
Identifying Immune-Specific Subtypes of Adrenocortical Carcinoma Based on Immunogenomic Profiling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Single-Sample Gene Set Enrichment Analysis
2.3. Clustering Analysis
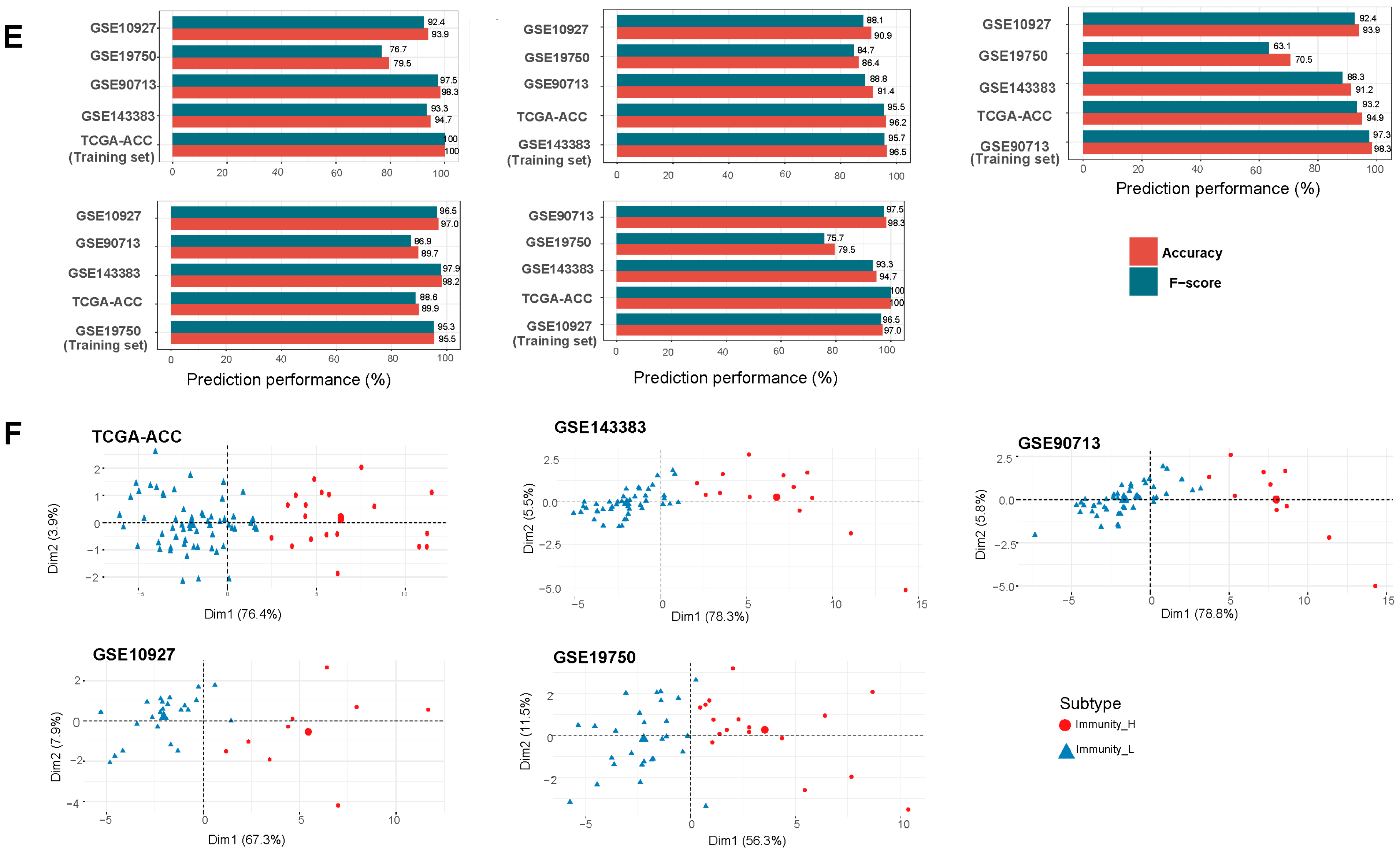
2.4. Prediction of the Immune-Specific Subtypes of ACC
2.5. Survival Analysis
2.6. Evaluation of Tumor Immune Score, Stromal