2.3. General Procedure for the Synthesis of Olefinic Diarylmethanes
To a suspension of zinc (2.86 mmol, 182 mg, 6 eq) in anhydrous tetrahydrofuran (THF) (2.6 mL) under argon was added dropwise TiCl4 (1.872 mmol, 0.2 mL, 4 eq). The reaction mixture was stirred for 2 h at 85 °C. A solution of and (4-methoxyphenyl) (pyridin-2-yl)methanone 5 (0.468 mmol, 100 mg, 1 eq) and the corresponding aromatic aldehyde 2 (0.486 mmol, 1.04 eq) in THF (1 mL) was then added dropwise via a syringe. After reaction completion, the mixture was cooled at room temperature and then poured into the water and extracted with dichloromethane (DCM). The combined organic extracts were dried over anhydrous MgSO4, filtered and concentrated. The crude was purified by FCC on silica gel.
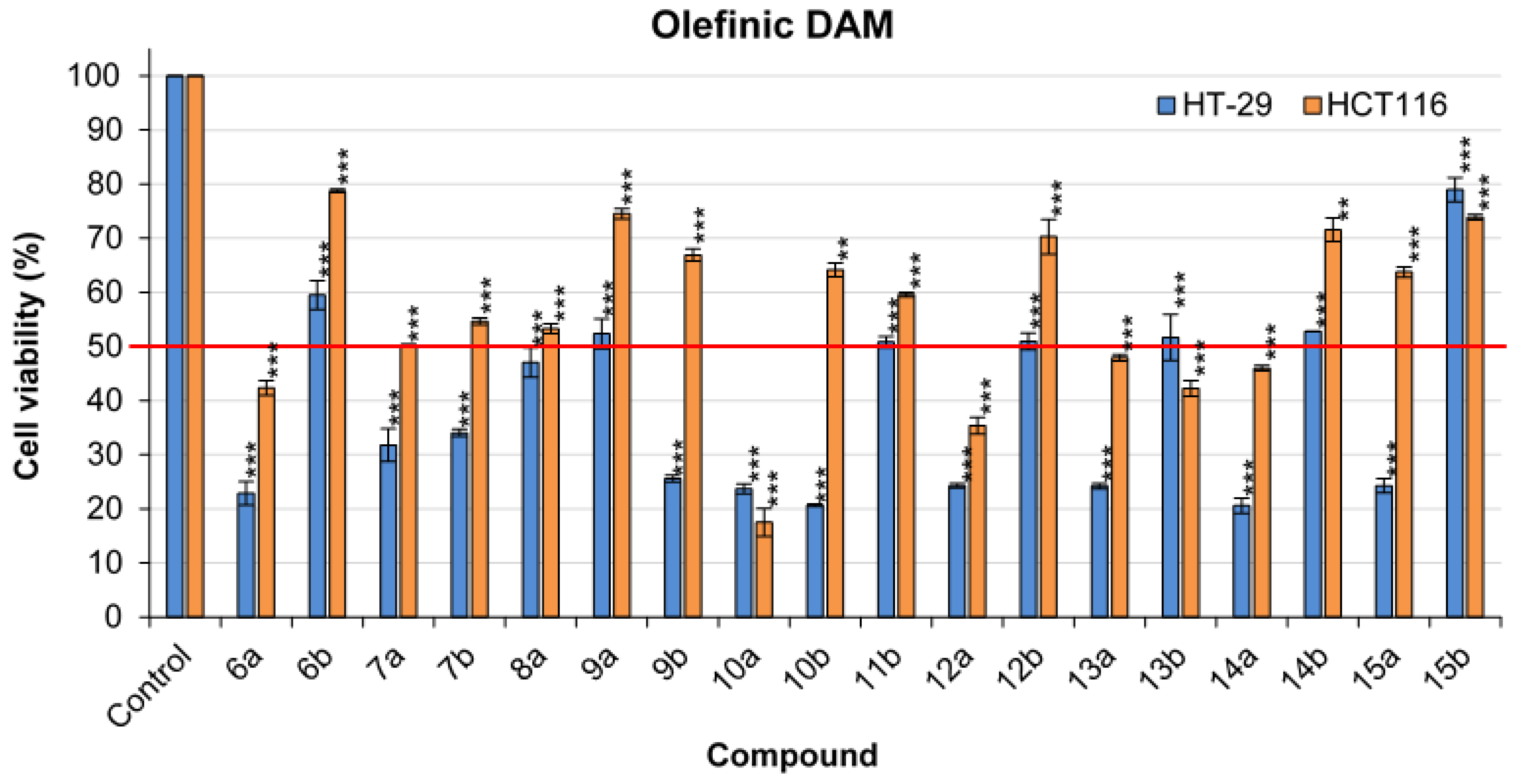
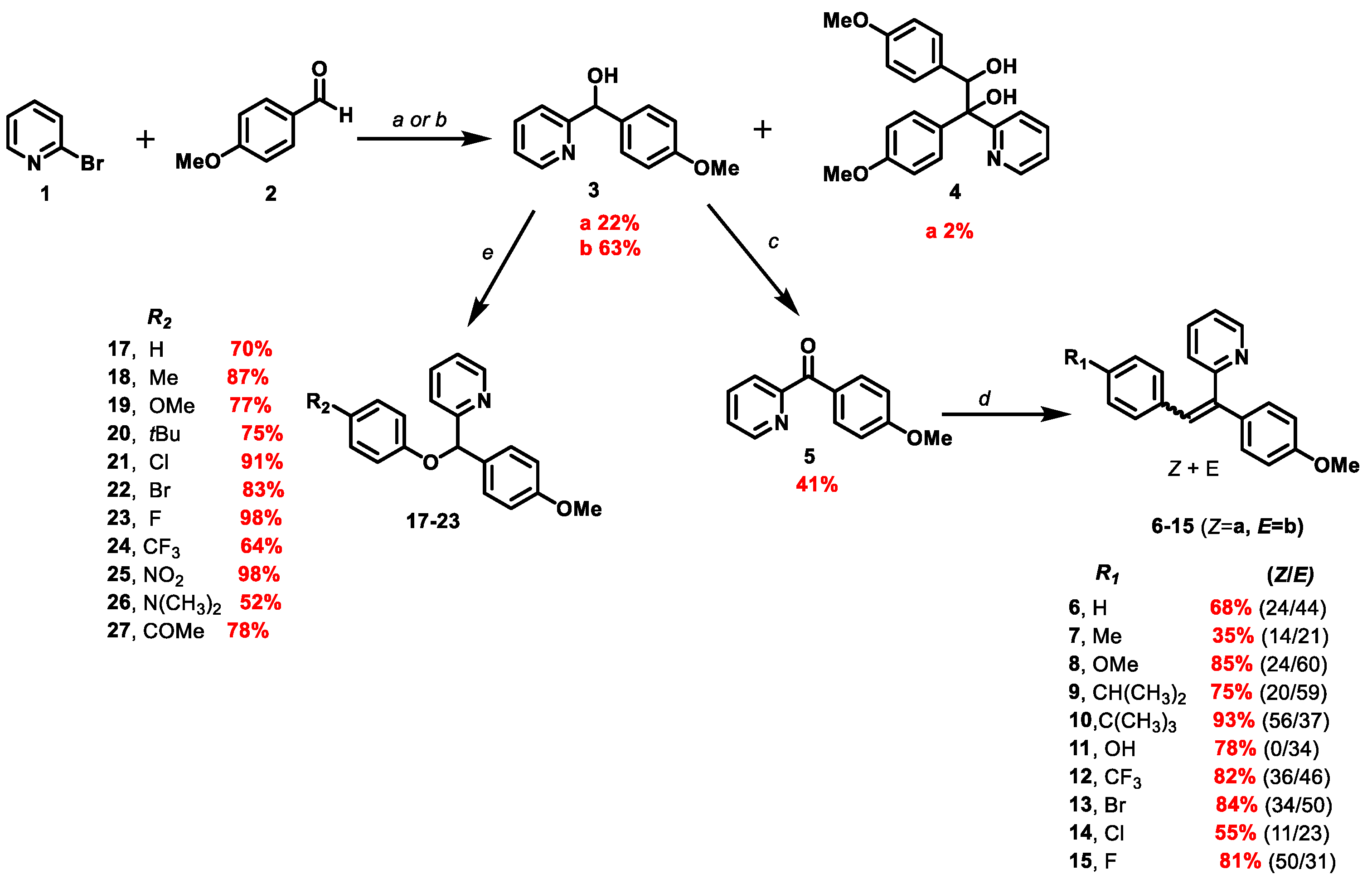
2.3.1. 2-(1-(4-methoxyphenyl)-2-arylvinyl)pyridine 6
0.05 mL of benzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 25 min. The crude product was purified by FCC using the CyHex/EtOAc: 95/5 and the 2-(1-(4-méthoxyphenyl) -2-phenylvinyl) pyridine was afforded 6 in two separable E and Z isomers.
