
Complexed Crystal Structure of the Dihydroorotase Domain of Human CAD Protein with the Anticancer Drug 5-Fluorouracil


Abstract
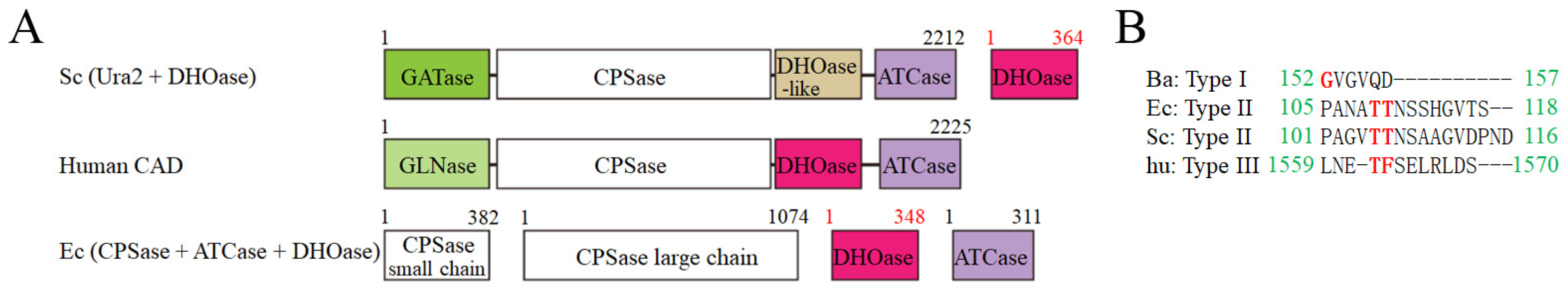
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Site-Directed Mutagenesis
2.3. Crystallization Experiments
2.4. X-ray Diffraction Data and Structure Determination
2.5. Determination of the Dissociation Constant (Kd)
3. Results
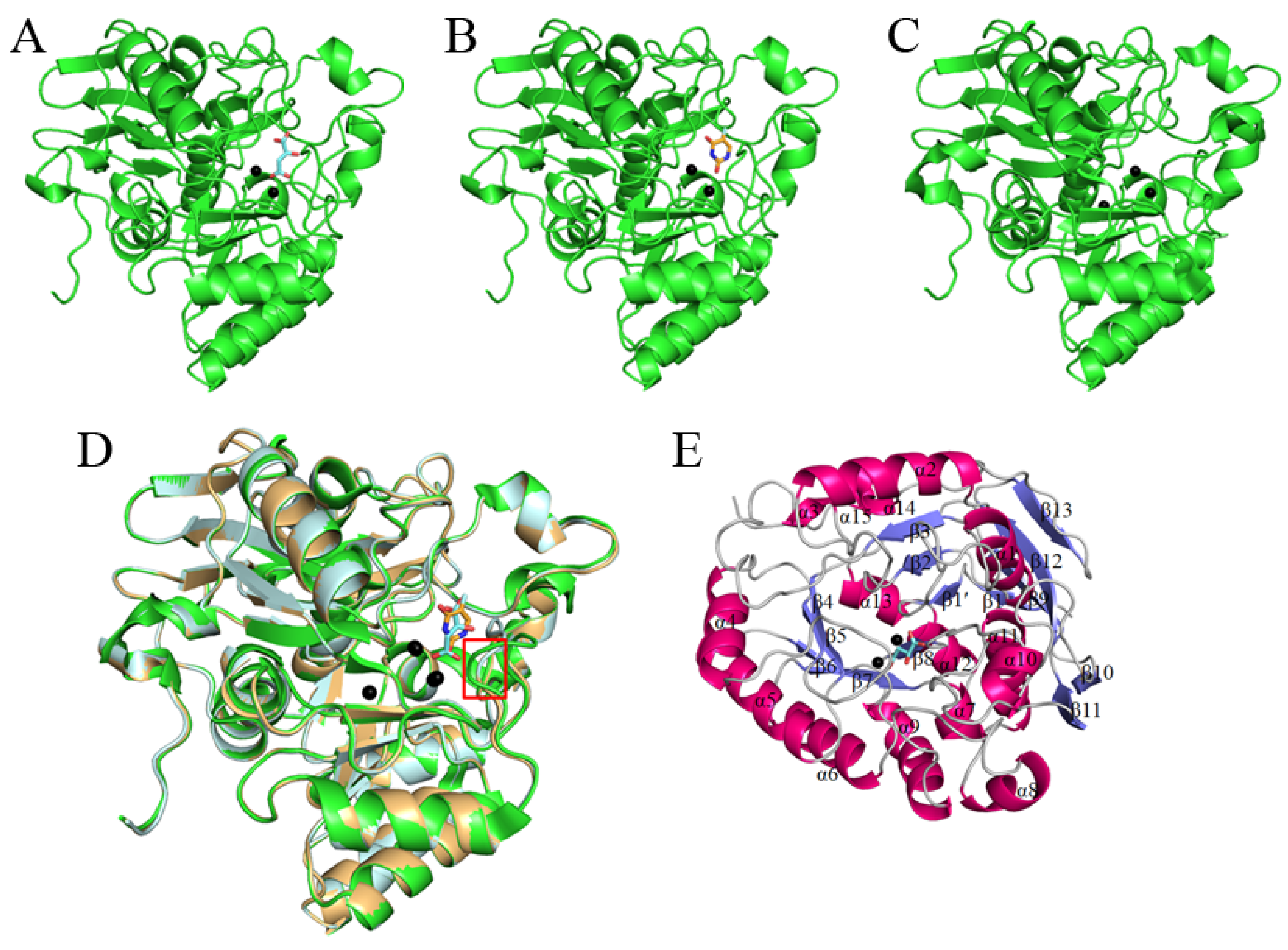
3.1. Crystallization of huDHOase in Complex with Malate and 5-FU
3.2. Overall Structure of the huDHOase Complexes
3.3. Potential Monomer–Monomer Interface of the huDHOase Complexes
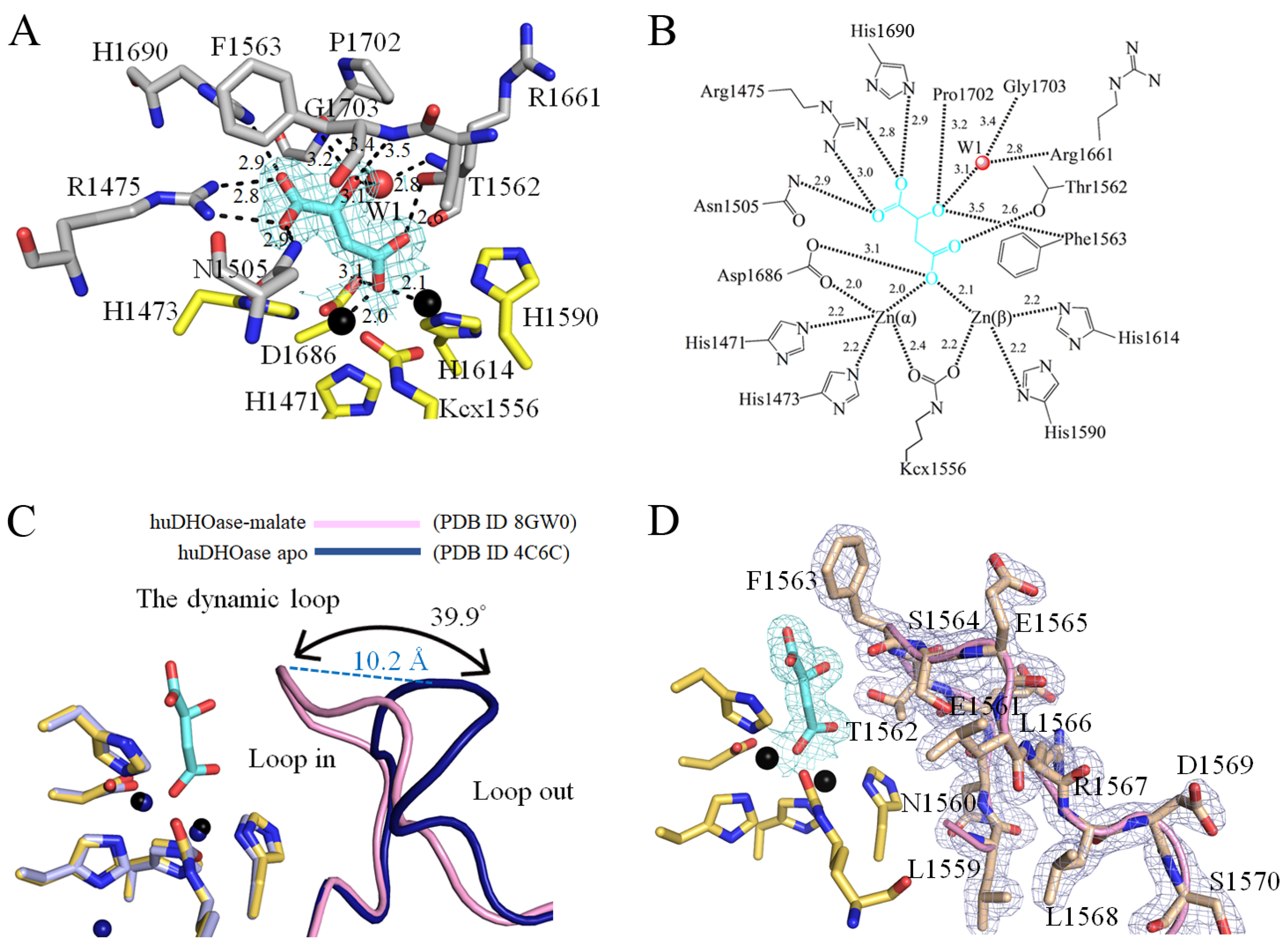
3.4. Malate Binding Mode of huDHOase
3.5. 5-FU Binding Mode of huDHOase
3.6. Structure-Based Mutational Analysis
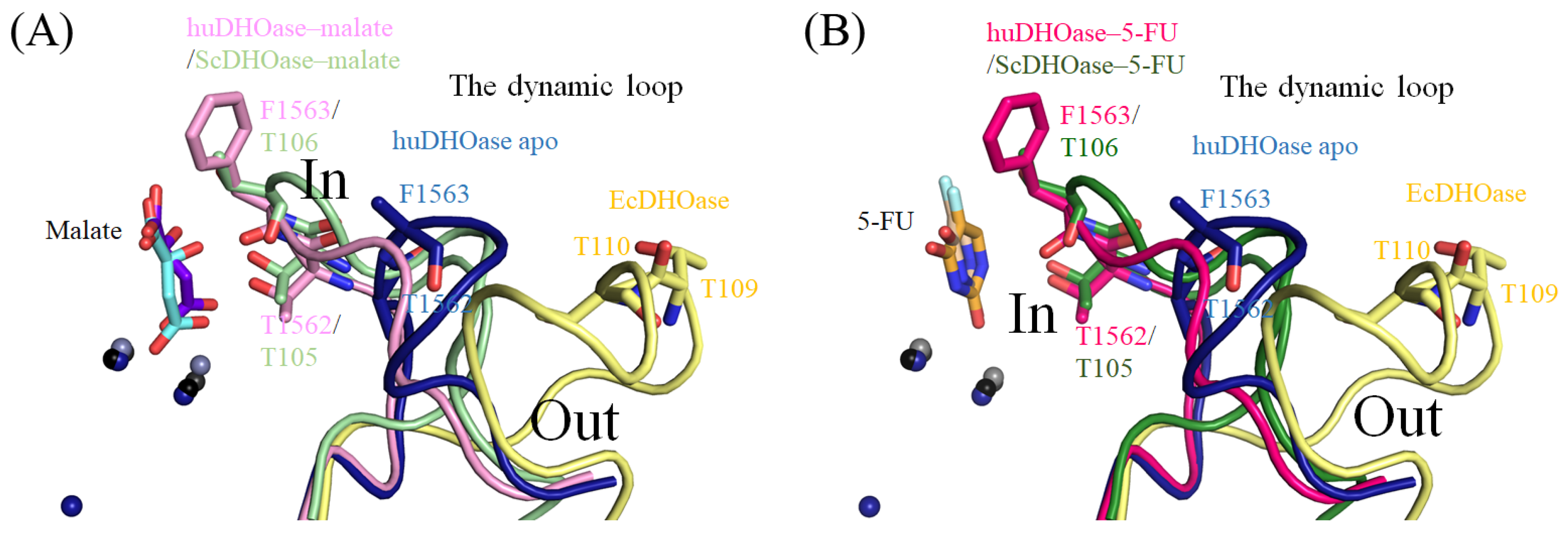
3.7. Structural Comparison of the Active Sites among the 5-FU Bound States of huDHOase, ScDHOase, and PaDHPase
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diasio, R.B.; Offer, S.M. Testing for Dihydropyrimidine Dehydrogenase Deficiency to Individualize 5-Fluorouracil Therapy. Cancers 2022, 14, 3207. [Google Scholar] [CrossRef]
- Kalasabail, S.; Engelman, J.; Zhang, L.Y.; El-Omar, E.; Yim, H.C.H. A Perspective on the Role of Microbiome for Colorectal Cancer Treatment. Cancers 2021, 13, 4623. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.-J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 11, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Ning, Z.-J.; Huang, C.-Y. Crystal structure of dihydropyrimidinase in complex with anticancer drug 5-fluorouracil. Biochem. Biophys. Res. Commun. 2019, 519, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Sumi, S.; Imaeda, M.; Kidouchi, K.; Ohba, S.; Hamajima, N.; Kodama, K.; Togari, H.; Wada, Y. Population and family studies of dihydropyrimidinuria: Prevalence, inheritance mode, and risk of fluorouracil toxicity. Am. J. Med. Genet. 