
Autophagy in Parkinson’s Disease

Abstract
:1. Introduction
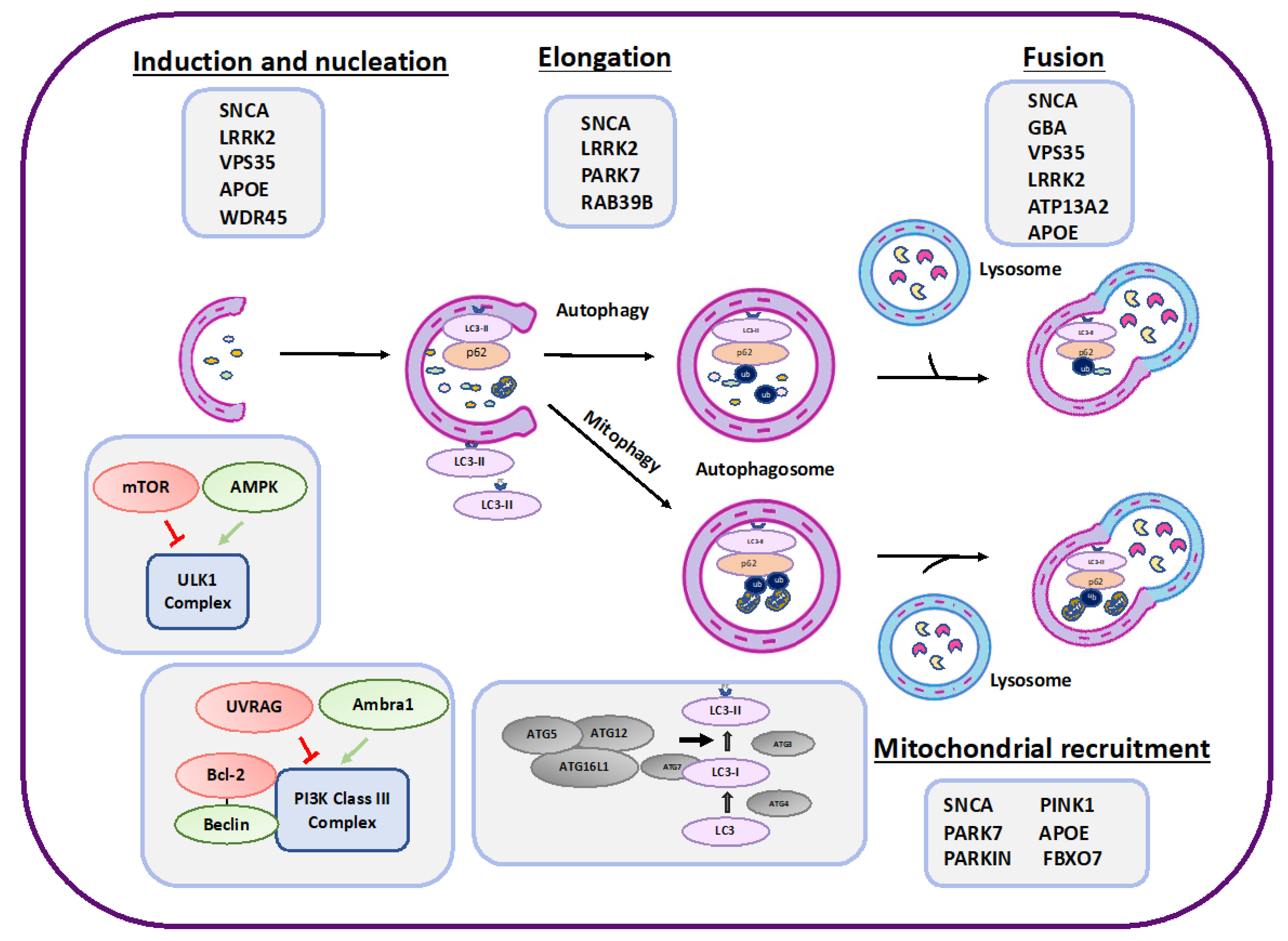
2. Autophagy
3. PD Risk Factors That Affect Autophagy
4. The Role of Autophagy Impairment and α-Syn Pathology in PD
5. Targeting Autophagy as Therapeutic Approach in PD
6. Concluding Remarks

{kind=link}
| Protein | Connection to PD | Autophagy/Mitophagy/CMA | Bibliography |
|---|---|---|---|
| α-synuclein | Mutations in α-synuclein are affiliated with early PD onset and Lewy bodies formation. | Inhibits autophagy in the nucleation and maturation steps. Inhibits mitophagy. | [31,103,104,105,106,107,109,110,111,112,113,114,115,116,117,118,119] |
| DJ-1 | Deletions and point mutations of DJ-1 cause autosomal recessive PD. | Regulates autophagy/mitophagy. | [66,67,68,69,70,71,72,73,74] |
| LRRK2 | LRRK2 mutations are found in families with late-onset autosomal-dominant PD. | Affects autophagy, mitophagy, and CMA. | [27,32,33,34,35,36,37,38,39,40,41,42,43,44,45] |
| Apolipoprotein E4 | APOE4 isoform was suggested to be involved in PD. | Impairs autophagy and mitophagy. | [79,80,81,82,84,85,86,87,88,89,90,91,92,93,154] |
| PTEN-induced kinase 1 (PINK1) and PARKIN | Loss of function mutations of PINK1 and PARKIN genes are the most common causes of autosomal recessive and early-onset PD. | Regulate mitophagy. | [54,55,56,57,58,59,60,61,62,63] |
| GBA | Mutations in the GBA gene lead to lysosomal dysfunction and impaired α-synuclein metabolism. | Affects lysosomal activity. | [46,47,48,49,50,51,52,53] |
| VPS35 | Mutations in the vacuolar protein sorting 35 ortholog (VPS35) gene cause late-onset autosomal dominant familial PD. | Inhibits autophagy in the nucleation step. | [94] |
| RAB39B | RAB39B gene mutations were associated with X-linked neurodevelopmental defects including early-onset Parkinson’s disease (PD). | Affects autophagy/mitophagy. | [77,78,155,156] |
| ATP13A2 (PARK9). | ATP13A2 (PARK9) gene mutations cause Kufor–Rakeb syndrome (Parkinson’s disease 9), an autosomal recessive form of Parkinsonism with dementia. | Affects lysosome activity and α-syn degradation. | [34,75,76,157,158,159,160] |
| WDR45 | WDR45 (WD Repeat Domain 45) is a component of the autophagy machinery that controls the major intracellular degradation process. | Regulates autophagy. | [95,96,97,98] |
| FBXO7 | FBXO7 (F-box-only protein 7) gene mutations have been identified in a number of families with severe autosomal recessive early-onset Parkinson’s disease. | Affects mitophagy. | [99,100] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-Syn | α-synuclein |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| Ambra1 | Activating molecule in Beclin1-regulated autophagy |
| AD | Alzheimer’s disease |
| AMPK | AMP-activated protein kinase |
| APOE | Apolipoprotein E |
| ALP | Autophagy-lysosomal pathway |
| ALR | Autophagic lysosome reformation |
| Atgs | Autophagy-related proteins |
| AV | Autophagic vacuoles |
| BafA1 | Bafilomycin A1 |
| CSF | Cerebrospinal fluid |
| CMA | Chaperone-mediated autophagy |
| CTSD | Protease cathepsin D |
| FBXO7 | F-box-only protein 7 |
| GCase | Glucocerebrosidase |
| GBA | Glucosylceramidase beta |
| HSC70 | Heat shock cognate 70 |
| LRRK2 | Leucine-rich repeat kinase 2 |
| LB | Lewy bodies |
| LC3 | Light chain 3 |
| LAG3 | Lymphocyte activation gene 3 |