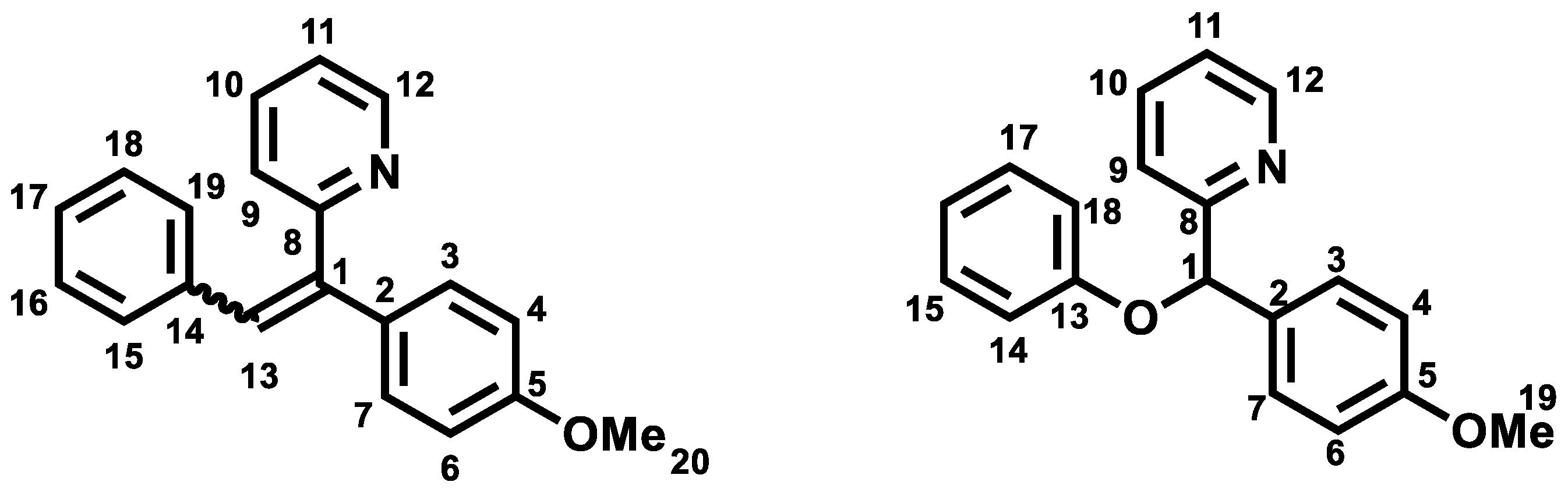
Z isomer (6a): 31.6 mg of a yellow oil in 24% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.36. IR ν (cm−1): 3061, 3029 (νCsp2-H); 2922 (νCsp3-H); 2857 (νOMe); 1604, 1582, 1511 and 1462 (νC=C); 1246 (νasym C-O-C); 1031 (νsym C-O-C); 860 (δCsp2-H p-substitution). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.78 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12, 1H, H12), 8.12–8.08 (m, 4H, H15, H16, H18, H19), 7.66–7.62 (m, 1H, H10), 7.48–7.44 (m, 3H, H3, H7, H17), 7.29–7.27 (m, 1H, H11), 7.07(s, 1H, H13), 6.96–6.93 (m, 1H, H9), 6.85 (d, J4–3, 6–7 = 8 Hz, 2H, H4, H6), 3.79 (s, 3H, H20, OMe). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.4 (C5), 159.1 (C8), 149.7 (C12), 140.9 (C1), 137.1 (C10), 134.4 (C2), 133.6 (C17), 130.1 (C15, C16, C18, C19), 129.8 (C14), 129.4 (C9), 128.8 (C13), 128.5 (C3, C7), 120.0 (C11), 113.9 (C4, C6), 55.3 (C20). LRMS: (ES+, CV = 30). m/z = 575 [2M + H]+, 288 [M + H]+, 257 [M − OMe]+. HRMS: calcd. for C20H17NOH [M + H]+ (288.1383); found (288.1383).
E isomer (6b): 31.8 mg of white amorphous solid 44% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.58. IR ν (cm−1): 3061, 3029 (νCsp2-H); 2922 (νCsp3-H); 2857 (νOMe); 1604, 1582, 1511 and 1462 (νC=C); 1246 (νasym C-O-C); 1031 (νsym C-O-C). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.65 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.76 (s, 1H, H13), 7.55 (td, J10–9,10–11 = 7.8 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.17- 7.09 (m, 8H, H3, H7, H11, H15, H16, H17, H18, H19), 7.02 (d, J9–10 = 7.9 Hz, 1H, H9), 6.95–6.92 (m, 2H, H4, H6), 3.85 (s, 3H, H20, OMe). 13C NMR (100 MHz, CDCl3) δ (ppm): 159.4 (C5), 159.2 (C8), 149.2 (C12), 140.1 (C1), 137.0 (C13), 136.5 (C10), 131.5 (C3, C7), 131.3 (C2), 130.1 (C15, C19), 129.2 (C14), 128.0 (C16, C18), 127.3 (C17), 122.6 (C9), 122.0 (C11), 114.5 (C4, C6), 55.3 (C20). LRMS: (ES+, CV = 30) m/z = 288 [M + H]+, 271 [M − Me]+.
2.3.2. 2-(1-(4-methoxyphenyl)-2-(p-tolyl)vinyl)pyridine 7
0.059 mL of p-tolylaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 12 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 1 to 5% of EtOAc), the 2-(1,2-bis(4 methoxyphenyl)vinyl)pyridine 7 was afforded in two separable E and Z isomers.
Z isomer (7a): 20 mg of yellow oil in 14% yield. TLC: CyHex/EtOAc:80:20, Rf = 0.2. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.66 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.76 (td, J10–9, 10–11 = 7.7 Hz, J10–12 = 1.7 Hz, 1H, H10), 7.36 (ddd, J11–10 = 7.5 Hz, J11–12 = 4.9 Hz, J11–9 = 1.2 Hz, 1H, H11), 7.20–7.12 (m, 3H, H9, H15, H19), 7.06 (s, 1H, H13), 6.95–6.86 (m, 4H, H3, H7, H16, H18), 6.75 (d, J4–3, 6–7 = 7.9 Hz, 2H, H4, H6), 3.76 (s, 3H, H20, OMe), 2.22–2.16 (s, 3H, H21, Me). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.12 (C5), 158.78 (C8), 149.95 (C12), 140.09 (C1), 136.96 (C10), 136.21 (C17), 133.98 (C2), 133.89 (C4, C6), 128.87 (C14), 128.63 (C3, C7), 127.99 (C15, C19), 126.81 (C13), 125.18 (C9), 122.55 (C11), 113.77 (C16, C18), 55.15 (C20), 28.68 (C21). LRMS: (ES+, CV = 30). m/z = 324.25 [M + Na]+, 303.23 [M + 2H]+, 302.24 [M + H]+. HRMS: calcd. for C21H19NOH [M + H]+ (302.1539); found (302.1540).
E isomer (7b): 30 mg of yellow amorphous solid in 21% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.4. IR ν (cm−1): 3000 (νCsp2-H); 2940, 2921 (ν Csp3-H); 2854 (νOMe); 1604, 1580, 1509 and 1462 (νC=C); 1240 (νC-O); 1174 (νC-N); 815 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.60 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.74 (s, 1H, H13), 7.68 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.28–7.24 (m, 1H, H11), 7.10 (d, J3–4,7–8 = 8.6 Hz, 2H, H3, H7), 7.02–6.95 (m, 4H, H15, H19, H16, H18), 6.94–6.93 (m, 1H, H9), 3.80 (s, 3H, H20, OMe), 2.08 (s, 3H, H21, Me). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.6 (C5), 158.2 (C8), 149.0 (C12), 136.8 (C1), 136.7 (C10), 134.5 (C2), 133.6 (C17), 130.9 (C3, C7), 129.8 (C16, C18), 129.5 (C13), 128.7 (C14), 124.4 (C15, C19), 122.1 (C9), 121.7 (C11), 114.6 (C6, C4), 55.1 (C20), 30.7 (C21). LRMS: (ES+, CV = 30). m/z = 324 [M + Na]+, 303.23 [M + 2H]+, 302.17 [M + H]+, 211.12 [M − CH3C6H5]+.
2.3.3. 2-(1,2-bis(4-methoxyphenyl)vinyl)pyridine 8
0.056 mL of p-anisaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 3 h. The crude product was purified by FCC using the CyHex/EtOAc 95:5, the 2-(1,2-bis(4-methoxyphenyl)vinyl)pyridine 8 was afforded in two separable E and Z isomers.