1998, 78, 336–340. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P.; Meinsma, R.; Zonnenberg, B.A.; Zoetekouw, L.; Baas, F.; Matsuda, K.; Tamaki, N.; Van Gennip, A.H. Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity. Clin. Cancer Res. 2003, 9, 4363–4367. [Google Scholar] [PubMed]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.R.; Nicholson, J.K.; Kinross, J.M. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356–365. [Google Scholar] [CrossRef]
- Nakayama, H.; Kinouchi, T.; Kataoka, K.; Akimoto, S.; Matsuda, Y.; Ohnishi, Y. Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5-(E)-(2-bromovinyl) uracil that increases the level and toxicity of 5-fluorouracil. Pharmacogenetics 1997, 7, 35–43. [Google Scholar] [CrossRef]
- Heggie, G.D.; Sommadossi, J.P.; Cross, D.S.; Huster, W.J.; Diasio, R.B. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987, 47, 2203–2206. [Google Scholar] [PubMed]
- Huang, C.-Y. Structure, catalytic mechanism, posttranslational lysine carbamylation, and inhibition of dihydropyrimidinases. Adv. Protein Chem. Struct. Biol. 2020, 122, 63–96. [Google Scholar] [CrossRef]
- Lohkamp, B.; Andersen, B.; Piškur, J.; Dobritzsch, D. The Crystal Structures of Dihydropyrimidinases Reaffirm the Close Relationship between Cyclic Amidohydrolases and Explain Their Substrate Specificity. J. Biol. Chem. 2006, 281, 13762–13776. [Google Scholar] [CrossRef] [Green Version]
- Thoden, J.B.; Phillips, G.N., Jr.; Neal, T.M.; Raushel, F.M.; Holden, H.M. Molecular structure of dihydroorotase: A paradigm for catalysis through the use of a binuclear metal center. Biochemistry 2001, 40, 6989–6997. [Google Scholar] [CrossRef]
- Mulrooney, S.B.; Hausinger, R.P. Metal Ion Dependence of Recombinant Escherichia coli Allantoinase. J. Bacteriol. 2003, 185, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Jimenez, J.M.; Martinez-Rodriguez, S.; Rodriguez-Vico, F.; Heras-Vazquez, F.J. Optically pure alpha-amino acids production by the “Hydantoinase Process”. Recent Pat. Biotechnol. 2008, 2, 35–46. [Google Scholar] [PubMed]