Score, and Intratumor Heterogeneity (ITH) Level
2.7. Evaluation of TMB and CNAs
2.8. Logistic Regression Model
2.9. Pathway and Gene Ontology (GO) Analysis
2.10. Statistical Analysis
3. Results
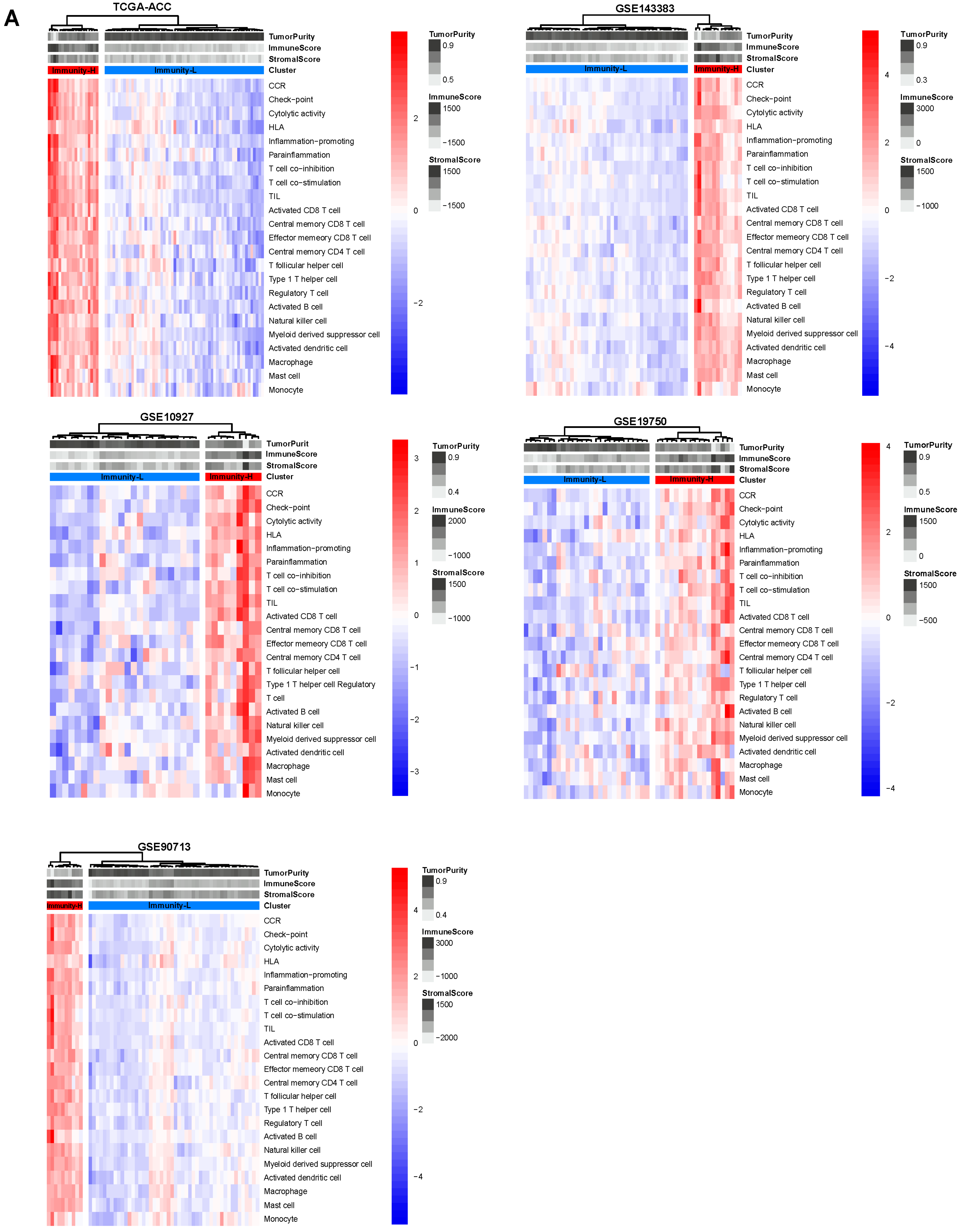
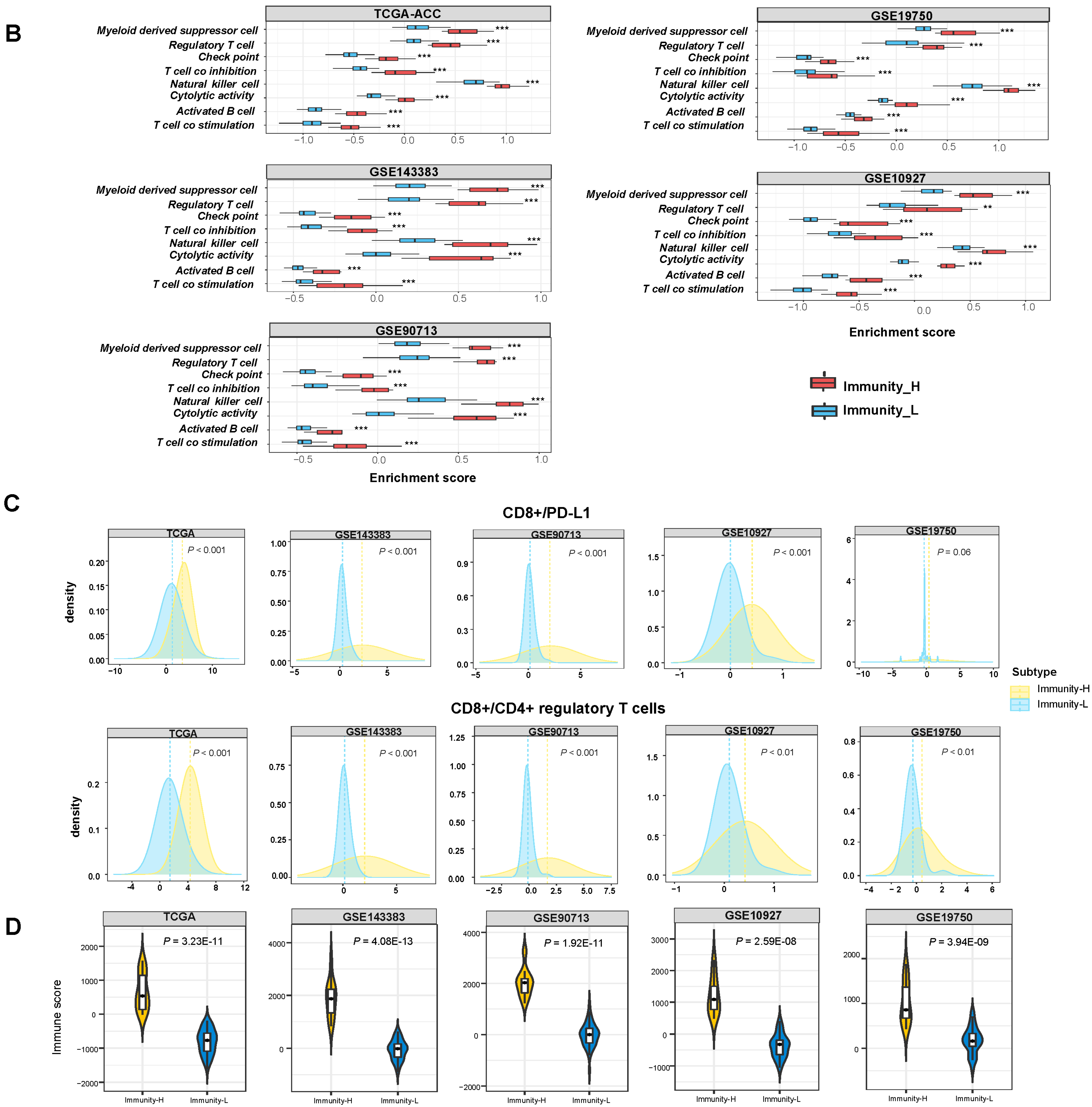
3.1. Clustering Analysis Identifies Two Immune Subtypes of ACC
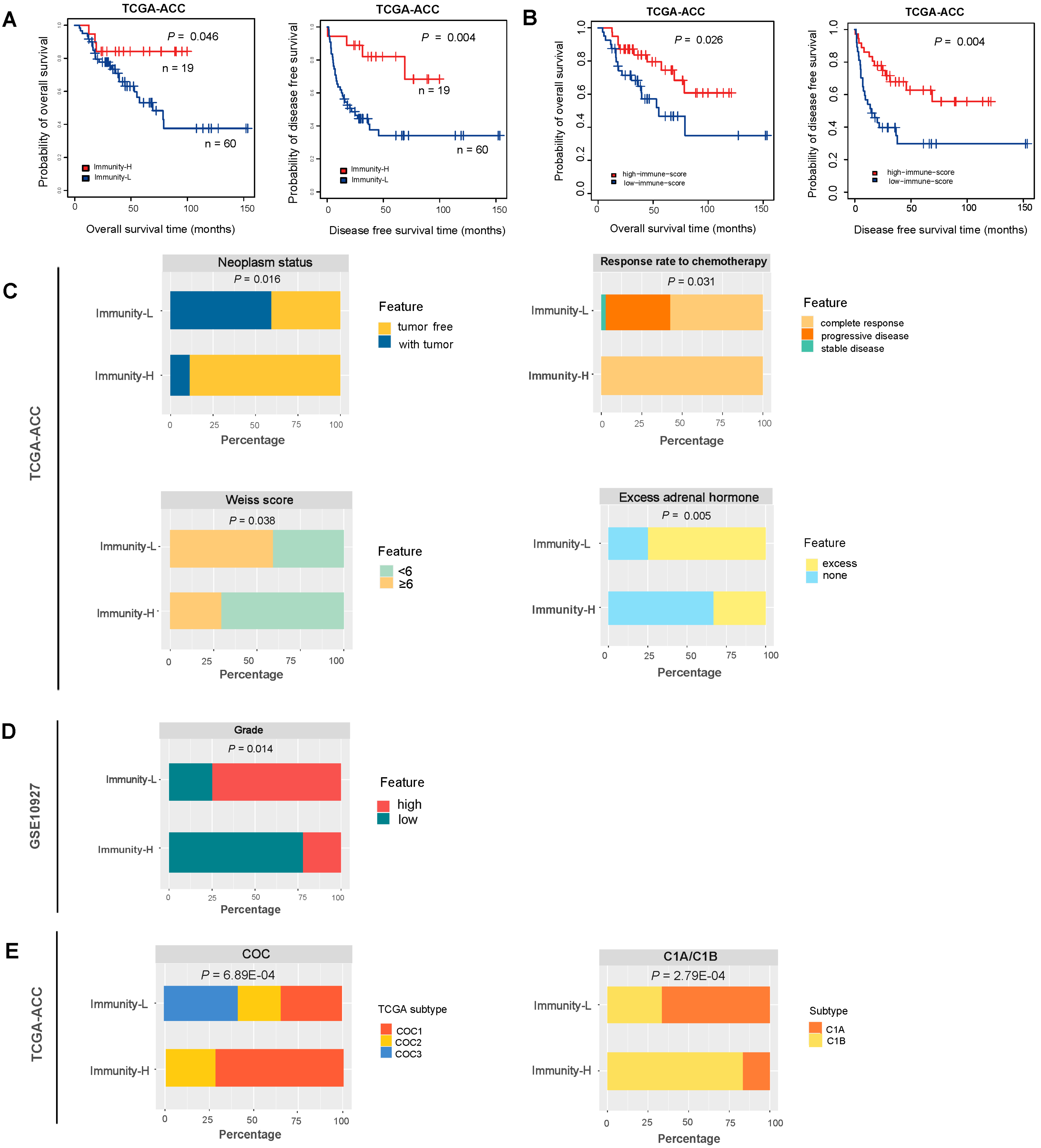
