
Screening of Tumor Antigens and Construction of Immune Subtypes for mRNA Vaccine Development in Head and Neck Squamous Cell Carcinoma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
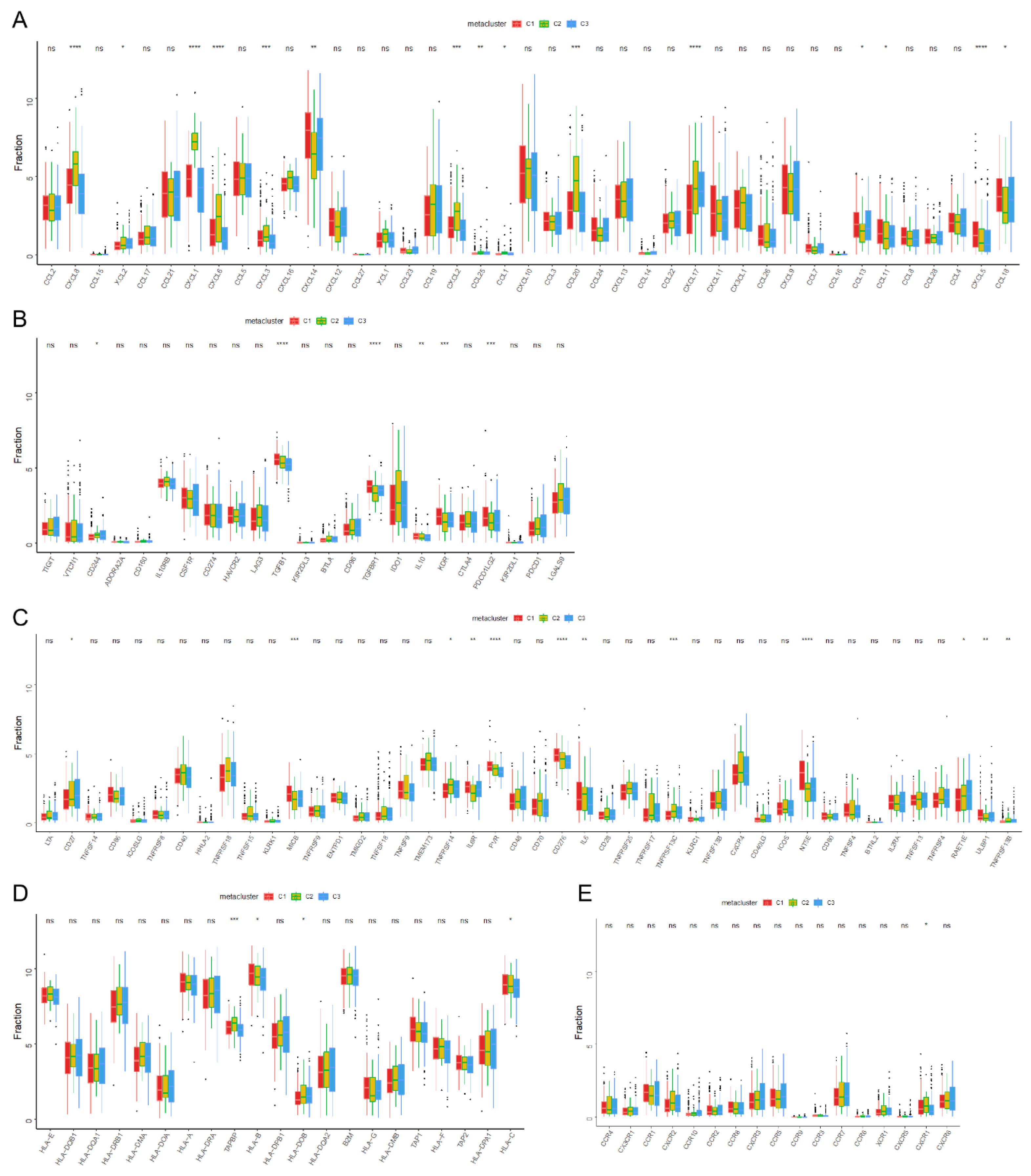
2.2. Tumor Immune Evaluation
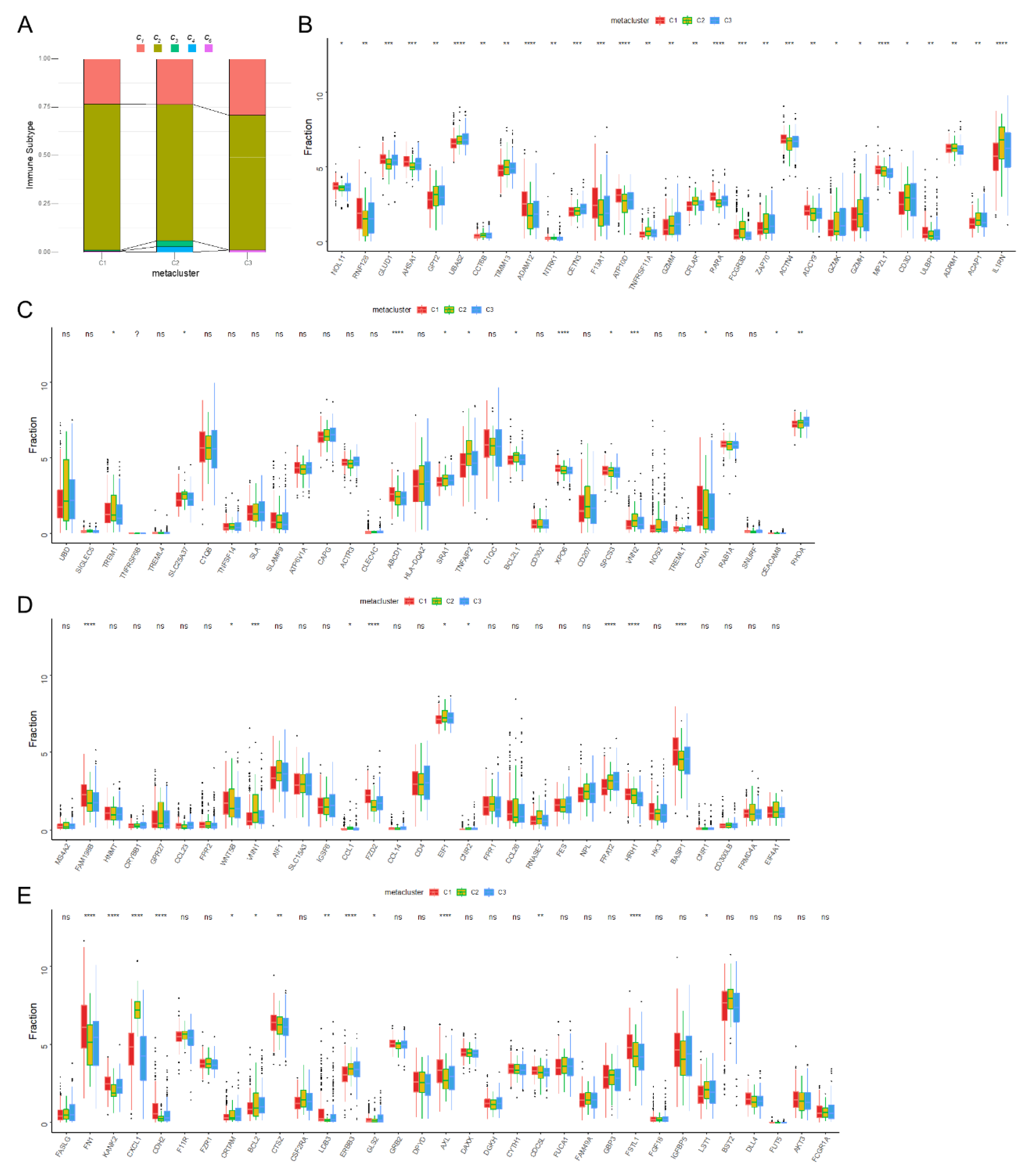
2.3. Classification of Immune Subtypes
2.4. Drug Sensitivity Analysis
2.5. Gene Set Variation Analysis
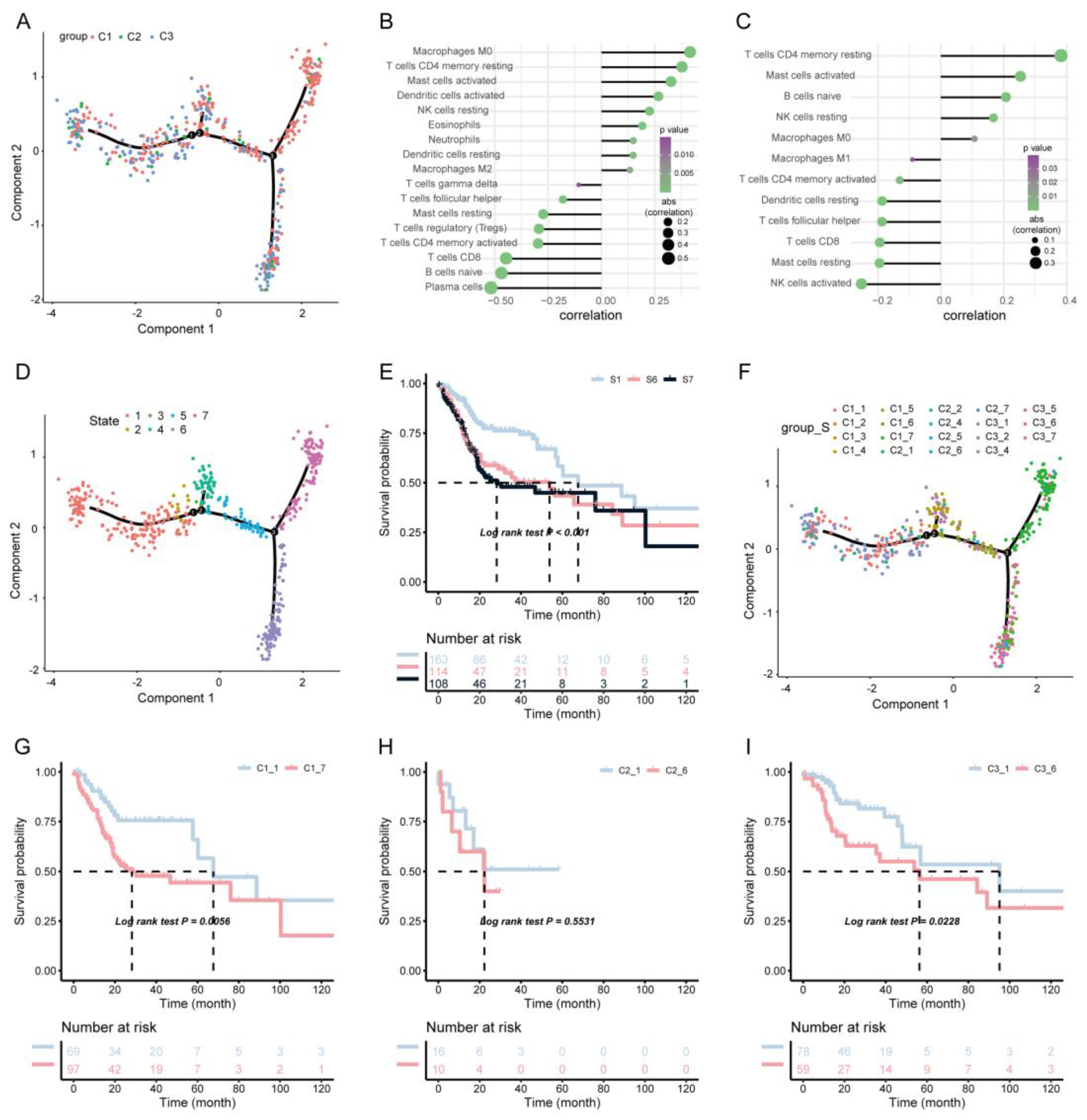
2.6. Immune Landscape Analysis
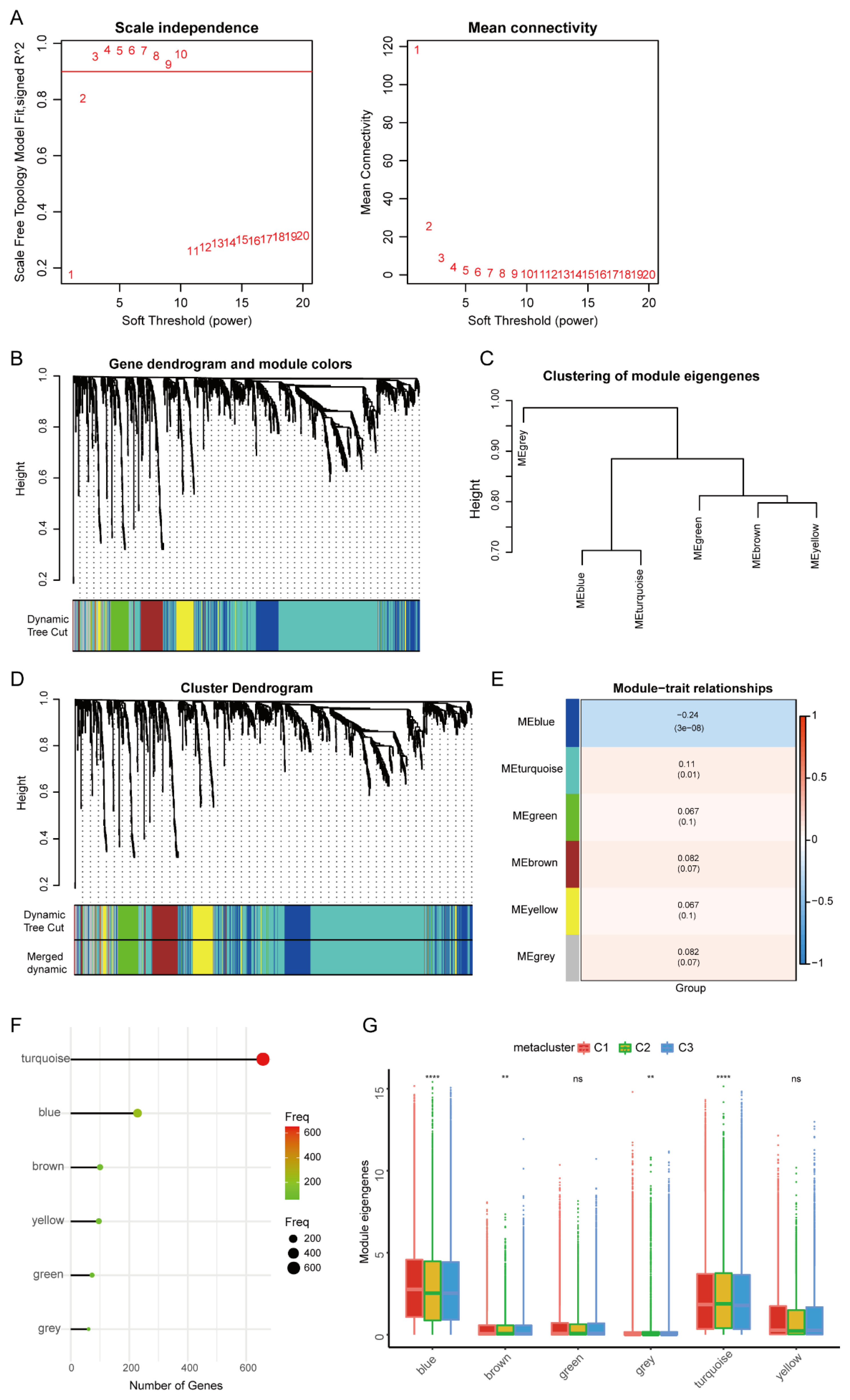
2.7. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.8. Analysis of GO and KEGG Functions
2.9. Subtype GSEA Analysis
2.10. Statistical Analysis
3. Results
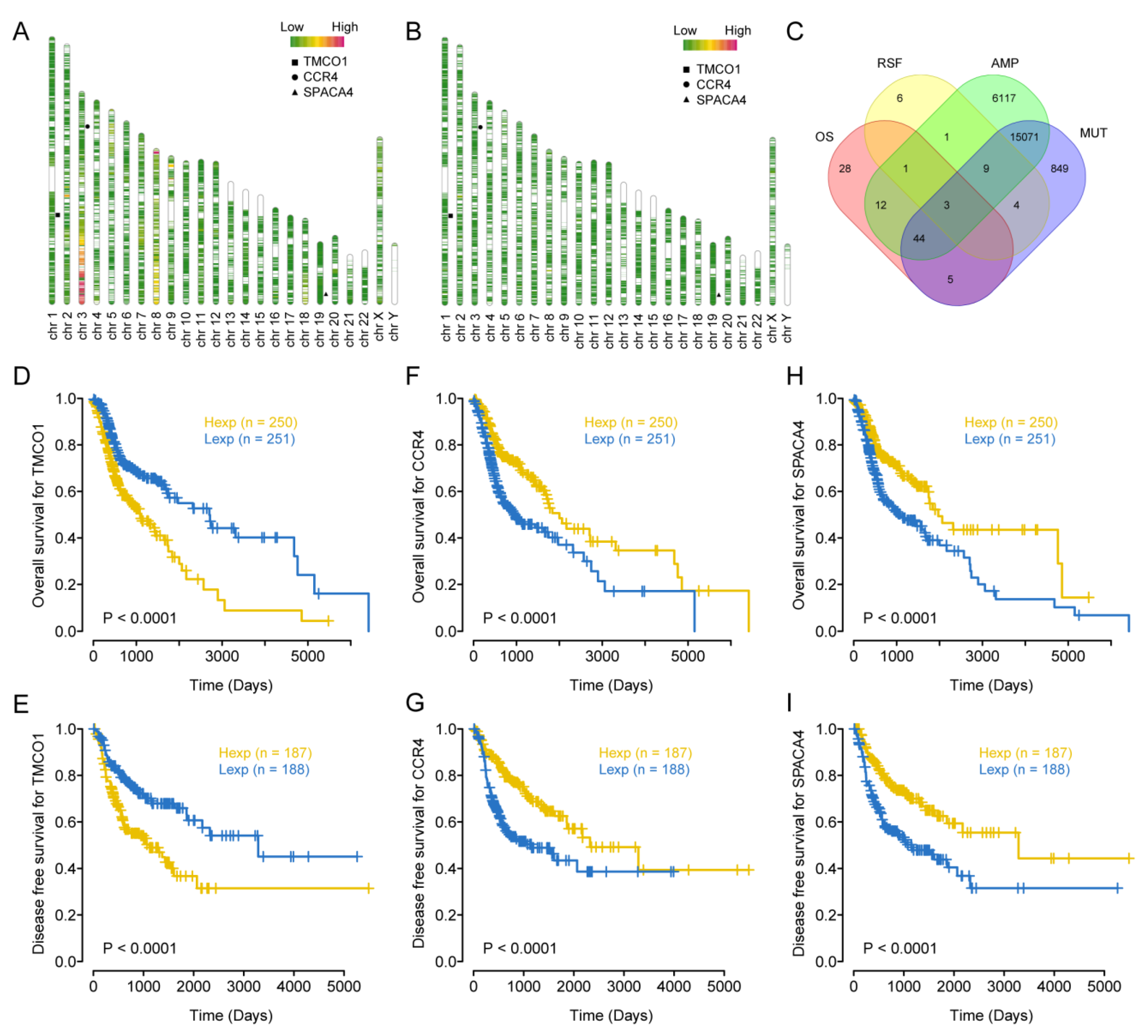
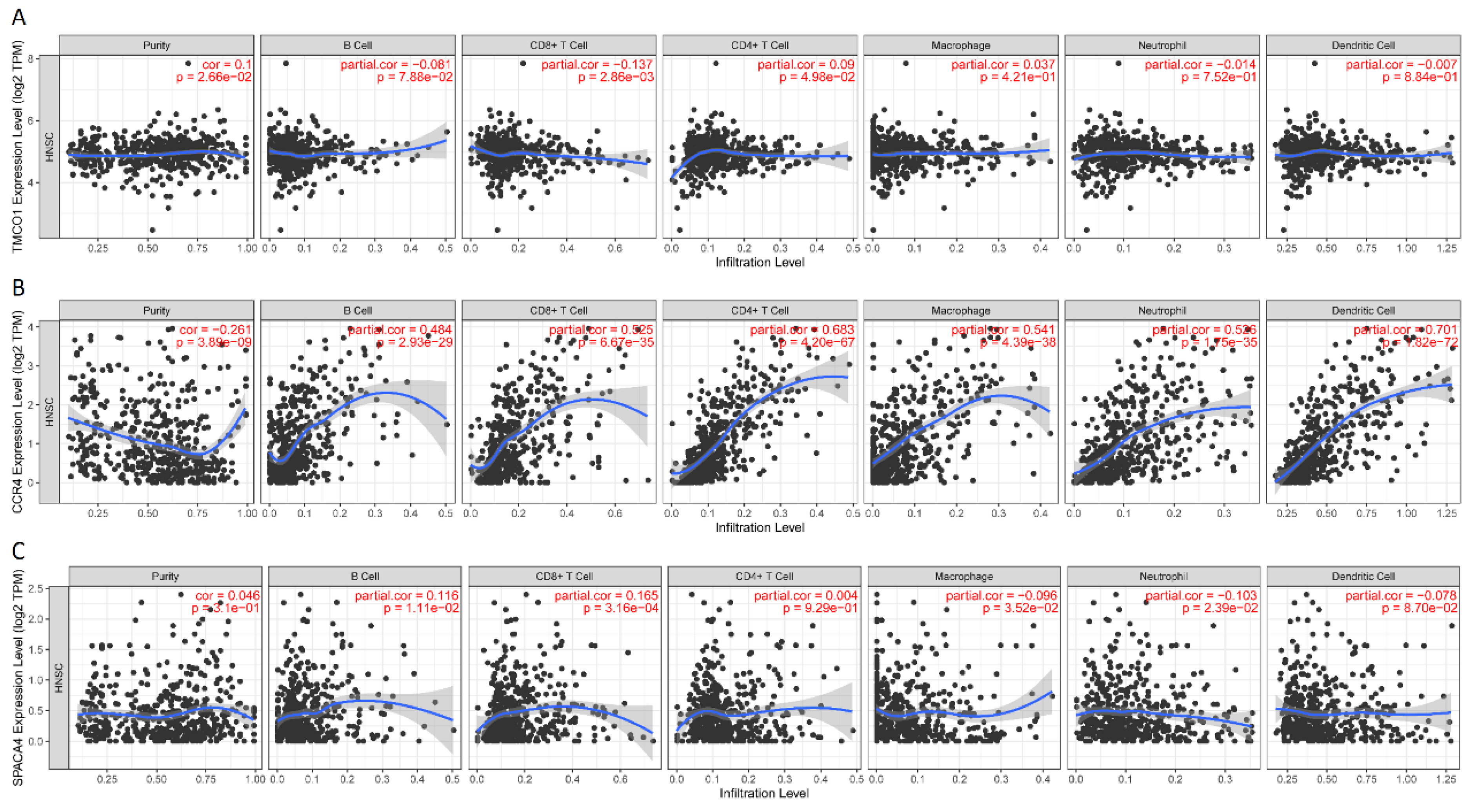
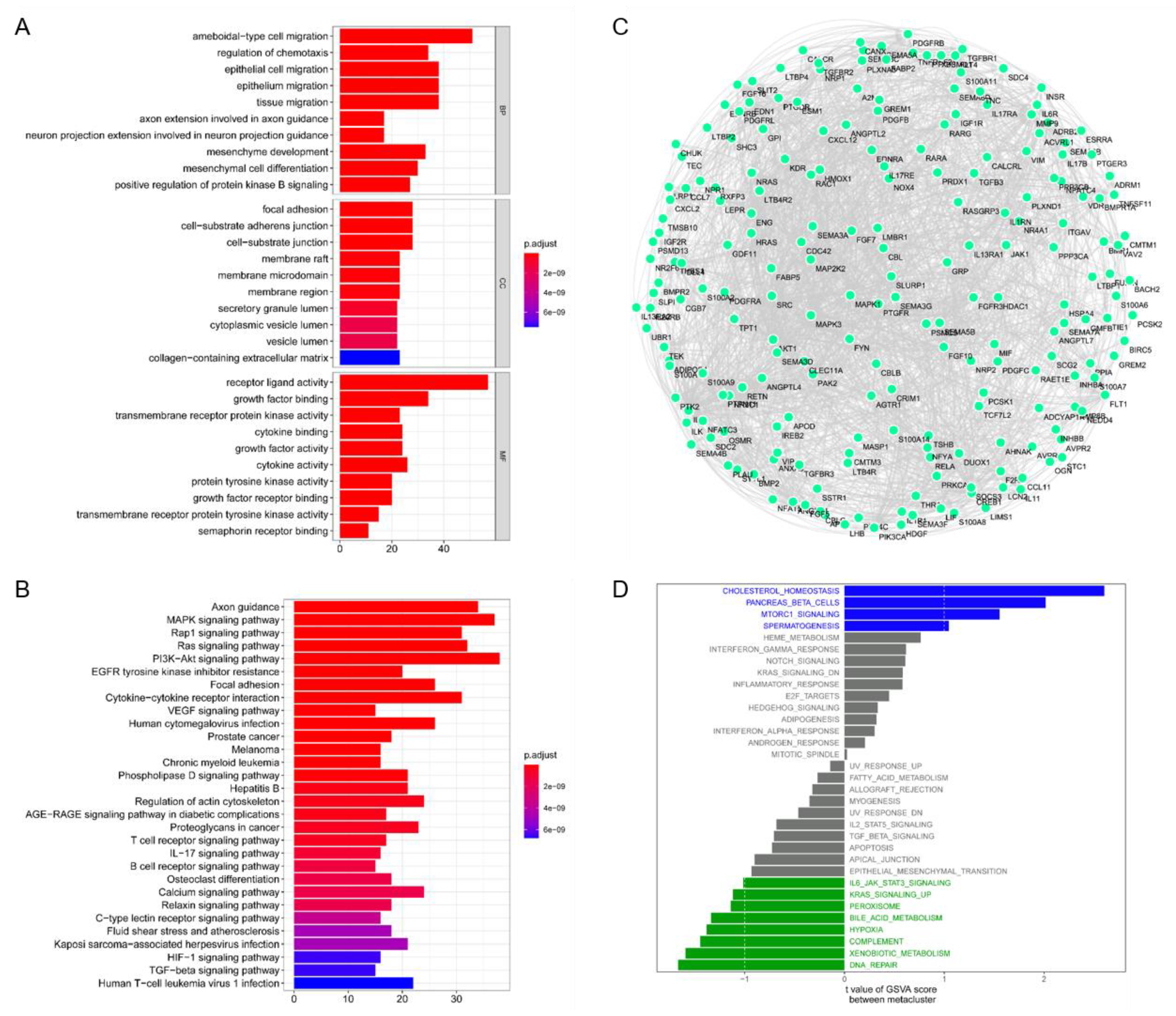
3.1. Identification of Immune-Critical Genes in HNSCC
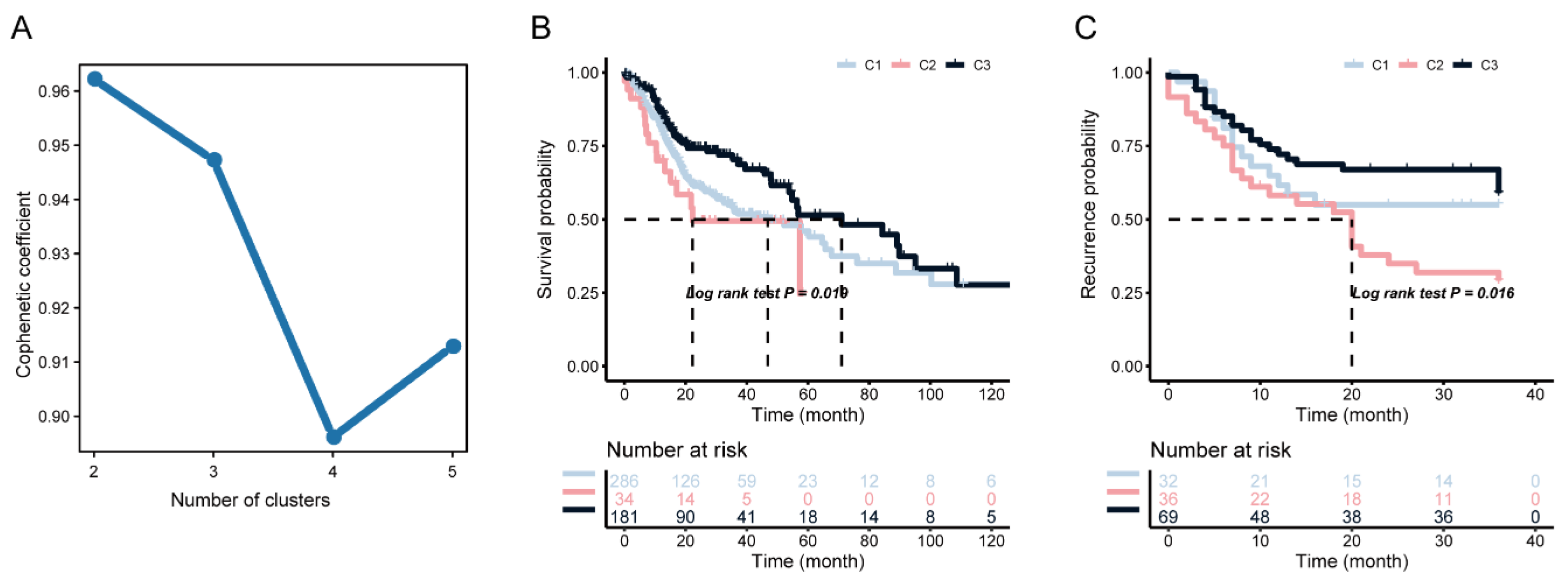
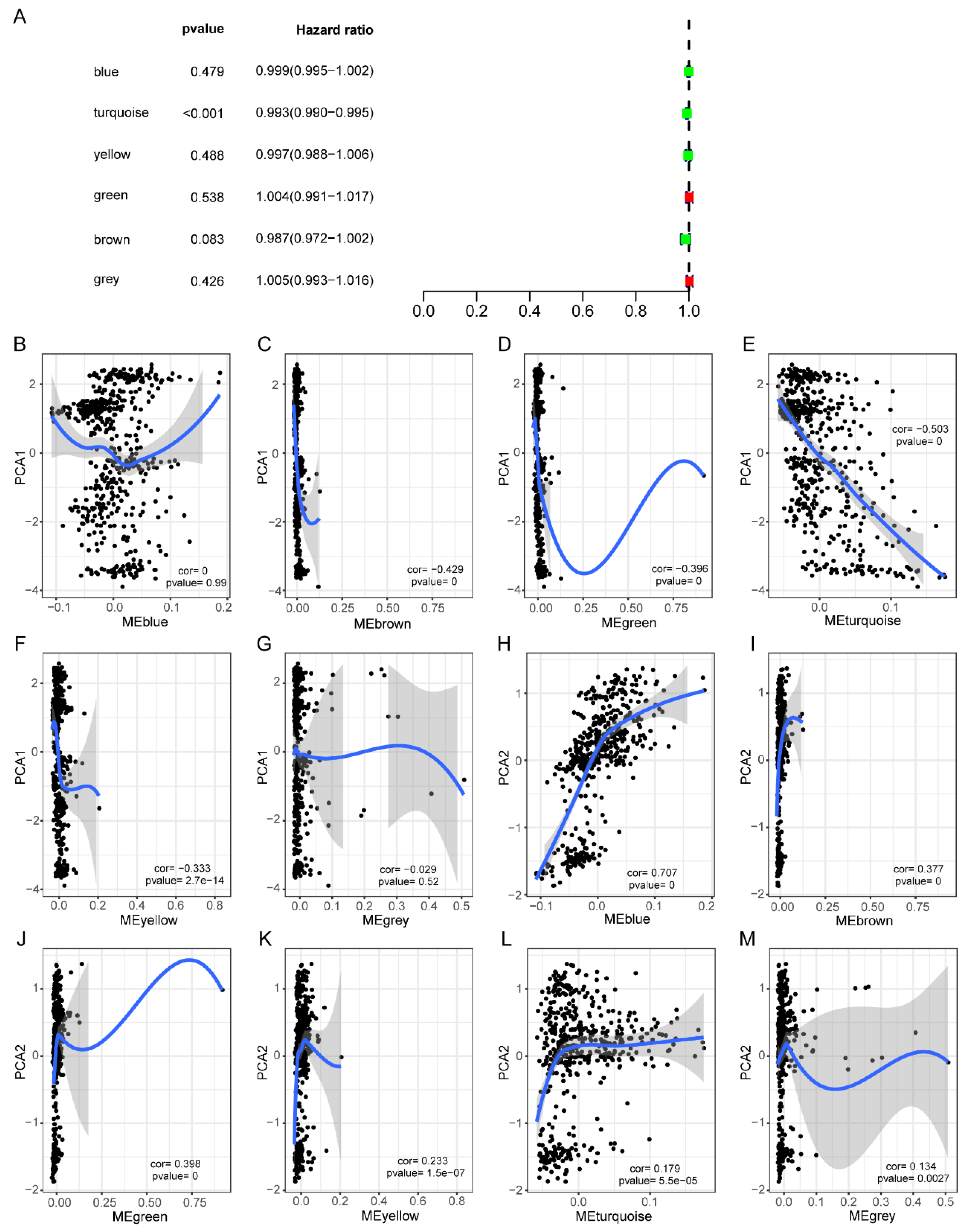
3.2. Identification of Potential Immune Subtypes of HNSC