| LAMP | Lysosome-associated membrane protein |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MMP | Mitochondrial Membrane Potential |
| NAADP | Nicotinic acid adenine dinucleotide phosphate |
| NLRP3 | Nucleotide-binding oligomerization domain-leucine-rich repeat-pyrin domain-containing 3 |
| PD | Parkinson’s disease |
| PE | Phosphatidylethanolamine |
| PINK1 | PTEN-induced kinase 1 |
| SN | Substantia Nigra |
| SNCA | Synuclein alpha |
| TLRs | Toll-like receptors |
| ULK1 | Unc-51-like autophagy-activating kinase 1 |
| VPS35 | Vacuolar protein sorting 35 ortholog |
| WDR45 | WD Repeat Domain 45 |
References
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- van Wamelen, D.J.; Martinez-Martin, P.; Weintraub, D.; Schrag, A.; Antonini, A.; Falup-Pecurariu, C.; Odin, P.; Ray Chaudhuri, K.; International Parkinson and Movement Disorder Society Parkinson’s Disease Non-Motor Study Group. The Non-Motor Symptoms Scale in Parkinson’s disease: Validation and use. Acta Neurol. Scand. 2021, 143, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef] [PubMed]
- Tambasco, N.; Nigro, P.; Romoli, M.; Prontera, P.; Simoni, S.; Calabresi, P. A53T in a parkinsonian family: A clinical update of the SNCA phenotypes. J. Neural Transm. 2016, 123, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson disease in 2015: Evolving basic, pathological and clinical concepts in PD. Nat. Rev. Neurol. 2016, 12, 65–66. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Julg, J.; Strohm, L.; Behrends, C. Canonical and Noncanonical Autophagy Pathways in Microglia. Mol. Cell Biol. 2021, 41, e0038920. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Kihara, A.; Kabeya, Y.; Ohsumi, Y.; Yoshimori, T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001, 2, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Liang, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef]
- Parys, J.B.; Decuypere, J.P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Blomgren, K.; Poletti, A.; Beart, P.M. Autophagy in neurodegeneration: New insights underpinning therapy for neurological diseases. J. Neurochem. 2020, 154, 354–371. [Google Scholar] [CrossRef]
- Bonam, S.R.; Tranchant, C.; Muller, S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells 2021, 10, 3547. [Google Scholar] [CrossRef]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Li, J.Q.; Tan, L.; Yu, J.T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Marin, I. Ancient origin of the Parkinson disease gene LRRK2. J. Mol. Evol. 2008, 67, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Nagamori, S.; Wiriyasermkul, P.; Zheng, S.; Yago, A.; Shimizu, T.; Tabuchi, Y.; Okumura, T.; Fujii, T.; Takeshima, H.; et al. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H(+),K(+)-ATPase. Nat. Commun. 2023, 14, 2174. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Pang, S.Y.; Lo, R.C.N.; Ho, P.W.; Liu, H.F.; Chang, E.E.S.; Leung, C.T.; Malki, Y.; Choi, Z.Y.; Wong, W.Y.; Kung, M.H.; et al. LRRK2, GBA and their interaction in the regulation of autophagy: Implications on therapeutics in Parkinson’s disease. Transl. Neurodegener. 2022, 11, 5. [Google Scholar] [CrossRef]