- Schoemaker, H.E.; Mink, D.; Wubbolts, M.G. Dispelling the Myths-Biocatalysis in Industrial Synthesis. Science 2003, 299, 1694–1697. [Google Scholar] [CrossRef]
- Huang, C.Y.; Yang, Y.S. A novel cold-adapted imidase from fish Oreochromis niloticus that catalyzes hydrolysis of maleimide. Biochem. Biophys. Res. Commun. 2003, 312, 467–472. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Yang, Y.-S. The role of metal on imide hydrolysis: Metal content and pH profiles of metal ion-replaced mammalian imidase. Biochem. Biophys. Res. Commun. 2002, 297, 1027–1032. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Babbitt, P.C. Divergent Evolution of Enzymatic Function: Mechanistically Diverse Superfamilies and Functionally Distinct Suprafamilies. Annu. Rev. Biochem. 2001, 70, 209–246. [Google Scholar] [CrossRef]
- Peng, W.-F.; Huang, C.-Y. Allantoinase and dihydroorotase binding and inhibition by flavonols and the substrates of cyclic amidohydrolases. Biochimie 2014, 101, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Del Caño-Ochoa, F.; Ramón-Maiques, S. Deciphering CAD: Structure and function of a mega-enzymatic pyrimidine factory in health and disease. Protein Sci. 2021, 30, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Grande-García, A.; Lallous, N.; Díaz-Tejada, C.; Ramón-Maiques, S. Structure, Functional Characterization, and Evolution of the Dihydroorotase Domain of Human CAD. Structure 2014, 22, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.J.; Lei, H.; Santarsiero, B.D.; Lee, H.; Johnson, M.E. Ca-asp bound X-ray structure and inhibition of Bacillus anthracis dihydroorotase (DHOase). Bioorg. Med. Chem. 2016, 24, 4536–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahuja, A.; Purcarea, C.; Ebert, R.; Sadecki, S.; Guy, H.I.; Evans, D.R. Aquifex aeolicus dihydroorotase: Association with aspartate transcarbamoylase switches on catalytic activity. J. Biol. Chem. 2004, 279, 53136–53144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souciet, J.L.; Nagy, M.; Le Gouar, M.; Lacroute, F.; Potier, S. Organization of the yeast URA2 gene: Identification of a defective dihydroorotase-like domain in the multifunctional carbamoylphosphate synthetase-aspartate transcarbamylase complex. Gene 1989, 79, 59–70. [Google Scholar] [CrossRef]
- Washabaugh, M.W.; Collins, K.D. Dihydroorotase from Escherichia coli. Purification and characterization. J. Biol. Chem. 1984, 259, 3293–3298. [Google Scholar] [CrossRef]
- Guan, H.-H.; Huang, Y.-H.; Lin, E.-S.; Chen, C.-J.; Huang, C.-Y. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5-fluorouracil and 5-aminouracil. Biochem. Biophys. Res. Commun. 2021, 551, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chan, C.W.; Graham, S.C.; Christopherson, R.I.; Guss, J.M.; Maher, M.J. Structures of Ligand-free and Inhibitor Complexes of Dihydroorotase from Escherichia coli: Implications for Loop Movement in Inhibitor Design. J. Mol. Biol. 2007, 370, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Maher, M.J.; Christopherson, R.I.; Guss, J.M. Kinetic and Structural Analysis of Mutant Escherichia coli Dihydroorotases: A Flexible Loop Stabilizes the Transition State. Biochemistry 2007, 46, 10538–10550. [Google Scholar] [CrossRef]
- Guan, H.-H.; Huang, Y.-H.; Lin, E.-S.; Chen, C.-J.; Huang, C.-Y. Structural Analysis of Saccharomyces cerevisiae Dihydroorotase Reveals Molecular Insights into the Tetramerization Mechanism. Molecules 2021, 26, 7249. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.-H.; Huang, Y.-H.; Lin, E.-S.; Chen, C.-J.; Huang, C.-Y. Complexed Crystal Structure of Saccharomyces cerevisiae Dihydroorotase with Inhibitor 5-Fluoroorotate Reveals a New Binding Mode. Bioinorg. Chem. Appl. 2021, 2021, 2572844. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.-H.; Huang, Y.-H.; Lin, E.-S.; Chen, C.-J.; Huang, C.-Y. Plumbagin, a Natural Product with Potent Anticancer Activities, Binds to and Inhibits Dihydroorotase, a Key Enzyme in Pyrimidine Biosynthesis. Int. J. Mol. Sci. 2021, 22, 6861. [Google Scholar] [CrossRef]
- Liu, F.; Gai, X.; Wu, Y.; Zhang, B.; Wu, X.; Cheng, R.; Tang, B.; Shang, K.; Zhao, N.; Deng, W.; et al. Oncogenic β-catenin stimulation of AKT2–CAD-mediated pyrimidine synthesis is targetable vulnerability in liver cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2202157119. [Google Scholar] [CrossRef] [PubMed]
- Ridder, D.A.; Schindeldecker, M.; Weinmann, A.; Berndt, K.; Urbansky, L.; Witzel, H.R.; Heinrich, S.; Roth, W.; Straub, B.K. Key Enzymes in Pyrimidine Synthesis, CAD and CPS1, Predict Prognosis in Hepatocellular Carcinoma. Cancers 2021, 13, 744. [Google Scholar] [CrossRef]
- Li, G.; Li, D.; Wang, T.; He, S. Pyrimidine Biosynthetic Enzyme CAD: Its Function, Regulation, and Diagnostic Potential. Int. J. Mol. Sci. 2021, 22, 10253. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Chiang, W.-Y.; Chen, P.-J.; Lin, E.-S.; Huang, C.-Y. Anticancer and Antioxidant Activities of the Root Extract of the Carnivorous Pitcher Plant Sarracenia purpurea. Plants 2022, 11, 1668. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Adams, P.D.; Read, R.J.; McCoy, A.J.; Moriarty, N.W.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Zwart, P.H.; Hung, L.-W. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 582–601. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.-W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.-S.; Luo, R.-H.; Yang, Y.-C.; Huang, C.-Y. Molecular Insights into How the Dimetal Center in Dihydropyrimidinase Can Bind the Thymine Antagonist 5-Aminouracil: A Different Binding Mode from the Anticancer Drug 5-Fluorouracil. Bioinorg. Chem. Appl. 2022, 2022, 1817745. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y. Inhibition of a Putative Dihydropyrimidinase from Pseudomonas aeruginosa PAO1 by Flavonoids and Substrates of Cyclic Amidohydrolases. PLoS ONE 2015, 10, e0127634. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- del Caño-Ochoa, F.; Grande-García, A.; Reverte-López, M.; D’Abramo, M.; Ramón-Maiques, S. Characterization of the catalytic flexible loop in the dihydroorotase domain of the human multi-enzymatic protein CAD. J. Biol. Chem. 2018, 293, 18903–18913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef] [PubMed]
- Diasio, R.B.; Harris, B.E. Clinical Pharmacology of 5-Fluorouracil. Clin. Pharmacokinet. 1989, 16, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Edwards, B.F.; Fernando, R.; Martin, P.D.; Grimley, E.; Cordes, M.; Vaishnav, A.; Brunzelle, J.S.; Evans, H.G.; Evans, D.R. The mononuclear metal center of type-I dihydroorotase from Aquifex aeolicus. BMC Biochem. 2013, 14, 36. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.D.; Purcarea, C.; Zhang, P.; Vaishnav, A.; Sadecki, S.; Guy-Evans, H.I.; Evans, D.R.; Edwards, B.F. The Crystal Structure of a Novel, Latent Dihydroorotase from Aquifex aeolicus at 1.7Å Resolution. J. Mol. Biol. 2005, 348, 535–547. [Google Scholar] [CrossRef]
- Huang, Y.H.; Huang, C.Y. Creation of a putative third metal binding site in type II dihydroorotases significantly enhances enzyme activity. Protein Pept. Lett. 2015, 22, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Cheon, Y.-H.; Kim, H.-S.; Han, K.-H.; Abendroth, J.; Niefind, K.; Schomburg, D.; Wang, J.; Kim, Y. Crystal Structure of D-Hydantoinase from Bacillus stearothermophilus: Insight into the Stereochemistry of Enantioselectivity. Biochemistry 2002, 41, 9410–9417. [Google Scholar] [CrossRef] [PubMed]
- Abendroth, J.; Niefind, K.; Schomburg, D. X-ray Structure of a Dihydropyrimidinase from Thermus sp. at 1.3Å Resolution. J. Mol. Biol. 2002, 320, 143–156. [Google Scholar] [CrossRef]