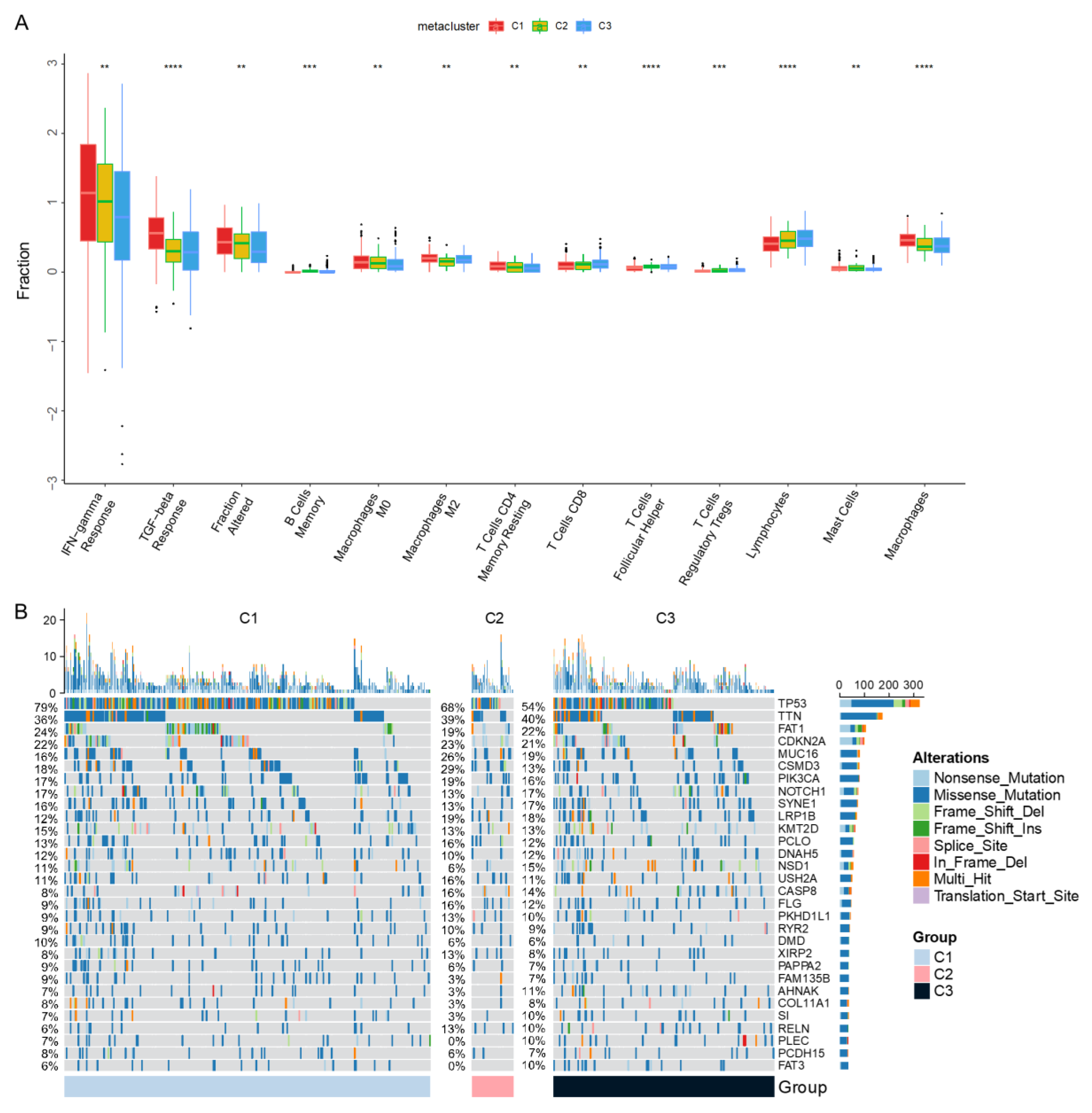
3.3. Molecular Characteristics of Immune Subtypes
3.4. Immune Landscape of HNSCC
3.5. HNSCC Immune Gene Co-Expression Module
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulte, D.; Brenner, H. Changes in survival in head and neck cancers in the late 20th and early 21st century: A period analysis. Oncol. 2010, 15, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, R.L.; Blumenschein, G., Jr. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiwert, T.Y.; Burtness, B. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Thess, A.; Grund, S. Sequence-engineered mRNA without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Muramatsu, H. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Campillo-Davo, D.; Versteven, M. Rapid Assessment of Functional Avidity of Tumor-Specific T Cell Receptors Using an Antigen-Presenting Tumor Cell Line Electroporated with Full-Length Tumor Antigen mRNA. Cancers 2020, 12, 256. [Google Scholar] [CrossRef] [Green Version]