- Sanchez-Danes, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jimenez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Niso-Santano, M.; Gomez-Sanchez, R.; Pizarro-Estrella, E.; Aiastui-Pujana, A.; Gorostidi, A.; Climent, V.; Lopez de Maturana, R.; Sanchez-Pernaute, R.; Lopez de Munain, A.; et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol. Life Sci. 2013, 70, 121–136. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Luzon-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Hilfiker, S. LRRK2 as a modulator of lysosomal calcium homeostasis with downstream effects on autophagy. Autophagy 2012, 8, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Araki, M.; Katai, Y.; Nishimura, Y.; Imotani, S.; Inoue, H.; Ito, G.; Tomita, T. Pathogenic LRRK2 compromises the subcellular distribution of lysosomes in a Rab12-RILPL1-dependent manner. FASEB J. 2023, 37, e22930. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Beilina, A.; Williamson, C.D.; Lewis, P.A.; Cookson, M.R.; Bonet-Ponce, L. Lysosomal positioning regulates Rab10 phosphorylation at LRRK2(+) lysosomes. Proc. Natl. Acad. Sci. USA 2022, 119, e2205492119. [Google Scholar] [CrossRef]
- Mantegazza, A.R. LRRK2 Suppresses Lysosome Degradative Activity in Macrophages and Microglia via Transcription Factor E3 Inhibition. Mol. Biol. Cell 2023, 34, P23. [Google Scholar] [CrossRef]
- Albanese, F.; Mercatelli, D.; Finetti, L.; Lamonaca, G.; Pizzi, S.; Shimshek, D.R.; Bernacchia, G.; Morari, M. Constitutive silencing of LRRK2 kinase activity leads to early glucocerebrosidase deregulation and late impairment of autophagy in vivo. Neurobiol. Dis. 2021, 159, 105487. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef]
- Kuo, S.H.; Tasset, I.; Cuervo, A.M.; Sulzer, D. Misfolded GBA/beta-glucocerebrosidase impairs ER-quality control by chaperone-mediated autophagy in Parkinson disease. Autophagy 2022, 18, 3050–3052. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Murphy, K.E.; Halliday, G.M. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy 2014, 10, 1350–1351. [Google Scholar] [CrossRef]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Nguyen, M.; Young, T.J.; Severino, A.; Schwake, M.; Merchant, K.; Krainc, D. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat. Commun. 2019, 10, 5570. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef]
- Rub, C.; Wilkening, A.; Voos, W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017, 367, 111–123. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J.F. PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19, e44981. [Google Scholar] [CrossRef]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Zhu, J.H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003, 13, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Oostra, B.A.; Heutink, P. Linking DJ-1 to neurodegeneration offers novel insights for understanding the pathogenesis of Parkinson’s disease. J. Mol. Med. 2004, 82, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Lev, N.; Roncevic, D.; Ickowicz, D.; Melamed, E.; Offen, D. Role of DJ-1 in Parkinson’s disease. J. Mol. Neurosci. 2006, 29, 215–225. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-Synuclein Aggregation by Regulating Chaperone-Mediated Autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef]