3.2. Immunity-H Has More Favorable Clinical Outcomes Than Immunity-L
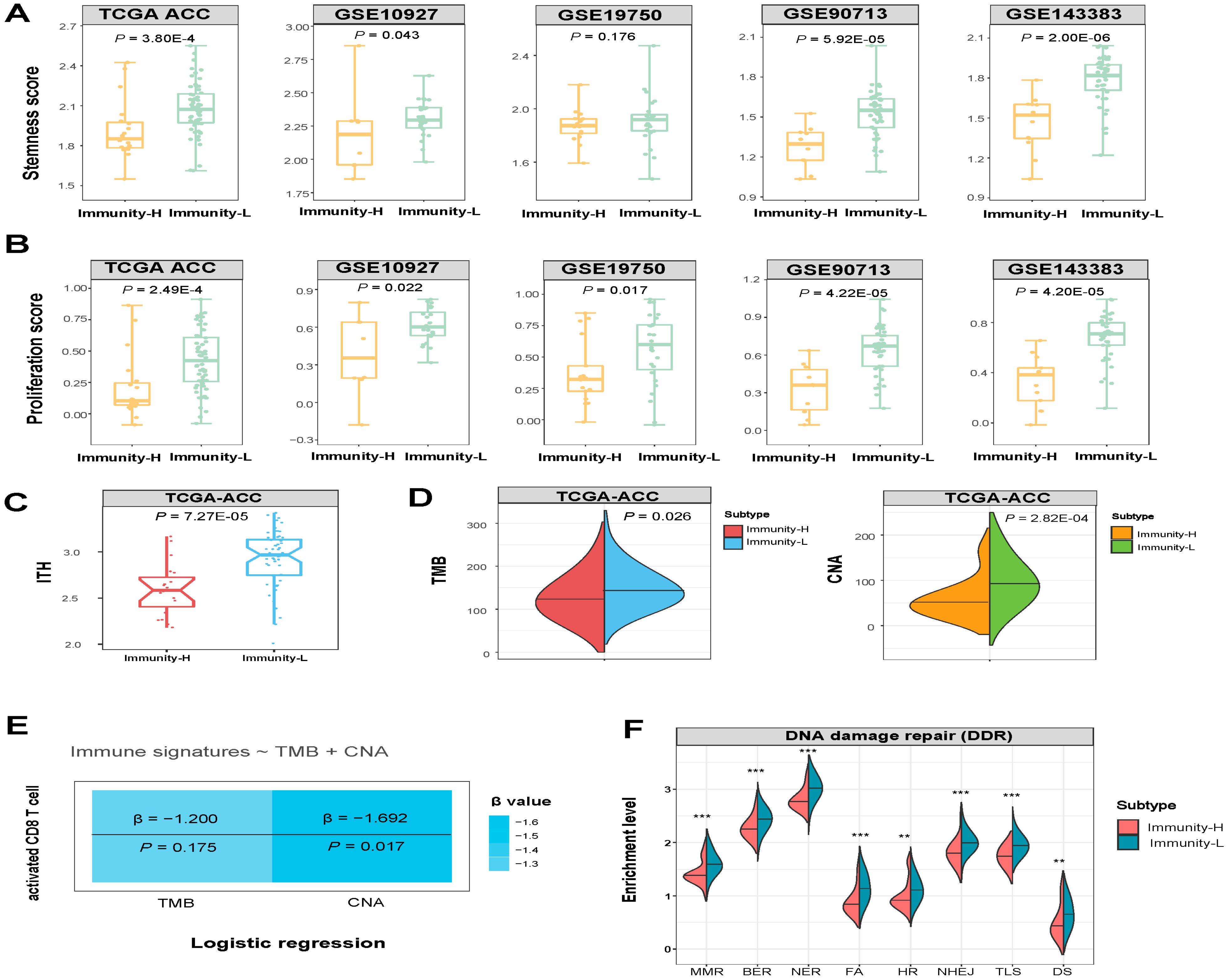
3.3. Immunity-H has More Favorable Tumor Progression Phenotypes and Lower Levels