Z Isomer (8a): 34.4 mg of a yellow colored oil in 24% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.5. IR ν (cm−1): 2932 (ν Csp3-H); 2855 (νOMe); 1604, 1509 and 1461 (νC=C); 1244 (νC-O); 1176 (νC-N); 829 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.66 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (td, J10–11, 10–9 = 7.9 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.38–7.35 (m, 1H, H9), 7.17 (m, 3H, H11, H3, H7), 7.03 (s, 1H, H13), 6.89 (d, J4–3, 6–7 = 7.2 Hz, 2H, H4, H6), 6.80 (d, J16–15, 18–19 = 8.6 Hz, 1H, H16, H18), 6.70 (d, J15–16, 19–18 = 7.4 Hz, 2H, H15, H19), 3.74 (s, 3H, H21, Me), 3.67 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.7 (C17), 159.1 (C5), 158.6 (C8), 150.4 (C12), 139.2 (C1), 137.5 (C10), 134.5 (C2), 130.7 (C16, C18), 129.7 (C14), 128.2 (C3, C7), 126.9 (C13), 125.6 (C11), 123.0 (C9), 114.21 (C15, C19), 113.9 (C6, C4), 55.8 (C20), 54.7 (C21). LRMS: (ES+, CV = 30). m/z = 657.28 [2M + Na]+, 635.35 [2M + H]+, 318.18 [M + H]+, 302.24 [M − Me]+. HRMS: calcd. for C21H19NO2H [M + H]+ (318.1489); found (318.1489).
E isomer (8b): 92.3 mg of a yellow-colored oil in 60% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.6. IR ν (cm−1): 2932 (ν Csp3-H); 2855 (νOMe); 1604, 1509 and 1461 (νC=C); 1244 (νC-O); 829 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ 8.59 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.76 (s, 1H, H13), 7.66 (td, J10–9, 10–11 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.24–7.21 (m, 1H, H11), 7.11 (d, J3–4, 7–6 = 8.7 Hz, 2H, H3, H7), 7.04–6.98 (m, 4H, H6, H4, H16, H18), 6.91 (d, J9–10 = 8 Hz, 1H, H9), 6.47 (d, J15–16, 19–18 = 8.8 Hz, 2H, H15, H19), 3.81 (s, 3H, H21, Me), 3.63 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.1 (C17), 159.0 (C5), 158.7 (C8), 149.4 (C12), 138.0 (C1), 137.1 (C10), 134.5 (C2), 131.4 (C3, C7, C16, C18), 129.9 (13), 129.4 (C14), 122.3 (C11), 121.9 (C9), 115.2 (C6, C4), 114.1 (C15, C19), 55.5 (C20, C21). LRMS: (ES+, CV = 30). m/z = 340.19 [M + Na]+, 308.18 [M + H]+.
2.3.4. 2-(2-(4-isopropylphenyl)-1-(4-methoxyphenyl)vinyl)pyridine 9
0.059 mL of 4-isopropylbenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 15 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 2 to 10% of EtOAc), the 2-(1,2-bis(4 methoxyphenyl)vinyl)pyridine 9 was afforded in two separable E and Z isomers.
Z isomer (9a): 100 mg of yellow amorphous solid in 65% yield. TLC: CyHex/EtOAc:80/20, Rf = 0.30. IR ν (cm−1): 3001 (νCsp2-H); 2958, 2927 (νCsp3-H); 2835 (νOMe); 1602, 1583, 1509 and 1460 (νC=C); 1244 (νC-O); 1179 (νC-N); 826 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.68 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12, 1H, H12), 7.80 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.40 (ddd, J9–10 = 7.6 Hz, J9–11 = 4.9 Hz, J9–12 = 1.2 Hz, 1H, H9), 7.21–7.16 (m, 3H, H11, H15, H19), 7.07 (s, 1H, H13), 7.02–6.98 (m, 2H, H3, H7), 6.93–6.88 (m, 2H, H16, H18), 6.78 (d, J4–3 = J6–7 = 8.3 Hz, 2H, H4, H6), 3.76 (s, 3H, H20, OMe), 2.85–2.71 (m, 1H, H21), 1.23 (d, J22–21= 6.9 Hz, 6H, H22). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.56 (C5), 159.25 (C8), 150.45 (C12), 147.67 (C17), 140.41 (C1), 137.55 (C10), 134.74 (C14), 134.33 (C3, C7), 129.41 (C2), 128.41 (C15, C19), 127.17 (C13), 126.44 (C16, C18), 125.60 (C9), 123.10 (C11), 114.26 (C4, C6), 55.62 (C20), 33.48 (C21), 29.46 (C22). LRMS: (ES+, CV = 30) m/z: 681.58 [2M + Na]+; 352.25 [M + Na]+; 331.31 [M + 2H]+; 330.24 [M + H]+. HRMS: calcd. for C23H23NOH [M + H]+ (308.1281); found (308.1258).
E isomer (9b): 46 mg of a yellow-colored oil in 30% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.7. IR ν (cm−1): 3001 (νCsp2-H); 2958, 2927 (νCsp3-H); 2835 (νOMe); 1602, 1583, 1509 and 1460 (νC=C); 1244 (νC-O); 1179 (νC-N); 826 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.60 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12, 1H, H12), 7.78 (s, 1H, H13), 7.63 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.27–7.23 (m, 1H, H11), 7.12 (d, J3–4,7–8 = 8.7 Hz, 2H, H3, H7), 7.06–7.02 (m, 4H, H15, H19, H16, H18), 6.95 (d, J4–3, 6–7 = 8.2 Hz, 2H, H4, H6), 6.94–6.91 (m, 1H, H9), 3.81 (s, 3H, H20, OMe), 2.82–2.80 (m, 1H, H21), 1.13 (d, J22–21 = 6.8 Hz, 6H, H22). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.3 (C5), 158.1 (C8), 150.6 (C12), 145.5 (C17), 139.0 (C1), 136.5 (C10), 135.7 (C2), 134.8 (C3, C7), 129.6 (C14), 129.1 (C15, C19), 127.2 (C13), 127.0 (C16, C18), 125.6 (C9), 123.1 (C11), 113.8 (C4, C6), 55.5 (C20), 33.8 (C21), 29.5 (C22). LRMS: (ES+, CV = 30) m/z: 681.58 [2M + Na]+; 352.25 [M + Na]+; 331.23 [M + 2H]+; 330.24 [M + H]+.
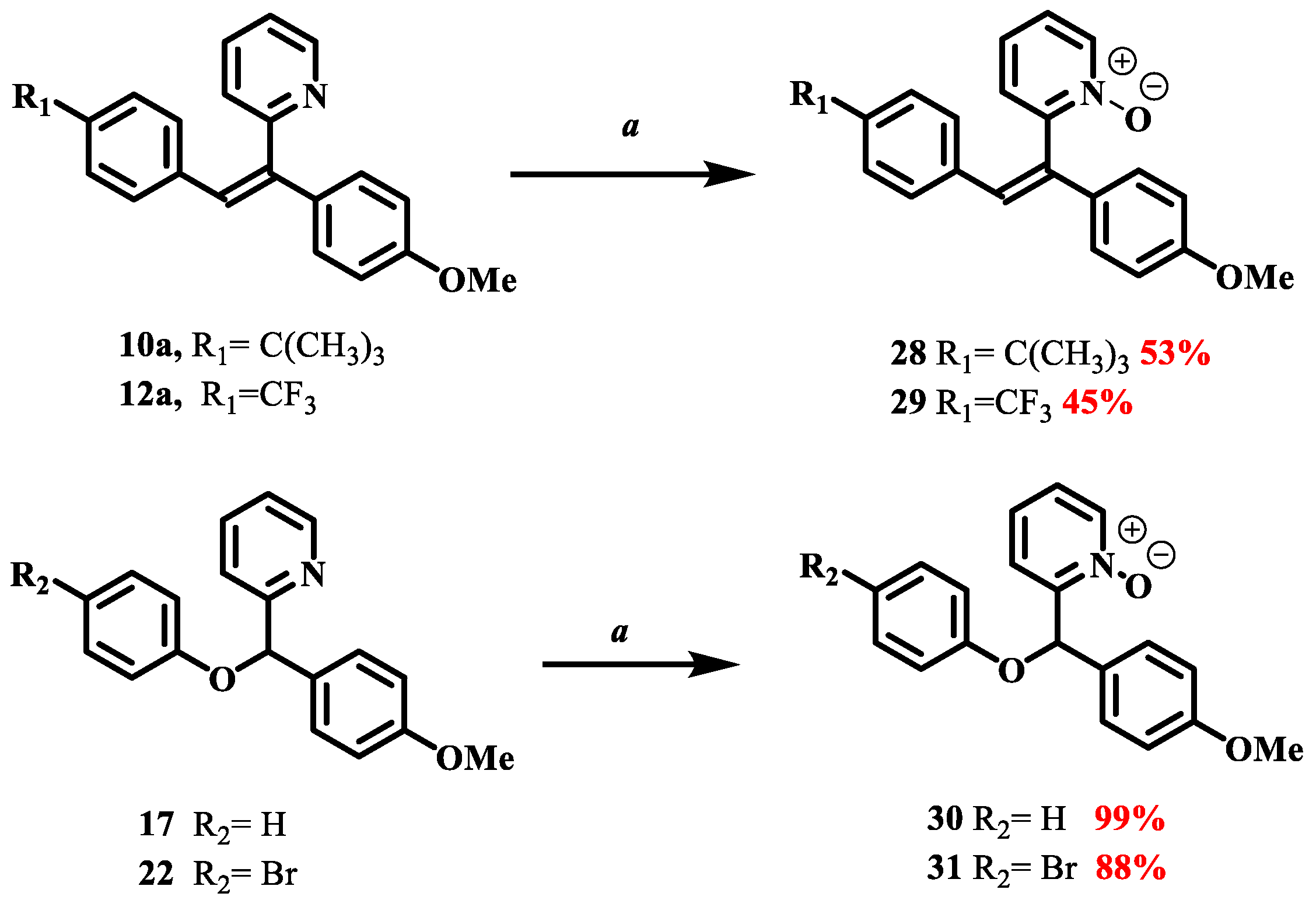
2.3.5. 2-(2-(4-(tert-butyl)phenyl)-1-(4-methoxyphenyl)vinyl)pyridine 10
0.084 mL of 4-tert-butylbenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 20 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 2 to 10% of EtOAc), the 2-(1,2-bis(4 methoxyphenyl)vinyl)pyridine 10 was afforded in two separable E and Z isomers.