- Zhao, N.; Li, Y.; Wang, C.; Xue, Y.; Peng, L.; Wang, T.; Zhao, Y.; Xu, G.; Yu, S. DJ-1 activates the Atg5-Atg12-Atg16L1 complex via Sirt1 to influence microglial polarization and alleviate cerebral ischemia/reperfusion-induced inflammatory injury. Neurochem. Int. 2022, 157, 105341. [Google Scholar] [CrossRef] [PubMed]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.J.; Zhang, L.; Troncoso, J.; Lee, M.K.; Hattori, N.; Mizuno, Y.; Dawson, T.M.; Dawson, V.L. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum. Mol. Genet. 2005, 14, 71–84. [Google Scholar] [CrossRef]
- Imberechts, D.; Kinnart, I.; Wauters, F.; Terbeek, J.; Manders, L.; Wierda, K.; Eggermont, K.; Madeiro, R.F.; Sue, C.; Verfaillie, C.; et al. DJ-1 is an essential downstream mediator in PINK1/parkin-dependent mitophagy. Brain 2022, 145, 4368–4384. [Google Scholar] [CrossRef]
- Dhanushkodi, N.R.; Abul Khair, S.B.; Ardah, M.T.; Haque, M.E. ATP13A2 Gene Silencing in Drosophila Affects Autophagic Degradation of A53T Mutant alpha-Synuclein. Int. J. Mol. Sci. 2023, 24, 1775. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef]
- Koss, D.J.; Campesan, S.; Giorgini, F.; Outeiro, T.F. Dysfunction of RAB39B-Mediated Vesicular Trafficking in Lewy Body Diseases. Mov. Disord. 2021, 36, 1744–1758. [Google Scholar] [CrossRef]
- Chiu, C.C.; Weng, Y.H.; Yeh, T.H.; Lu, J.C.; Chen, W.S.; Li, A.H.; Chen, Y.L.; Wei, K.C.; Wang, H.L. Deficiency of RAB39B Activates ER Stress-Induced Pro-apoptotic Pathway and Causes Mitochondrial Dysfunction and Oxidative Stress in Dopaminergic Neurons by Impairing Autophagy and Upregulating alpha-Synuclein. Mol. Neurobiol. 2023, 60, 2706–2728. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimer’s Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef]
- Parcon, P.A.; Balasubramaniam, M.; Ayyadevara, S.; Jones, R.A.; Liu, L.; Shmookler Reis, R.J.; Barger, S.W.; Mrak, R.E.; Griffin, W.S.T. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimer’s Dement. 2017, 14, 230–242. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The Effects of APOE4 on Mitochondrial Dynamics and Proteins in vivo. J. Alzheimer’s Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Michaelson, D.M.; Pinkas-Kramarski, R. The Interplay Between Apolipoprotein E4 and the Autophagic-Endocytic-Lysosomal Axis. Mol. Neurobiol. 2018, 55, 6863–6880. [Google Scholar] [CrossRef]
- Schmukler, E.; Solomon, S.; Simonovitch, S.; Goldshmit, Y.; Wolfson, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 2020, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, D.M. APOE epsilon4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 861–868. [Google Scholar] [CrossRef]
- Ghebremedhin, E.; Del Tredici, K.; Vuksic, M.; Rub, U.; Thal, D.R.; Burbach, G.J.; Rosenberger, A.; Bickeboller, H.; Deller, T.; de Vos, R.A.; et al. Relationship of apolipoprotein E and age at onset to Parkinson disease neuropathology. J. Neuropathol. Exp. Neurol. 2006, 65, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Hauser, M.A.; Scott, W.K.; Martin, E.R.; Booze, M.W.; Qin, X.J.; Walter, J.W.; Nance, M.A.; Hubble, J.P.; Koller, W.C.; et al. Apolipoprotein E controls the risk and age at onset of Parkinson disease. Neurology 2004, 62, 2005–2009. [Google Scholar] [CrossRef]
- Kim, R.; Shin, J.H.; Park, S.; Kim, H.J.; Jeon, B. Apolipoprotein E epsilon4 genotype and risk of freezing of gait in Parkinson’s disease. Park. Relat. Disord. 2020, 81, 173–178. [Google Scholar] [CrossRef]