- Hess, P.R.; Boczkowski, D. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol. Immunother. CII 2006, 55, 672–683. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Z. mRNA vaccine: A potential therapeutic strategy. Mol. Cancer 2021, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberli, M.A.; Reichmuth, A.M. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Boudewijns, S.; Bloemendal, M. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: A prospective, randomized phase 2 trial. Cancer Immunol. Immunother. CII 2020, 69, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westdorp, H.; Creemers, J.H.A. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weide, B.; Pascolo, S. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Simon, N.; Friedman, J. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011, 39, 1–13. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Cacchiarelli, D. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Mao, Q. Probabilistic Dimensionality Reduction via Structure Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2019, 41, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.G. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef] [Green Version]
- Oliva, M.; Spreafico, A. Immune biomarkers of response to immune-checkpoint inhibitors in head and neck squamous cell carcinoma. Ann. Oncol. 2019, 30, 57–67. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulieres, D. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.; Yin, Y. Tumor RNA-loaded nanoliposomes increases the anti-tumor immune response in colorectal cancer. Drug Deliv. 2021, 28, 1548–1561. [Google Scholar] [CrossRef] [PubMed]
- Cafri, G.; Gartner, J.J. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; He, X. CCR4, CCR8, and P2RY14 as Prognostic Factors in Head and Neck Squamous Cell Carcinoma Are Involved in the Remodeling of the Tumor Microenvironment. Front. Oncol. 