Pohland, D.; Timmann, C.; Horstmann, R.D. Selection for deafness? Nat. Med. 2002, 8, 1332–1333. [Google Scholar] [CrossRef] [PubMed]
- Common, J.E.; Di, W.L.; Davies, D.; Kelsell, D.P. Further evidence for heterozygote advantage of GJB2 deafness mutations: A link with cell survival. J. Med. Genet. 2004, 41, 573–575. [Google Scholar] [CrossRef] [PubMed]
- Man, Y.K.; Trolove, C.; Tattersall, D.; Thomas, A.C.; Papakonstantinopoulou, A.; Patel, D.; Scott, C.; Chong, J.; Jagger, D.J.; O’Toole, E.A.; et al. A deafness-associated mutant human connexin 26 improves the epithelial barrier in vitro. J. Membr. Biol. 2007, 218, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.D.; Caria, H.; Simões-Teixeira, H.; Aasen, T.; Dias, O.; Andrea, M.; Kelsell, D.P.; Fialho, G. A novel M163L mutation in connexin 26 causing cell death and associated with autosomal dominant hearing loss. Hear. Res. 2008, 240, 87–92. [Google Scholar] [CrossRef] [PubMed]
- de Zwart-Storm, E.A.; van Geel, M.; van Neer, P.A.; Steijlen, P.M.; Martin, P.E.; van Steensel, M.A. A novel missense mutation in the second extracellular domain of GJB2, p.Ser183Phe, causes a syndrome of focal palmoplantar keratoderma with deafness. Am. J. Pathol. 2008, 173, 1113–1119. [Google Scholar] [CrossRef]
- Su, C.C.; Li, S.Y.; Su, M.C.; Chen, W.C.; Yang, J.J. Mutation R184Q of connexin 26 in hearing loss patients has a dominant-negative effect on connexin 26 and connexin 30. Eur. J. Hum. Genet. 2010, 18, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Primignani, P.; Castorina, P.; Sironi, F.; Curcio, C.; Ambrosetti, U.; Coviello, D.A. A novel dominant missense mutation--D179N--in the GJB2 gene (Connexin 26) associated with non-syndromic hearing loss. Clin. Genet. 2003, 63, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Huang, S.H.; Chou, K.H.; Liao, P.J.; Su, C.C.; Li, S.Y. Identification of mutations in members of the connexin gene family as a cause of nonsyndromic deafness in Taiwan. Audiol. Neurootol. 2007, 12, 198–208. [Google Scholar] [CrossRef]
- Zonta, F.; Girotto, G.; Buratto, D.; Crispino, G.; Morgan, A.; Abdulhadi, K.; Alkowari, M.; Badii, R.; Gasparini, P.; Mammano, F. The p.Cys169Tyr variant of connexin 26 is not a polymorphism. Hum. Mol. Genet. 2015, 24, 2641–2648. [Google Scholar] [CrossRef]
- Maslova, E.A.; Orishchenko, K.E.; Posukh, O.L. Functional Evaluation of a Rare Variant c.516G>C (p.Trp172Cys) in the GJB2 (Connexin 26) Gene Associated with Nonsyndromic Hearing Loss. Biomolecules 2021, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, C.; Walker, A.E.; Depriest, A.D.; Cone, A.C.; Lu, C.; Badger, J.; Skerrett, I.M.; Sosinsky, G.E. Analysis of trafficking, stability and function of human connexin 26 gap junction channels with deafness-causing mutations in the fourth transmembrane helix. PLoS ONE 2013, 8, e70916. [Google Scholar] [CrossRef] [PubMed]
- Birkenhäger, R.; Prera, N.; Aschendorff, A.; Laszig, R.; Arndt, S. A novel homozygous mutation in the EC1/EC2 interaction domain of the gap junction complex connexon 26 leads to profound hearing impairment. Biomed. Res. Int. 2014, 2014, 307976. [Google Scholar] [CrossRef]
- Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique Mutational Spectrum of the GJB2 Gene and its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys). Genes 2019, 10, 429. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posukh, O.L.; Maslova, E.A.; Danilchenko, V.Y.; Zytsar, M.V.; Orishchenko, K.E. Functional Consequences of Pathogenic Variants of the GJB2 Gene (Cx26) Localized in Different Cx26 Domains. Biomolecules 2023, 13, 1521. https://doi.org/10.3390/biom13101521
Posukh OL, Maslova EA, Danilchenko VY, Zytsar MV, Orishchenko KE. Functional Consequences of Pathogenic Variants of the GJB2 Gene (Cx26) Localized in Different Cx26 Domains. Biomolecules. 2023; 13(10):1521. https://doi.org/10.3390/biom13101521
Chicago/Turabian StylePosukh, Olga L., Ekaterina A. Maslova, Valeriia Yu. Danilchenko, Marina V. Zytsar, and Konstantin E. Orishchenko. 2023. "Functional Consequences of Pathogenic Variants of the GJB2 Gene (Cx26) Localized in Different Cx26 Domains" Biomolecules 13, no. 10: 1521. https://doi.org/10.3390/biom13101521