- Pu, J.L.; Jin, C.Y.; Wang, Z.X.; Fang, Y.; Li, Y.L.; Xue, N.J.; Zheng, R.; Lin, Z.H.; Yan, Y.Q.; Si, X.L.; et al. Apolipoprotein E Genotype Contributes to Motor Progression in Parkinson’s Disease. Mov. Disord. 2022, 37, 196–200. [Google Scholar] [CrossRef]
- Pang, S.; Li, J.; Zhang, Y.; Chen, J. Meta-Analysis of the Relationship between the APOE Gene and the Onset of Parkinson’s Disease Dementia. Park. Dis. 2018, 2018, 9497147. [Google Scholar] [CrossRef]
- Liu, J.Y.; Ma, L.Z.; Wang, J.; Cui, X.J.; Sheng, Z.H.; Fu, Y.; Li, M.; Ou, Y.N.; Yu, J.T.; Tan, L.; et al. Age-Related Association Between APOE varepsilon4 and Cognitive Progression in de novo Parkinson’s Disease. J. Alzheimer’s Dis. 2023, 91, 1121–1132. [Google Scholar] [CrossRef]
- Real, R.; Martinez-Carrasco, A.; Reynolds, R.H.; Lawton, M.A.; Tan, M.M.X.; Shoai, M.; Corvol, J.C.; Ryten, M.; Bresner, C.; Hubbard, L.; et al. Association between the LRP1B and APOE loci and the development of Parkinson’s disease dementia. Brain 2023, 146, 1873–1887. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Attrebi, O.N.; Ren, Y.; Qiao, W.; Sonustun, B.; Martens, Y.A.; Meneses, A.D.; Li, F.; Shue, F.; Zheng, J.; et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 2020, 12, aay1809. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.A.; Inman, C.E.; Wargel, Z.M.; Dube, U.; Freeberg, B.M.; Galluppi, A.; Haines, J.N.; Dhavale, D.D.; Miller, R.; Choudhury, F.A.; et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 2020, 12, eaay3069. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.; Rubinsztein, D.C. VPS35 Parkinson mutation impairs autophagy via WASH. Cell Cycle 2014, 13, 2155–2156. [Google Scholar] [CrossRef]
- Singer, H.S.; Mink, J.W.; Gilbert, D.L.; Jankovic, J. Chapter 17—Inherited Metabolic Disorders with Associated Movement Abnormalities. In Movement Disorders in Childhood, 2nd ed.; Elsevier: London, UK, 2016; pp. 337–407. [Google Scholar]
- Cong, Y.; So, V.; Tijssen, M.A.J.; Verbeek, D.S.; Reggiori, F.; Mauthe, M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy 2021, 17, 3908–3923. [Google Scholar] [CrossRef] [PubMed]
- Aring, L.; Choi, E.K.; Kopera, H.; Lanigan, T.; Iwase, S.; Klionsky, D.J.; Seo, Y.A. A neurodegeneration gene, WDR45, links impaired ferritinophagy to iron accumulation. J. Neurochem. 2022, 160, 356–375. [Google Scholar] [CrossRef] [PubMed]
- Manti, F.; Panteghini, C.; Garavaglia, B.; Leuzzi, V. Neurodevelopmental Disorder and Late-Onset Degenerative Parkinsonism in a Patient with a WDR45 Defect. Mov. Disord. Clin. Pract. 2022, 9, 110–112. [Google Scholar] [CrossRef]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef]
- Liu, Y.; Lear, T.B.; Verma, M.; Wang, K.Z.; Otero, P.A.; McKelvey, A.C.; Dunn, S.R.; Steer, E.; Bateman, N.W.; Wu, C.; et al. Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 2020, 5, e131834. [Google Scholar] [CrossRef]
- Surgucheva, I.; Newell, K.L.; Burns, J.; Surguchov, A. New alpha- and gamma-synuclein immunopathological lesions in human brain. Acta Neuropathol. Commun. 2014, 2, 132. [Google Scholar]
- Hayashi, J.; Carver, J.A. beta-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules 2022, 12, 142. [Google Scholar] [CrossRef]
- Gallegos, S.; Pacheco, C.; Peters, C.; Opazo, C.M.; Aguayo, L.G. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef]