2021, 11, 618187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K. CCR4 is a prognostic biomarker and correlated with immune infiltrates in head and neck squamous cell carcinoma. Ann. Transl. Med. 2021, 9, 1443. [Google Scholar] [CrossRef]
- Ling, Z.; Li, W. Targeting CCL2-CCR4 axis suppress cell migration of head and neck squamous cell carcinoma. Cell Death Dis. 2022, 13, 158. [Google Scholar] [CrossRef]
- Tan, Y.S.; Sansanaphongpricha, K. Engineering Vaccines to Reprogram Immunity against Head and Neck Cancer. J. Dent. Res. 2018, 97, 627–634. [Google Scholar] [CrossRef]
- Li, Q.; Ren, J. CpG Oligodeoxynucleotide Developed to Activate Primate Immune Responses Promotes Antitumoral Effects in Combination with a Neoantigen-Based mRNA Cancer Vaccine. Drug Des. Devel. 2021, 15, 3953–3963. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef]
- Ribbat-Idel, J.; Perner, S. Immunologic “Cold” Squamous Cell Carcinomas of the Head and Neck Are Associated with an Unfavorable Prognosis. Front. Med. 2021, 8, 622330. [Google Scholar] [CrossRef]
- Zhang, L.; Li, B. The prognostic value of TMB and the relationship between TMB and immune infiltration in head and neck squamous cell carcinoma: A gene expression-based study. Oral Oncol. 2020, 110, 104943. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, D. Comprehensive Analysis of Myeloid Signature Genes in Head and Neck Squamous Cell Carcinoma to Predict the Prognosis and Immune Infiltration. Front. Immunol. 2021, 12, 659184. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Zheng, Z.Q. Comprehensive characterization of the alternative splicing landscape in head and neck squamous cell carcinoma reveals novel events associated with tumorigenesis and the immune microenvironment. Theranostics 2019, 9, 7648–7665. [Google Scholar] [CrossRef] [PubMed]