Z isomer (10a): 89 mg of a white amorphous solid in 56%. TLC: CyHex/EtOAc:80/20, Rf = 0.2. IR ν (cm−1): 3047, 3005 (νCsp2-H); 2956, 2931 (ν Csp3-H); 2866 (νOMe); 1603, 1593, 1509 and 1462 (νC=C); 1244 (νC-O); 1181 (νC-N); 827 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.68 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.80 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.40 (m, 1H, H11), 7.20–7.14 (m, 5H, H9, H3, H7, H15, H19), 7.06 (s, 1H, H13), 6.92–6.89 (m, 2H, H6, H4), 6.79 (d, J16–17, 18–19 = 8.4 Hz, 2H, H16, H18), 3.75 (s, 3H, H20, OMe), 1.20 (s, 9H, H22). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.91 (C5), 158.57 (C8), 149.80 (C17), 149.21(C12), 139.76 (C1), 136.87 (C10), 133.67 (C2), 129.14 (C14), 128.48 (C3, C7), 127.72 (C16, C18), 126.94 (C13), 126.34 (C15, C19), 124.90 (C9), 123.89 (C11), 122.43, 113.58 (C4, C6), 54.96 (C20), 34.00 (C21), 30.76 (C22). LRMS: (ES+, CV = 30) m/z: 709.37 [2M + Na]+; 687.42 [2M + H]+; 366.22 [M + Na]+; 345.27 [M + 2H]+; 344.28 [M + H]+. HRMS: calcd. for C24H25NOH [M + H]+ (344.2009); found (344.2009).
E isomer (10b): 59 mg of a yellow amorphous solid 37%. TLC: CyHex/EtOAc:80/20, Rf = 0.5. IR ν (cm−1): 3050, 3005 (νCsp2-H); 2957, 2926 (νCsp3-H); 2858 (νOMe); 1602, 1581, 1509 and 1461 (νC=C); 1240 (νC-O); 1178 (νC-N); 832 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.61 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12, 1H, H12), 7.80 (s, 1H, H13), 7.68 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.26 (m, 1H, H11), 7.20 (d, J15–16, 19–18 = 8.5 Hz, 2H, H15, H19), 7.15–7.11 (m, 2H, H3, H17), 7.07–7.03 (m, 2H, H4, H6), 6.99 (d, J16–17, 18–19 = 8.5 Hz, 2H, H16, H18), 6.92 (d, J9–10 = 8.0 Hz, 1H, H9), 3.82 (s, 3H, H20, OMe), 1.21 (s, 9H, H22). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.68 (C5), 158.12 (C8), 150.09 (C17), 149.07 (C12), 138.92 (C1), 136.72 (C10), 133.54 (C2), 131.72 (C3, C7), 130.84 (C14), 129.55 (C13), 129.41 (C16, C18), 124.99 (C15, C19), 122.12 (C9), 121.65 (C11), 114.75 (C6, C4), 55.09 (C20), 34.30 (C21), 30.95 (C22). LRMS: (ES+, CV = 30) m/z: 709.37 [2M + Na]+; 366.22 [M + Na]+; 345.27 [M + 2H]+; 344.28 [M + H]+, 214.16 [M − (CH3)3CHC6H4]+.
2.3.6. 4-(2-(4-methoxyphenyl)-2-(pyridin-2-yl)vinyl)phenol 11b
61.2 mg of 4-hydroxybenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 10 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 1 to 10% of EtOAc), the 4-(2-(4-methoxyphenyl)-2-(pyridin-2-yl)vinyl)phenol 11b was afforded in two E and Z isomers but only E isomer was isolated.
E isomer (11b): 48 mg of a yellow amorphous solid in 34% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.24. IR ν (cm−1): 3512 (νOH); 3062, 3002 (νCsp2-H); 2957, 2920 (νCsp3-H); 2853 (νOMe); 1602, 1586, 1507 and 1462 (νC=C); 1242 (νC-O); 1171 (νC-N); 831 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.58 (s, 1H, OH), 8.59 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.72 (s, 1H, H13), 7.70–7.62 (m, 1H, H10), 7.24–7.19 (m, 1H, H11), 7.12–7.08 (m, 2H, H15, H19), 7.06–7.01 (m, 2H, H, H16, H18 ), 6.92–6.87 (m, 3H, H9, H3, H7), 6.61–6.50 (m, 2H, H4, H6), 3.85–3.78 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.57 (C5), 158.48 (C17), 157.03 (C8), 149.00 (C12), 136.63 (C1), 131.5 (C2), 131.12 (C3, C7, C15, C19), 129.90 (C13), 127.40 (C14), 121.75 (C9), 121.32 (C11), 115.05 (C16, C18), 114.72 (C4, C6), 113.84 (C14), 55.09 (C20). LRMS: (ES+, CV = 30) m/z: 629.26 [2M + Na]+; 326.15 [M + Na]+; 3034.21 [M + H]+; 200.20 [M − C6H6OHCH]+. HRMS: calcd. for C20H17NO2H [M + H]+ (304.1332); found (304.1333).
2.3.7. 2-(1-(4-methoxyphenyl)-2-(4-(trifluoromethyl)phenyl)vinyl)pyridine 12
0.064 mL of 4-(trifluoromethyl) benzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 15 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 1 to 10% of EtOAc), the 2-(1-(4-methoxyphenyl)-2-(4-(trifluoromethyl)phenyl) vinyl)pyridine 12 was afforded in two separable E and Z isomers.
Z isomer (12a): 60 mg of yellow oil in 36% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.25. IR ν (cm−1): 3020, 3013 (νCsp2-H); 2921 (ν Csp3-H); 2846 (νOMe); 1605, 1579, 1510 and 1464 (νC=C); 1244 (νC-O); 1111 (νC-N); 834 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.67 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.79 (td, J10–11= 7.7, 1.8 Hz, 1H, H10), 7.51 (d, J15–16, 19–18 = 8.2 Hz, 2H, H15, H19), 7.42–7.37 (m, 1H, H11), 7.27–7.15 (m, 4H, H13, H9, H3, H7), 7.05 (d, J16–15, 18–19 = 8.1 Hz, 2H, H16, H18), 6.92 (d, J4–3, 6–7 = 8.9 Hz, 2H, H4, H6), 3.82–3.66 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.96 (C5), 157.64 (C8), 149.25 (C12), 142.50 (C1), 140.84 (C10), 136.88 (C2), 131.9 (C17), 130.95 (C16, C19, C3, C7), 129.89 (C14), 128.22 (C14), 126.00 (C13), 124.93 (C16, C18), 124.89 (C21), 122.29 (C9), 121.51 (C11), 114.72 (C4, C6), 55.11 (C20). LRMS: (ES+, CV = 30) m/z: 733.29 [2M + Na]+; 378.13 [M + Na]+; 357.19 [M + 2H]+; 307.17 [M + 2H]+; 356.16 [M + H]+; 288.89 [M − CF3]+. HRMS: calcd. for C21H16F3NOH [M + H]+ (356.1256); found (396.1257).