- Gojkovic, Z.; Rislund, L.; Andersen, B.; Sandrini, M.P.; Cook, P.F.; Schnackerz, K.D.; Piskur, J. Dihydropyrimidine amidohydrolases and dihydroorotases share the same origin and several enzymatic properties. Nucleic Acids Res. 2003, 31, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, M.-I.; Chung, J.; Ahn, J.-H.; Rhee, S. Crystal Structure of Metal-Dependent Allantoinase from Escherichia coli. J. Mol. Biol. 2009, 387, 1067–1074. [Google Scholar] [CrossRef]
- Huang, Y.-H.; Lien, Y.; Chen, J.-H.; Lin, E.-S.; Huang, C.-Y. Identification and characterization of dihydropyrimidinase inhibited by plumbagin isolated from Nepenthes miranda extract. Biochimie 2020, 171–172, 124–135. [Google Scholar] [CrossRef]
- del Caño-Ochoa, F.; Moreno-Morcillo, M.; Ramón-Maiques, S. CAD, A Multienzymatic Protein at the Head of de Novo Pyrimidine Biosynthesis. Macromol. Protein Complexes II Struct. Funct. 2019, 93, 505–538. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | ||
|---|---|---|
| Crystal | huDHOase–Malate | huDHOase–5-FU |
| Wavelength (Å) | 1.00 | 1.00 |
| Resolution (Å) | 30–1.64 | 30–1.97 |
| Space group | C2221 | C2221 |
| Cell dimension (Å) | a = 82.05 α = 90° | a = 81.78 α = 90° |
| b = 158.25 β = 90° | b = 157.79 β = 90° | |
| c = 61.19 γ = 90° | c = 61.83 γ = 90° | |
| Completeness (%) | 98.9 (98.6) * | 99.9 (99.8) * |
| <I/σI> | 11.51 (2.41) | 29.84 (6.63) |
| CC1/2 | 0.998 (0.807) | 0.991 (0.972) |
| Redundancy | 7.0 (7.0) | 7.0 (7.2) |
| Refinement | ||
| Resolution (Å) | 28.53–1.64 | 28.78–1.97 |
| No. reflections | 48510 | 28310 |
| Rwork/Rfree | 0.172/0.191 | 0.174/0.214 |
| No. atoms | ||
| Protein | 3072 | 3063 |
| Ligand | 15 | 14 |
| Zinc | 2 | 2 |
| Water | 269 | 260 |
| R.m.s deviation | ||
| Bond lengths (Å) | 0.006 | 0.008 |
| Bond angles (°) | 0.951 | 1.012 |
| Ramachandran plot | ||
| In preferred regions | 96.36% | 96.01% |
| In allowed regions | 3.08% | 3.43% |