- Grozdanov, V.; Danzer, K.M. Release and uptake of pathologic alpha-synuclein. Cell Tissue Res. 2018, 373, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Dave, K.D.; Kordower, J.H. Therapeutic approaches to target alpha-synuclein pathology. Exp. Neurol. 2017, 298, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Bras, I.C.; Xylaki, M.; Outeiro, T.F. Mechanisms of alpha-synuclein toxicity: An update and outlook. Prog. Brain Res. 2020, 252, 91–129. [Google Scholar] [PubMed]
- Emmenegger, M.; De Cecco, E.; Hruska-Plochan, M.; Eninger, T.; Schneider, M.M.; Barth, M.; Tantardini, E.; de Rossi, P.; Bacioglu, M.; Langston, R.G.; et al. LAG3 is not expressed in human and murine neurons and does not modulate alpha-synucleinopathies. EMBO Mol. Med. 2021, 13, e14745. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson’s disease pathogenesis: The environment takes a toll on the gut. Transl. Neurodegener. 2021, 10, 47. [Google Scholar] [CrossRef]
- Nguyen, L.T.N.; Nguyen, H.D.; Kim, Y.J.; Nguyen, T.T.; Lai, T.T.; Lee, Y.K.; Ma, H.I.; Kim, Y.E. Role of NLRP3 Inflammasome in Parkinson’s Disease and Therapeutic Considerations. J. Park. Dis. 2022, 12, 2117–2133. [Google Scholar] [CrossRef]
- Wu, A.G.; Zhou, X.G.; Qiao, G.; Yu, L.; Tang, Y.; Yan, L.; Qiu, W.Q.; Pan, R.; Yu, C.L.; Law, B.Y.; et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res. Rev. 2021, 65, 101202. [Google Scholar] [CrossRef]
- Zhao, S.; Li, X.; Wang, J.; Wang, H. The Role of the Effects of Autophagy on NLRP3 Inflammasome in Inflammatory Nervous System Diseases. Front. Cell Dev. Biol. 2021, 9, 657478. [Google Scholar] [CrossRef]
- Amo-Aparicio, J.; Daly, J.; Hojen, J.F.; Dinarello, C.A. Pharmacologic inhibition of NLRP3 reduces the levels of alpha-synuclein and protects dopaminergic neurons in a model of Parkinson’s disease. J. Neuroinflamm. 2023, 20, 147. [Google Scholar] [CrossRef] [PubMed]
- Song, J.X.; Lu, J.H.; Liu, L.F.; Chen, L.L.; Durairajan, S.S.; Yue, Z.; Zhang, H.Q.; Li, M. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014, 10, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.Y.; Yuan, B.S.; Hou, X.O.; Zhang, X.J.; Pei, C.S.; Ma, Y.T.; Yang, Y.P.; Fan, Y.; Qin, Z.H.; Liu, C.F.; et al. Alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Chen, T.H.; Koga, S.; Bredenberg, J.M.; Faroqi, A.H.; Delenclos, M.; Bu, G.; Wszolek, Z.K.; Carr, J.A.; Ross, O.A.; et al. Alpha-synuclein-associated changes in PINK1-PRKN-mediated mitophagy are disease context dependent. Brain Pathol. 2023, 33, e13175. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef]
- Sarkar, S.; Olsen, A.L.; Sygnecka, K.; Lohr, K.M.; Feany, M.B. alpha-synuclein impairs autophagosome maturation through abnormal actin stabilization. PLoS Genet. 2021, 17, e1009359. [Google Scholar] [CrossRef]
- Tang, Q.; Gao, P.; Arzberger, T.; Hollerhage, M.; Herms, J.; Hoglinger, G.; Koeglsperger, T. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis. 2021, 12, 854. [Google Scholar] [CrossRef]
- Nascimento, A.C.; Erustes, A.G.; Reckziegel, P.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. alpha-Synuclein Overexpression Induces Lysosomal Dysfunction and Autophagy Impairment in Human Neuroblastoma SH-SY5Y. Neurochem. Res. 2020, 45, 2749–2761. [Google Scholar] [CrossRef]