E isomer (12b): 76 mg of a white amorphous solid in 46% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.44. IR ν (cm−1): 3043, 3005 (νCsp2-H); 2920 (ν Csp3-H); 2846 (νOMe); 1605, 1579, 1509 and 1464 (νC=C); 1244 (νC-O); 1111 ((νC-N); 831 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.63 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.82 (s, 1H, H13), 7.73 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.54 (d, J15–16 = J19–18 = 8.3 Hz, 2H, H15, H19), 7.33 (ddd, J11–10 = 7.5 Hz, J11–12 = 4.7, J11–9 = 1.1 Hz, 1H, H11), 7.29–7.25 (m, 2H, H16, H18), 7.13 (d, J3–4 = J7–6 = 8.7 Hz, 2H, H3, H7), 7.07–6.99 (m, 3H, H9, H4, H6), 3.80–3.79 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.96 (C5), 157.64 (C8), 149.25 (C12), 142.50 (C1), 140.84 (C10), 136.88 (C2), 131.57 (C17), 130.95 (C3, C7), 130.01 (C16, C18), 129.89 (C21), 129.20 (C14), 128.22 (C13), 124.97 (C15, C19), 122.82 (C9), 122.29 (C11), 114.72 (C4, C6), 55.11 (C20). LRMS: (ES+, CV = 30) m/z: 378.07 [M + Na]+; 357.12 [M + 2H]+; 365.05 [M + H]+; 212.15 [M − CF3C6H5]+.
2.3.8. 2-(2-(4-bromophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 13
76.8 mg of 4-bromobenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 15 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (90/10), the 2-(2-(4-bromophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 13 was afforded in two separable E and Z isomers.
Z isomer (13a): 57.5 mg of a yellow oil 34% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.34. IR ν (cm−1): 3048, 3000 (νCsp2-H); 2950, 2927 (νCsp3-H); 2837 (νOMe); 1601, 1582, 1509 and 1473 (νC=C); 1245 (νC-O); 1179 (νC-N); 818 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.65 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (t, J10–11, 10–9 = 7.7 Hz, 1H, H10), 7.40–7.31 (m, 1H, H11), 7.32 (d, J15–16, 19–18 = 8.5 Hz, H15, H19), 7.19 (d, J3–4, 7–6 = 8.8 Hz, 2H, H3, H7), 7.16 (d, J9–10 = 7.8 Hz, 1H, H9), 7.10 (s, 1H, H13), 6.91 (d, J4–5, 6–7 = 8.8 Hz, 4H, H4, H6), 6.81 (d, J16–15, 18–19 = 8.5 Hz, 2H, 2H, H16, H18), 3.75 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.0 (C5), 158.5 (C8), 150.0 (C12), 141.7 (C1), 137.1 (C10), 136.2 (C2), 133.4 (14), 130.9 (C15, C19), 130.8 (C16, C18), 128.2 (C3, C7), 125.6 (C13), 125.1 (C9), 122.8 (C11), 119.9 (C17), 113.8 (C4, C6), 55.1 (C20). LRMS: (ES+, CV = 30) m/z: 754.92 [2M + Na]+; 366.92 [M]+; 278.98 [M − Br]+. HRMS: calcd. for C20H16BrNOH [M + H]+ (366.0488); found (366.0490).
E isomer (13b): 84.7 mg of a white amorphous solid product in 50% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.53. IR ν (cm−1): 3065, 3001 (νCsp2-H); 2957, 2931 (ν Csp3-H); 2838 (νOMe); 1602, 1576, 1507 and 1461 (νC=C); 1240 (νC-O); 1173 (νC-N); 818 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.61 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.73–7.69 (m, 2H, H10, H13), 7.29 (d, J15–16, 19–18 = 8.5 Hz, 1H, H15, H19), 7.29–7.27 (m, 1H, H11), 7.10 (d, J3–4, 7–6 = 8.7 Hz, 2H, H3, H7), 7.02–6.98 (m, 5H, H9, H4, H6, H16, H18), 6.95–6.92 (m, 2H, H4, H6), 3.80 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.8 (C5), 157.8 (C8), 149.2 (C12), 140.8 (C1), 136.8 (C10), 135.8 (C2), 131.4 (C15,C19), 131.1 (C16, C18), 130.9 (C3, C7), 130.1 (C14), 128.5 (C13), 125.1 (C9), 122.5 (C11), 122.0 (C17), 114.7 (C4, C6), 55.1 (C20). LRMS: (ES+, CV = 30) m/z: 369.12 [M+3H]+; 367.93 [M + H]+.
2.3.9. 2-(2-(4-chlorophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 14
70.4 mg of 4-chlorobenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 10 min. The crude was purified by FCC using a gradient system of CyHex/EtOAc (from 1 to 5% of EtOAc). The 2-(2-(4-bromophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 14 was afforded in two separable E and Z isomers.
Z isomer (14a) 17.8 mg of an orange-colored oil 12% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.37. IR ν (cm−1): 3080, 3011 (νCsp2-H); 2940, 2926 (νCsp3-H); 2838 (νOMe); 1604, 1578, 1508 and 1461 (νC=C); 1241 (νC-O); 1174 (νC-N); 820 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.65 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (td, J10–9, 10–11 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.40–7.37 (m, 1H, H11), 7.20- 7.15 (m, 5H, H3, H7, H9, H15, H19), 7.10 (s, 1H, H13), 6.92–6.86 (m, 4H, H4, H6, H16, H18), 3.75 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.0 (C5), 158.5 (C8), 150.0 (C12), 141.6 (C1), 137.1 (C10), 135.8 (C2), 133.4 (C14), 131.2 (C17), 130.5 (C16, C18), 128.2 (C3, C7), 128.0 (C15, C19), 125.5 (C13), 125.0 (C9), 122.8 (C11), 113.8 (C6, C4), 55.2 (C20). LRMS: (ES+, CV = 30). m/z = 643.08 [2M]+; 344.01 [M + Na]+; 322.05 [M + H]+; 197.09 [M − C6H4CHCl]+. HRMS: calcd. for C20H16ClNOH [M + H]+ (322.0992); found (322.0995).
E isomer (14 b): 35 mg of a white amorphous solid 24% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.5. IR ν (cm−1): 3080, 3013 (νCsp2-H); 2940, 2926 (νCsp3-H); 2838 (νOMe); 1604, 1578, 1508 and 1461 (νC=C); 1241 (νC-O); 1174 (νC-N); 820 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.62 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.74 (s, 1H, H13), 7.61 (t, J10–9,10–11 = 7.7 Hz, 1H, H10), 7.30- 7.27 (m, 1H, H11), 7.24 (d, J15–16, 19–18 = 8.6 Hz, 2H, H15, H19), 7.11–7.00 (m, 6H, H3, H7, H16, H18, H4, H6), 6.98 (d, J9–10 = 6.9 Hz, 1H, H9), 3.80 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.8 (C5), 157.8 (C8), 149.1 (C12), 140.7 (C1), 136.8 (C10), 135.5 (C2), 131.7 (C14), 131.1 (C3, C7), 130.9 (C16,C18), 130.1 (C17), 128.4 (C13), 128.1 (C15,C19), 122.5 (C9), 121.9 (C11), 114.7 (C6, C4), 55.1 (C20). LRMS: (ES+, CV = 30). m/z = 344.06 [M + Na]+, 324.12 [M+3H]+, 322.12 [M + H]+.
2.3.10. 2-(2-(4-fluorophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 15
0.053 mL of 4-fluorobenzaldehyde was used (0.486 mmol, 1.04 eq). The reaction lasted 15 min. The crude product was purified by FCC using a gradient system of CyHex/EtOAc (from 1 to 5% of EtOAc), the 2-(2-(4-bromophenyl)-1-(4-methoxyphenyl)vinyl)pyridine 15 was afforded in two separable E and Z isomers.
Z isomer (15a): 71 mg of yellow amorphous solid in 50% yield. TLC: CyHex/EtOAc:80:20, Rf = 0.34. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.66 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.8 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.79 (td, J10–11, 10–9 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.39 (ddd, J11–10 = 7.6 Hz, J11–12 = 4.9 Hz, J11–9 = 1.2 Hz, 1H, H11), 7.24–7.15 (m, 3H, H9, H15, H19), 7.13 (s, 1H, H13), 7.03–6.95 (m, 2H, H3, H7), 6.95–6.88 (m, 4H, H4, H6, H16, H18), 3.75 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.2 (C5), 158.5 (C8), 149.9 (C12), 141.4 (C1), 137.8 (C10), 135.8 (C2), 133.5 (C14), 131.6 (C17), 130.5 (C16, C18), 128.2 (C3, C7), 128.0 (C15, C19), 125.5 (C13), 125.0 (C9), 122.8 (C11), 113.8 (C6, C4), 55.2 (C20). LRMS: (ES+, CV = 30) m/z: 633.29 [2M + Na]+; 328.20 [M + Na]+; 306.19 [M + H]+. HRMS: calcd. For C20H16FNOH [M + H]+ (306.1288); found (306.1289).