| Outliers | 0.56% | 0.56% |
| PDB entry | 8GW0 | 8GVZ |
| Subunit A | Subunit A′ | Distance [Å] (the Malate Complex) | Distance [Å] (the 5-FU Complex) |
|---|---|---|---|
| T1595 [OG1] | Q1594 [OE1] | 2.4 | 2.4 |
| R1630 [NE] | L1605 [O] | 2.7 | 3.0 |
| R1630 [NH2] | L1605 [O] | 2.8 | 3.1 |
| R1630 [NH2] | Q1607 [OE1] | 2.4 | 2.5 |
| V1571 [N] | E1619 [OE2] | 2.8 | 2.8 |
| Q1594 [OE1] | T1595 [OG1] | 2.4 | 2.4 |
| L1605 [O] | R1630 [NE] | 2.7 | 3.0 |
| L1605 [O] | R1630 [NH2] | 2.8 | 3.1 |
| Q1607 [OE1] | R1630 [NH2] | 2.4 | 2.5 |
| E1619 [OE2] | V1571 [N] | 2.8 | 2.8 |
| Protein | λmax (nm) | λem Shift (nm) | Quenching (%) | Kd Value (µM) |
|---|---|---|---|---|
| huDHOase | From 340 to 349 | 8.5 | 94.5 | 91.2 ± 1.7 |
| huDHOase-T1562A | From 340 to 346 | 6.0 | 89.0 | 146.5 ± 2.1 |
| huDHOase-R1475A | From 339.5 to 341.5 | 2.0 | 75.3 | 161.5 ± 1.6 |
| Oligonucleotide | Primer |
|---|---|
| huDHOase-R1475A-N | TCCATGTGCACCTGGCGGAACCAGGTGGGA |
| huDHOase-R1475A-C | CACCTGGTTCCGCCAGGTGCACATGGACAT |
| huDHOase-T1562A-N | CTTTACCTCAATGAGGCCTTCTCTGAGCTG |
| huDHOase-T1562A-C | GCCGCAGCTCAGAGAAGGCCTCATTGAGGT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, E.-S.; Huang, Y.-H.; Yang, P.-C.; Peng, W.-F.; Huang, C.-Y. Complexed Crystal Structure of the Dihydroorotase Domain of Human CAD Protein with the Anticancer Drug 5-Fluorouracil. Biomolecules 2023, 13, 149. https://doi.org/10.3390/biom13010149
Lin E-S, Huang Y-H, Yang P-C, Peng W-F, Huang C-Y. Complexed Crystal Structure of the Dihydroorotase Domain of Human CAD Protein with the Anticancer Drug 5-Fluorouracil. Biomolecules. 2023; 13(1):149. https://doi.org/10.3390/biom13010149
Chicago/Turabian StyleLin, En-Shyh, Yen-Hua Huang, Po-Chun Yang, Wei-Feng Peng, and Cheng-Yang Huang. 2023. "Complexed Crystal Structure of the Dihydroorotase Domain of Human CAD Protein with the Anticancer Drug 5-Fluorouracil" Biomolecules 13, no. 1: 149. https://doi.org/10.3390/biom13010149