- Oh, C.-k.; Nakamura, T.; Lipton, S.A. Inhibition of autophagic flux by S-nitrosylation of SQSTM1/p62 promotes neuronal secretion and cell-to-cell transmission of SNCA/α-synuclein in Parkinson disease and Lewy body dementia. Autophagy Rep. 2022, 1, 223–225. [Google Scholar] [CrossRef]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal Dysfunction and alpha-Synuclein Aggregation in Parkinson’s Disease: Diagnostic Links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [PubMed]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 2005, 25, 5544–5552. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, D.; Dybdal, N.; Donaldson, M.T.; Miller, L.; Cribb, A.E. Nitration and increased alpha-synuclein expression associated with dopaminergic neurodegeneration in equine pituitary pars intermedia dysfunction. J. Neuroendocrinol. 2005, 17, 73–80. [Google Scholar] [CrossRef]
- Klucken, J.; Poehler, A.M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlotzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; et al. Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef]
- Chavarria, C.; Ivagnes, R.; Souza, J.M. Extracellular Alpha-Synuclein: Mechanisms for Glial Cell Internalization and Activation. Biomolecules 2022, 12, 655. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from alpha-Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Sung, K.; Jimenez-Sanchez, M. Autophagy in Astrocytes and its Implications in Neurodegeneration. J. Mol. Biol. 2020, 432, 2605–2621. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mirasierra, I.; Ghimire, S.; Hernandez-Diaz, S.; Soukup, S.F. Targeting Macroautophagy as a Therapeutic Opportunity to Treat Parkinson’s Disease. Front. Cell Dev. Biol. 2022, 10, 921314. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Fan, M.; Xu, Z.; Cai, Y.; Chen, Y.; Yu, S.; Zeng, L. Neuroprotective effect of rapamycin against Parkinson’s disease in mice. Zhejiang Da Xue Xue Bao Yi Xue Ban 2018, 47, 465–472. [Google Scholar] [PubMed]
- Zhang, G.; Yin, L.; Luo, Z.; Chen, X.; He, Y.; Yu, X.; Wang, M.; Tian, F.; Luo, H. Effects and potential mechanisms of rapamycin on MPTP-induced acute Parkinson’s disease in mice. Ann. Palliat. Med. 2021, 10, 2889–2897. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, S.; Li, J.; Jiang, T.; Feng, L.; Pei, J.; Wang, G.; Ouyang, L.; Liu, B. Targeting autophagy using small-molecule compounds to improve potential therapy of Parkinson’s disease. Acta Pharm. Sin. B 2021, 11, 3015–3034. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Callegari, S.; Dewson, G. Therapeutic targeting of mitophagy in Parkinson’s disease. Biochem. Soc. Trans. 2022, 50, 783–797. [Google Scholar] [CrossRef]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef]
- Barini, E.; Miccoli, A.; Tinarelli, F.; Mulholland, K.; Kadri, H.; Khanim, F.; Stojanovski, L.; Read, K.D.; Burness, K.; Blow, J.J.; et al. The Anthelmintic Drug Niclosamide and Its Analogues Activate the Parkinson’s Disease Associated Protein Kinase PINK1. Chembiochem 2018, 19, 425–429. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, D.; Tian, K.; Ren, C.; Li, H.; Lin, C.; Huang, X.; Liu, J.; Mao, W.; Zhang, J. Small-molecule LRRK2 inhibitors for PD therapy: Current achievements and future perspectives. Eur. J. Med. Chem. 2023, 256, 115475. [Google Scholar] [CrossRef]