E isomer (15b): 43.7 mg of yellow oil in 31% yield. TLC: CyHex/EtOAc: 80/20, Rf = 0.5. IR ν (cm−1): 3056 (νCsp2-H); 2926 (νCsp3-H); 2857 (νOme); 1602, 1508 and 1463 (νC=C); 1224 (νC-O); 1156 (νC-N); 832 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.61 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.80 (s, 1H, H13), 7.7 (td, J10–9, 10–11 = 7.7 Hz, J10–12 = 1.9 Hz, 1H, H10), 7.34–7.25 (m, 1H, H11), 7.17–7.08 (m, 5H, H,9 H15, H17, H3, H7), 7.07–6.96 (m, 4H, H16, H18, H4, H6), 3.80 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 158.9 (C5), 157.8 (C8), 149.5 (C12), 140.7 (C1), 136.8 (C10), 135.7 (C2), 131.7 (C14), 131.1 (C3, C7), 130.4 (C16,C18), 130.3 (C17), 128.4 (C13), 128.7 (C15, C19), 122.5 (C9), 122.1 (C11), 114.7 (C6, C4), 55.5 (C20). LRMS: (ES+, CV = 30) m/z: 633.29 [2M + Na]+; 611.27 [2M + H]+; 328.20 [M + Na]+; 307.17 [M + 2H]+; 306.19 [M + H]+.
2.4. General Procedure for the Synthesis of aryloxyDAM
To a suspension of molecular sieves (480 mg) in anhydrous dichloromethane (4.8 mL) under argon was added carbinol (0.46 mmol, 100 mg), arylboronic acid (1.38 mmol, 3 eq), Cu(OAc)2 (0.46 mmol, 84.5 mg, 1 eq) and anhydrous pyridine (0.92 mmol, 0.074 mL, 2 eq). The reaction mixture was refluxed during 24 h at 40 °C under argon. After reaction completion, the mixture was cooled at room temperature, filtered under celite. To recover the product, the celite was washed using dichloromethane and ethyl acetate. The filtrate was concentrated under vacuum and the crude product was purified by FCC on silica gel (CyHex/EtOAc: 80/20).
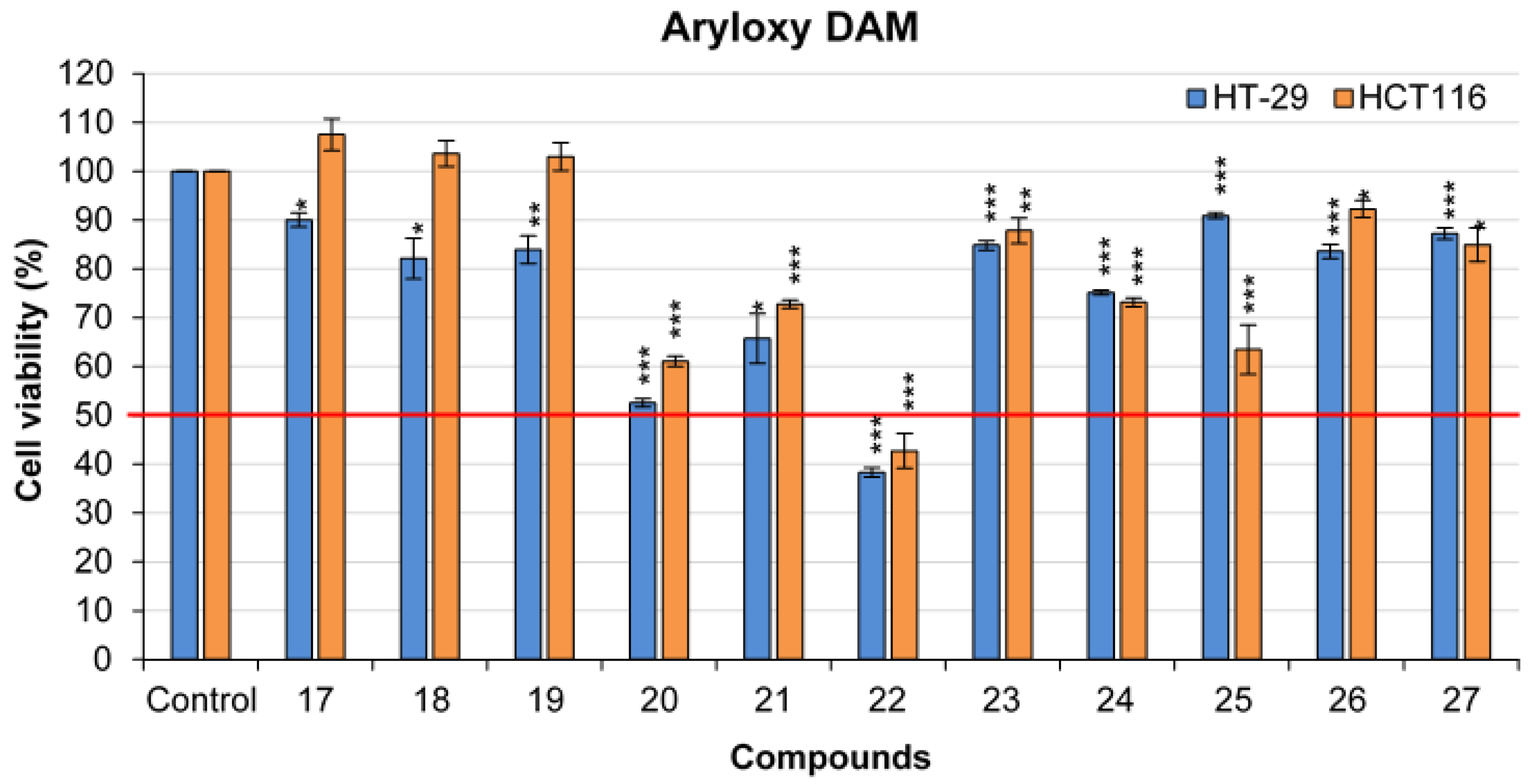
2.4.1. 2-((4-methoxyphenyl)(phenoxy)methyl)pyridine 17
168.2 mg of phenylboronic acid (1.38 mmol, 3 eq) was used. After FCC, the 2-((4-methoxyphenyl)(phenoxy)methyl)pyridine 17 was afforded as a yellow oil (94 mg) in 70% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.7. IR ν (cm−1): 3060, 3007 (νCsp2-H); 2932, 2927 (νCsp3-H); 2837 (νOMe); 1586, 1510 and 1491 and 1470 (νC=C); 1223 (νasym C-O-C); 1030 (νsym C-O-C); 749 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.51 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.79 (td, J10–9,10–11 = 7.6 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.58 (d, J9–10 = 8 Hz, 1H, H9), 7.42 (d, J3–4,7–6 = 8.7 Hz, 2H, H3, H7), 7.28–7.20 (m, 3H, H11, H15, H17), 6.98 (d, J14–15, 18–17 = 7.7 Hz, 2H, H14, H18), 6.90–6.86 (m, 3H, H4, H6, H16), 6.39 (s, 1H, H1), 3.70 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.4 (C13), 158.8 (C5), 157.2 (C8), 149 (C12), 137.2 (C10), 1321 (C2), 129.4 (C15, C17), 128.3 (C3, C7), 122.7 (C9), 120.9 (C16), 120.4 (C11), 115.8 (C14, C18), 113.8 (C4, C6), 80.9 (C1), 55 (C19). LRMS: (ES+, CV = 30) m/z: 314.04 [M + Na]+; 292.01 [M + H]+; 198.14 [M − C6H5O]+. HRMS: calcd. for C19H17NO2H [M + H]+ (292.1332); found (292.1752).
2.4.2. 2-((4-methoxyphenyl)(p-tolyloxy)methyl)pyridine 18
187.6 mg of 4-methylphenylboronic acid (1.38 mmol, 3 eq) was used. After FCC, the 2-((4-methoxyphenyl)(p-tolyloxy)methyl)pyridine 18 was afforded as a white solid (122.2 mg) in 87% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.7. IR ν (cm−1): 3065 (νCsp2-H); 2994, 2917 (νCsp3-H); 2853 (νOMe); 1610, 1588, 1510 and 1473 (νC=C); 1227 (νasym C-O-C); 1032 (νsym C-O-C); 810 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.62 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (td, J10–9,10–11 = 7.6 Hz, J10–12 = 1.7 Hz, 1H, H10), 7.57 (d, J9–10 = 8 Hz, 1H, H9), 7.42 (d, J3–4,7–6 = 8.6 Hz, 2H, H3, H7), 7.27–7.24 (m, 1H, H11), 7.01 (d, J15–16, 17–18 = 8.7 Hz, 2H, H15, H17), 6.98–6.85 (m, 4H, H4, H6, H14, H18), 6.41 (s, 1H, H1), 3.70 (s, 3H, H19, OMe), 2.17 (s, 3H, H20, Me). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.5 (C5), 158.7 (C8), 155 (C13), 149 (C12), 137.1 (C10), 132.2 (C16), 129.7 (C15, C17), 129.6 (C2), 128.2 (C3, C7), 122.7 (C9), 120.4 (C11), 115.7 (C4, C6), 113.8 (C14, C18), 81 (C1), 55 (C19), 20 (C20). LRMS (ES+, CV = 30) m/z: 328.16 [M + Na]+; 306.18 [M + H]+; 214.14 [M − C7H7]+; 198.14 [M − C7H7O]+. HRMS: calcd. for C20H19NO2H [M + H]+ (306.1489); found (306.1288).