- Budhwani, M.; Turrell, G. Immune-Inhibitory Gene Expression is Positively Correlated with Overall Immune Activity and Predicts Increased Survival Probability of Cervical and Head and Neck Cancer Patients. Front. Mol. Biosci. 2021, 8, 622643. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Takahashi, H. Molecular profiling of circulating tumor cells predicts clinical outcome in head and neck squamous cell carcinoma. Oral Oncol. 2020, 102, 104558. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, T. Analysis of the Prognosis and Therapeutic Value of the CXC Chemokine Family in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 570736. [Google Scholar] [CrossRef]
- Huang, X.; Tang, T. Identification of tumor antigens and immune subtypes of cholangiocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 50. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, G. Identification of tumor antigens and immune subtypes of pancreatic adenocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 44. [Google Scholar] [CrossRef]
- Shibata, H.; Zhou, L. Personalized cancer vaccination in head and neck cancer. Cancer Sci. 2021, 112, 978–988. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.-X.; Liu, T.-R.; Tu, Z.-X.; Xie, C.-B.; Wen, W.-P.; Sun, W. Screening of Tumor Antigens and Construction of Immune Subtypes for mRNA Vaccine Development in Head and Neck Squamous Cell Carcinoma. Biomolecules 2023, 13, 90. https://doi.org/10.3390/biom13010090
Li H-X, Liu T-R, Tu Z-X, Xie C-B, Wen W-P, Sun W. Screening of Tumor Antigens and Construction of Immune Subtypes for mRNA Vaccine Development in Head and Neck Squamous Cell Carcinoma. Biomolecules. 2023; 13(1):90. https://doi.org/10.3390/biom13010090
Chicago/Turabian StyleLi, Hong-Xia, Tian-Run Liu, Zhao-Xu Tu, Chu-Bo Xie, Wei-Ping Wen, and Wei Sun. 2023. "Screening of Tumor Antigens and Construction of Immune Subtypes for mRNA Vaccine Development in Head and Neck Squamous Cell Carcinoma" Biomolecules 13, no. 1: 90. https://doi.org/10.3390/biom13010090