- Taymans, J.M.; Fell, M.; Greenamyre, T.; Hirst, W.D.; Mamais, A.; Padmanabhan, S.; Peter, I.; Rideout, H.; Thaler, A. Perspective on the current state of the LRRK2 field. NPJ Park. Dis. 2023, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.; Prescott, A.R.; Rosewell, P.; Ball, G.; Reith, A.D.; Ganley, I.G. Pharmacological rescue of impaired mitophagy in Parkinson’s disease-related LRRK2 G2019S knock-in mice. Elife 2021, 10, e67604. [Google Scholar] [CrossRef] [PubMed]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and clinical evaluation of the LRRK2 inhibitor DNL201 for Parkinson’s disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef] [PubMed]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 2023, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Jennings, D.; Huntwork-Rodriguez, S.; Vissers, M.; Daryani, V.M.; Diaz, D.; Goo, M.S.; Chen, J.J.; Maciuca, R.; Fraser, K.; Mabrouk, O.S.; et al. LRRK2 Inhibition by BIIB122 in Healthy Participants and Patients with Parkinson’s Disease. Mov. Disord. 2023, 38, 386–398. [Google Scholar] [CrossRef]
- Wallings, R.; Connor-Robson, N.; Wade-Martins, R. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum. Mol. Genet. 2019, 28, 2696–2710. [Google Scholar] [CrossRef]
- Ma, Z.; Liang, H.; Hu, B.; Cai, S.; Yan, D. Autophagy-regulating miRNAs: Novel therapeutic targets for Parkinson’s disease (Review). Int. J. Mol. Med. 2023, 51, 50. [Google Scholar] [CrossRef]
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; Kerchner, G.A.; Ayalon, G.; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial Dysfunction and Mitophagy in Parkinson’s Disease: From Mechanism to Therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Autophagy and Alpha-Synuclein: Relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 2016, 31, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Schmukler, E.; Pinkas-Kramarski, R. Autophagy induction in the treatment of Alzheimer’s disease. Drug Dev. Res. 2020, 81, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Zheng, N.; Wang, Z.; Gao, Y.; Luo, X.; Chen, Z.; Fu, X.; Wang, Y.; Wang, T.; Liu, M.; et al. RAB39B Deficiency Impairs Learning and Memory Partially Through Compromising Autophagy. Front. Cell Dev. Biol. 2020, 8, 598622. [Google Scholar] [CrossRef]
- Diab, R.; Pilotto, F.; Saxena, S. Autophagy and neurodegeneration: Unraveling the role of C9ORF72 in the regulation of autophagy and its relationship to ALS-FTD pathology. Front. Cell Neurosci. 2023, 17, 1086895. [Google Scholar] [CrossRef]
- Yang, X.; Xu, Y. Mutations in the ATP13A2 gene and Parkinsonism: A preliminary review. BioMed Res. Int. 2014, 2014, 371256. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 612476. [Google Scholar] [CrossRef]
- Ramonet, D.; Podhajska, A.; Stafa, K.; Sonnay, S.; Trancikova, A.; Tsika, E.; Pletnikova, O.; Troncoso, J.C.; Glauser, L.; Moore, D.J. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Hum. Mol. Genet. 2012, 21, 1725–1743. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nechushtai, L.; Frenkel, D.; Pinkas-Kramarski, R. Autophagy in Parkinson’s Disease. Biomolecules 2023, 13, 1435. https://doi.org/10.3390/biom13101435
Nechushtai L, Frenkel D, Pinkas-Kramarski R. Autophagy in Parkinson’s Disease. Biomolecules. 2023; 13(10):1435. https://doi.org/10.3390/biom13101435
Chicago/Turabian StyleNechushtai, Lior, Dan Frenkel, and Ronit Pinkas-Kramarski. 2023. "Autophagy in Parkinson’s Disease" Biomolecules 13, no. 10: 1435. https://doi.org/10.3390/biom13101435