2.4.3. 2-((4-methoxyphenoxy)(4-methoxyphenyl)methyl)pyridine 19
210 mg of 4-methoxyphenylboronic (1.38 mmol, 3 eq) was used. After a FCC, the 2-((4-methoxyphenoxy)(4-methoxyphenyl)methyl)pyridine 19 as a transparent oil (110 mg) in 75% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.7. IR ν (cm−1): 3065 (νCsp2-H); 2990, 2930 (νCsp3-H); 2837 (νOMe); 1605, 1588, 1507 and 1463 (νC=C); 1216 (νasym C-O-C); 1031 (νsym C-O-C); 823 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.50 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (td, J10–9,10–11 = 7.7 Hz, J10–12 = 1.7 Hz, 1H, H10), 7.58 (d, J9–10 = 7.8 Hz, 1H, H9), 7.40 (d, J3–4,7–6 = 8.7 Hz, 2H, H3, H7), 7.27–7.24 (m, 1H, H11), 6.93–6.87 (m, 4H, H4, H6, H15, H17), 6.68 (d, J14–15,18–17 = 9.1 Hz, 2H, H14, H18), 6.28 (s, 1H, H1), 3.70 (s, 3H, H19, OMe), 3.64 (s, 3H, H20, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.6 (C5), 158.7 (C8), 153.5 (C13), 151.1 (C16), 149 (C12), 137.1 (C10), 132.3 (C2), 128.3 (C3, C7), 122.6 (C9), 120.4 (C11), 116.9 (C4, C6), 114.5 (C14, C18), 113.8 (C15, C17), 81.6 (C1), 55.2 (C20). 55 (C19). LRMS: (ES+, CV = 30). m/z: 323.24 [M + 2H]+; 322.16 [M + H]+; 214.14 [M − C7H7O]+; 198.15 [M − C7H7O2]+. HRMS: calcd. for C20H19NO3H [M + H]+ (322.1438); found (322.1439).
2.4.4. 2-((4-(tert-butyl)phenoxy)(4-methoxyphenyl)methyl)pyridine 20
245 mg of 4-tertbutylmethoxyphenylboronic acid (1.38 mmol, 3 eq) was used. After FCC, the 2-((4-(tert-butyl)phenoxy)(4-methoxyphenyl)methyl)pyridine 20 was afforded as a white solid (123.5 mg) in 77% yield. TLC: CyHex/EtOAc 60:40, Rf = 0.7. IR ν (cm−1): 2958, 2927, 2927 (νCsp3-H); 2847 (νOMe); 1609, 1585, 1512 and 1463 (νC=C); 1238 (νasym C-O-C); 1024 (νsym C-O-C); 822 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.51 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9= 0.9 Hz, 1H, H12), 7.79 (td, J10–9,10–11 = 7.8 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.58 (d, J9–10 = 7.8 Hz, 1H, H9), 7.41 (d, J15–14,17–18 = 8.4 Hz, 2H, H15, H17), 7.28–7.24 (m, 1H, H11), 7.22 (d, J3–4, 7–6 = 8.8 Hz, 2H, H3, H7), 6.89–6.85 (m, 4H, H4, H6, H14, H18), 6.53 (s, 1H, H1), 3.70 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.56 (C5), 158.7 (C8), 154.9 (C13), 149 (C12), 143 (C16), 137.2 (C10), 132.3 (C2), 128.2 (C15, C17), 126 (C3, C7), 122.7 (C9), 120.4 (C11), 115.2 (C4, C6), 113.8 (C14, C18), 80.9 (C1), 55 (C19), 33.7 (C20), 31.2 (C21). LRMS: (ES+, CV = 30) m/z: 349.27 [M + 2H]+; 348.21 [M + H]+; 214.12 [M − C10H13]+; 198.13 [M − C10H13O]+. HRMS: calcd. for C23H25NO2H [M + H]+ (348.1958); found (348.1959).
2.4.5. 2-((4-chlorophenoxy)(4-methoxyphenyl)methyl)pyridine 21
215.79 mg (1.38 mmol, 3 eq) of 4-chlorophenylboronic acid was used. After FCC, the 2-((4-chlorophenoxy)(4-methoxyphenyl)methyl)pyridine 21 was afforded as a white solid (135.2 mg) in 91% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.7. IR ν (cm−1): 3056, 3005 (νCsp2-H); 2931 (ν Csp3-H); 2837 (νOMe); 1604, 1588, 1511 and 1487 (νC=C); 1229 (νasym C-O-C); 1029 (νsym C-O-C); 820 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.62 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.79 (td, J10–9,10–11 = 7.8 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.57 (d, J9–10 = 7.8 Hz, 1H, H9), 7.42 (d, J3–4,7–6 = 8.7 Hz 2H, H3, H7), 7.29–7.25 (m, 3H, H11, H15, H17), 7.01 (d, J14–15, 18–17 = 9 Hz, 2H, H14, H18), 6.90 (d, J4–3, 6–7 = 8.7 Hz, 2H, H4, H6), 6.41 (s, 1H, H1), 3.70 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.9 (C5), 158.8 (C13), 156 (C8), 149.1 (C12), 137.2 (C10), 131.7 (C2), 129.2 (C16, C18), 128.3 (C3, C7), 124.6 (C16), 122.8 (C9), 120.5 (C11), 117.6 (C14, C18), 113.8 (C4, C6), 81.23 (C1), 55.0 (C19). LRMS: (ES+, CV = 30) m/z: 348.10 [M + Na]+; 326.11 [M + H]+; 214.14 [M − C6H4Cl]+; 198.14 [M − C6H4ClO]+. HRMS: calcd. for C20H16ClNO2H [M + H]+ (326.0942); found (326.0944).
2.4.6. 2-((4-bromophenoxy)(4-methoxyphenyl)methyl)pyridine 22
277.14 mg of 4-bromophenylboronic acid (1.38 mmol, 3 eq) was added. After FCC, the 2-((4-bromophenoxy)(4-methoxyphenyl)methyl)pyridine 22 was afforded as a white solid (140 mg) in 83% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.5. IR ν (cm−1): 3066 (νCsp2-H); 2974, 2919 (νCsp3-H); 2837 (νOMe); 1615, 1588, 1510 and 1488 (νC=C); 1228 (νasym C-O-C); 1019 (νsym C-O-C), 811 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.51 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.79 (td, J10–9,10–11 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.57 (d, J9–10 = 7.9 Hz, 1H, H9), 7.42–7.37 (m, 4H, H3, H7, H15, H17), 7.29–7.25 (m, 1H, H11), 6.96 (d, J14–15, 18–17 = 9 Hz, 2H, H14, H18), 6.89 (d, J4–3, 6–7 = 8.8 Hz, 2H, H4, H6), 6.40 (s, 1H, H1), 3.70 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 159.9 (C13), 158.9 (C5), 156.5 (C8), 149.1 (C12), 137.2 (C10), 132.1 (C15, C17), 131.7 (C2), 128.3 (C3, C7), 122.8 (C9), 120.5 (C11), 118.2 (C14, C18), 113.9 (C4, C6), 112.4 (C16), 81.17 (C1), 55.0 (C19). LRMS: (ES+, CV = 30) m/z: 370.02 [M + H]+; 214.1 [M − C6H4Br]+; 198.16 [M − C6H4OBr]+. HRMS: calcd. for C19H16BrNO2H [M + H]+ (370.0437); found (370.0439).
2.4.7. 2-((4-fluorophenoxy)(4-methoxyphenyl)methyl)pyridine 23
193 mg of 4-fluorophenylboronic acid (1.38 mmol, 3 eq) was added. After FCC, the 2-((4-fluorophenoxy)(4-methoxyphenyl)methyl)pyridine 23 was afforded as a transparent oil (139 mg) in 98% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.67. IR ν (cm−1): 3055, 3005 (νCsp2-H); 2920 (ν Csp3-H); 2838 (νOMe); 1600, 1588, 1501 and 1467 (νC=C); 1242 (νasym C-O-C); 1029 (νsym C-O-C); 823 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.50 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.78 (td, J10–9,10–11 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.58 (d, J9–10 = 7.9 Hz, 1H, H9), 7.40 (d, J3–4,7–6 = 8.7 Hz 2H, H3, H7), 7.28–7.24 (m, 1H, H11), 7.07–6.97 (m, 4H, H14, H15, H17, H18), 6.89 (d, J4–3, 6–7 = 8.7 Hz, 2H, H4, H6), 6.63 (s, 1H, H1), 3.70 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.2 (C5), 158.8 (C8), 155.4 (C13), 153.5 (C16), 149.1 (C12), 137.2 (C10), 131.9 (C2), 128.3 (C3, C7), 122.8 (C9), 120.5 (C11), 117.3 (C15, C18), 115.9 (C14, C7), 113.8 (C4, C6), 81.5 (C1), 55.0 (C19). LRMS: (ES+, CV = 30) m/z: 311.23 [M + 2H]+; 310.14 [M + H]+; 214.12 [M − C6H4F]+; 198.12 [M − C6H4FO]+. HRMS: calcd. for C19H16FNO2H [M + H]+ (310.1238); found (309.2037).
2.4.8. 2-((4-methoxyphenyl)(4-(trifluoromethyl)phenoxy)methyl)pyridine 24
215.79 mg of 4-trifluorophenylboronic acid (1.38 mmol, 3 eq) was added. After FCC, the 2-((4-methoxyphenyl)(4-(trifluoromethyl)phenoxy)methyl)pyridine 24 was afforded as a yellow oil (105.9 mg) in 64% yield. TLC: CyHex/EtOAc: 60/40, Rf = 0.6. IR ν (cm−1): 3058, 3009 (νCsp2-H); 2990, 2934 (νCsp3-H); 2839 (νOMe); 1613, 1588, 1511 and 1468 (νC=C); 1238 (νasym C-O-C); 1029 (νsym C-O-C); 834 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.53 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.80 (td, J10–9,10–11 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.61–7.58 (m, 3H, H9, H15, H17), 7.44 (d, J3–4,7–6 = 8.7 Hz 2H, H3, H7), 7.30–7.27 (m, 1H, H11), 7.17 (d, J14–15, 18–17 = 8.5 Hz, 2H, H14, H18), 6.91 (d, J4–3, 6–7 = 8.8 Hz, 2H, H4, H6), 6.54 (s, 1H, H1), 3.71 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160 (C13), 159.6 (C5), 158.9 (C8), 149.2 (C12), 137.3 (C10), 131.4 (C2), 128.3 (C3, C7), 126.9 (C16), 125,7 (C20), 122.9 (C9), 121.6 (C11), 120.5 (C15, C17), 116.3 (C14, C18), 113.9 (C4, C6), 81 (C1), 55.0 (C19). LRMS: (ES+, CV = 30) m/z: 361.19 [M + 2H]+; 360.12 [M + H]+; 214.13 [M − C7H4F3]+; 198.14 [M − C7H4F3O]+. HRMS: calcd. for C20H16F3NO2H [M + H]+ (360.1206); found (360.1207).
2.4.9. 2-((4-methoxyphenyl)(4-nitrophenoxy)methyl)pyridine 25
230 mg of 4-nitrophenylboronic acid (1.38 mmol, 3 eq) was added. After FCC, the 2-((4-methoxyphenyl)(4-nitrophenoxy)methyl)pyridine 25 was afforded as a yellow oil (151.5mg) in 98% yield. TLC: CyHex/EtOAc 60:40, Rf = 0.5. IR ν (cm−1): 3079, 3006 (νCsp2-H); 2932 (νCsp3-H); 2838 (νOMe); 1610, 1588, 1509 and 1467 (νC=C); 1238 (νasym C-O-C); 1025 (νsym C-O-C); 843 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.54 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 8.15 (d, J15–14, 17–18 = 9.2 Hz, 2H, H15, H17), 7.82 (td, J10–9,10–11 = 7.8 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.60 (d, J9–10 = 7.9 Hz, 1H, H9), 7.45 (d, J3–4,7–6 = 8.7 Hz, 2H, H3, H7), 7.32–7.28 (m, 1H, H11), 7.21 (d, J14–15, 18–19 = 9.2 Hz, 2H, H14, H18), 6.91 (d, J4–3, 6–7 = 8.7 Hz, 2H, H4, H6), 6.64 (s, 1H, H1), 3.71 (s, 3H, H19, OMe). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 162.5 (C13), 159.1 (C5), 159 (C8), 149.3 (C12), 141.0 (C16), 137.4 (C10), 131 (C2), 128.4 (C3, C7), 125.8 (C15, C17), 123 (C9), 120.7 (C11), 116.3 (C14, C18), 114 (C4, C6), 81.4 (C1), 55.1 (C19). LRMS: (ES+, CV = 30) m/z: 359.09 [M + Na]+; 337.16 [M + H]+; 214.16 [M − C6H4NO2]+; 198.15 [M − C6H4NO3]+. HRMS: calcd. for C19H16N2O4H [M + H]+ (337.1183); found (337.1184).
2.4.10. 4-((4-methoxyphenyl)(pyridin-2-yl)methoxy)-N,N dimethylaniline 26
227.7 mg of 4-dimethylaminophenylboronic acid (1.38 mmol, 3 eq) was added. After FCC, the 4-((4-methoxyphenyl)(pyridin-2-yl)methoxy)-N,N-dimethylaniline 26 was afforded as a blue oil (80 mg) in 52% yield. TLC: CyHex/EtOAc 60:40, Rf = 0.6. IR ν (cm−1): 3049 (νCsp2-H); 2933, 2898 (νCsp3-H); 2837 (νOMe); 1610, 1589, 1509 and 1457 (νC=C); 1338 (νC-N-C); 1225 (νasym C-O-C); 1025 (νsym C-O-C); 813 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.49 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.77 (td, J10–9,10–11 = 7.6 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.58 (d, J9–10 = 7.8 Hz, 1H, H9), 7.38 (d, J3–4, 7–6 = 8.6 Hz, 2H, H3, H7), 7.26–7.23 (m, 1H, H11), 6.89–6.83 (m, 4H, H6, H4, H14, H18), 6.61 (d, J15–14, 17–18 = 9.1 Hz, 2H, H15, H17), 6.2 (s, 1H, H1), 3.70 (s, 3H, H19, OMe), 2.70 (s, 6H, H20). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 160.9 (C5), 158.7 (C8), 150.87 (C13), 148.9 (C12), 145.6 (C16), 137 (C10), 132.6 (C2), 128.2 (C3, C7), 122.6 (C9), 120.4 (C11), 116.8 (C14, C18), 113.9 (C15, C17), 113.7 (C4, C6), 81.7 (C1), 55 (C19), 40.9 (C20). LRMS: (ES+, CV = 30) m/z: 336.26 [M + 2H]+; 335.19 [M + H]+; 214.14 [M − C8H10N]+; 198.16 [M − C8H10NO]+. HRMS: calcd. for C21H19NO3H [M + H]+ (335.1754); found (335.1755).
2.4.11. 1-(4-((4-methoxyphenyl)(pyridin-2-yl)methoxy)phenyl)ethanone 27
189 mg of 4-acetylphenylboronic acid (1.38 mmol, 3 eq) was used. After FCC, the 1-(4-((4-methoxyphenyl)(pyridin-2-yl)methoxy)phenyl)ethanone 27 was afforded as a transparent oil (131 mg) in 78% yield. TLC: CyHex/EtOAc 60:40, Rf = 0.3. IR ν (cm−1): 3054, 3003 (νCsp2-H); 2931 (νCsp3-H); 2837 (νOMe); 1673 (νC=O); 1604, 1580, 1507 and 1467 (νC=C); 1234 (νasym C-O-C); 1025 (νsym C-O-C); 831 (δCsp2-H p-substitution). 1H NMR (400 MHz, DMSO-d6 ) δ (ppm): 8.53 (ddd, J12–11 = 4.7 Hz, J12–10 = 1.9 Hz, J12–9 = 0.9 Hz, 1H, H12), 7.86 (d, J15–14, 17–18 = 8.9 Hz, 2H, H15, H17), 7.80 (td, J10–9,10–11 = 7.7 Hz, J10–12 = 1.8 Hz, 1H, H10), 7.58 (d, J9–10 = 7.9 Hz, 1H, H9), 7.44 (d, J3–4,7–6 = 8.7 Hz 2H, H3, H7), 7.30–7.27 (m, 1H, H11), 7.09 (d, J14–15, 18–17 = 8.9 Hz, 2H, H14, H18), 6.90 (d, J4–3, 6–7 = 8.8 Hz, 2H, H4, H6), 6.55 (s, 1H, H1), 3.71 (s, 3H, H19, OMe), 2.46 (s, 3H, H21). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 196.2 (C20), 161 (C13), 159.7 (C5), 158.9 (C8), 149.2 (C12), 137.3 (C10), 131.5 (C2), 130.3 (C15, C17), 130.1 (C16), 128.4 (C3, C7), 122.9 (C9), 120.5 (C11), 115.6 (C14, C18), 113.9 (C4, C6), 81 (C1), 55 (C19), 26.4 (C21). LRMS: (ES+, CV = 30) m/z: 335.19 [M + 2H] +; 214.14 [M − C8H7O]+; 198.14 [M − C8H7O2]+. HRMS: calcd. for C21H19NO3H [M + H]+ (334.1438); found (334.1439).


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