of Genomic Instability Than Immunity-L
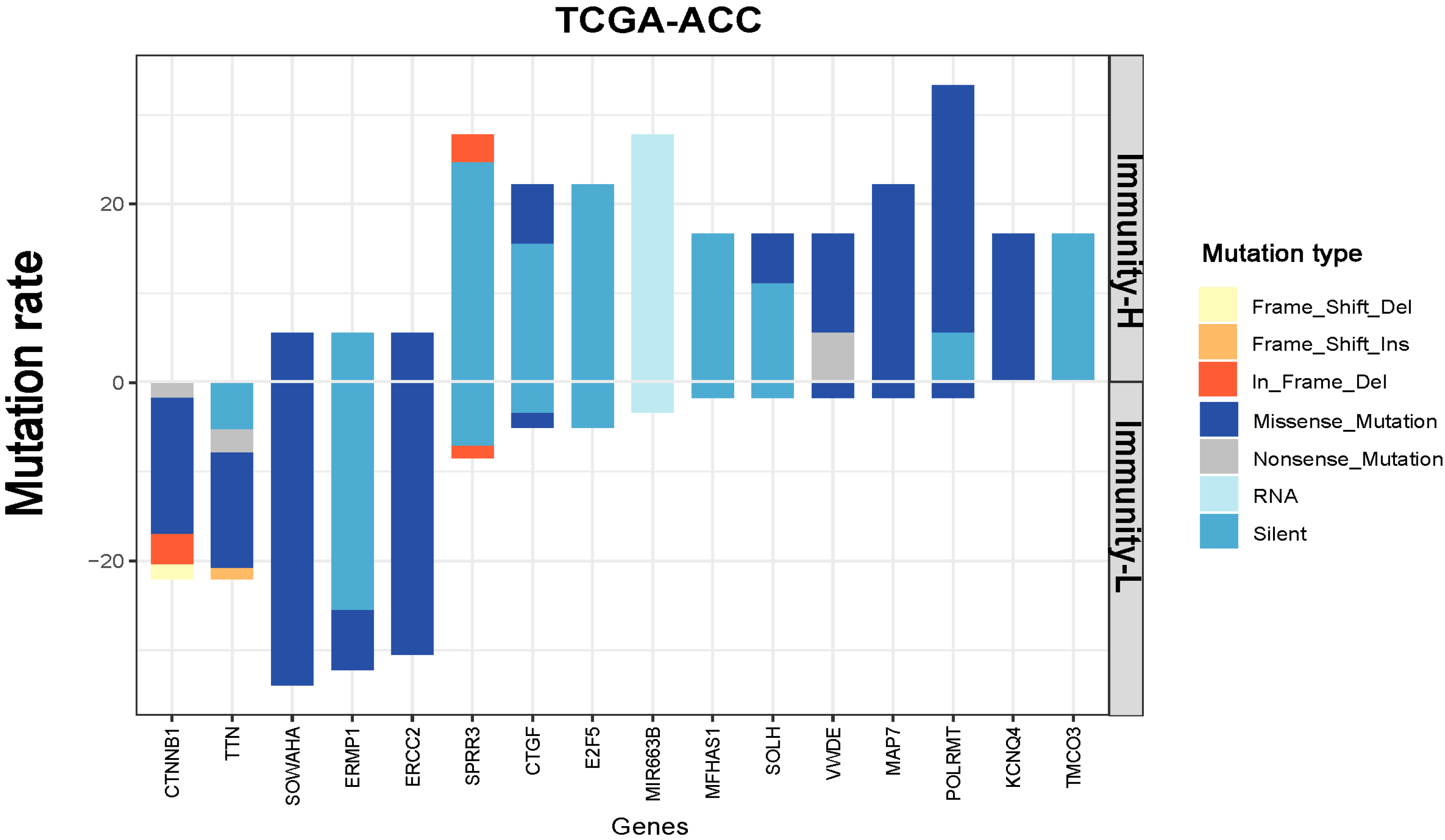
3.4. Identification of Genes with Significantly Different Mutation Rates between the Immune Subtypes of ACC
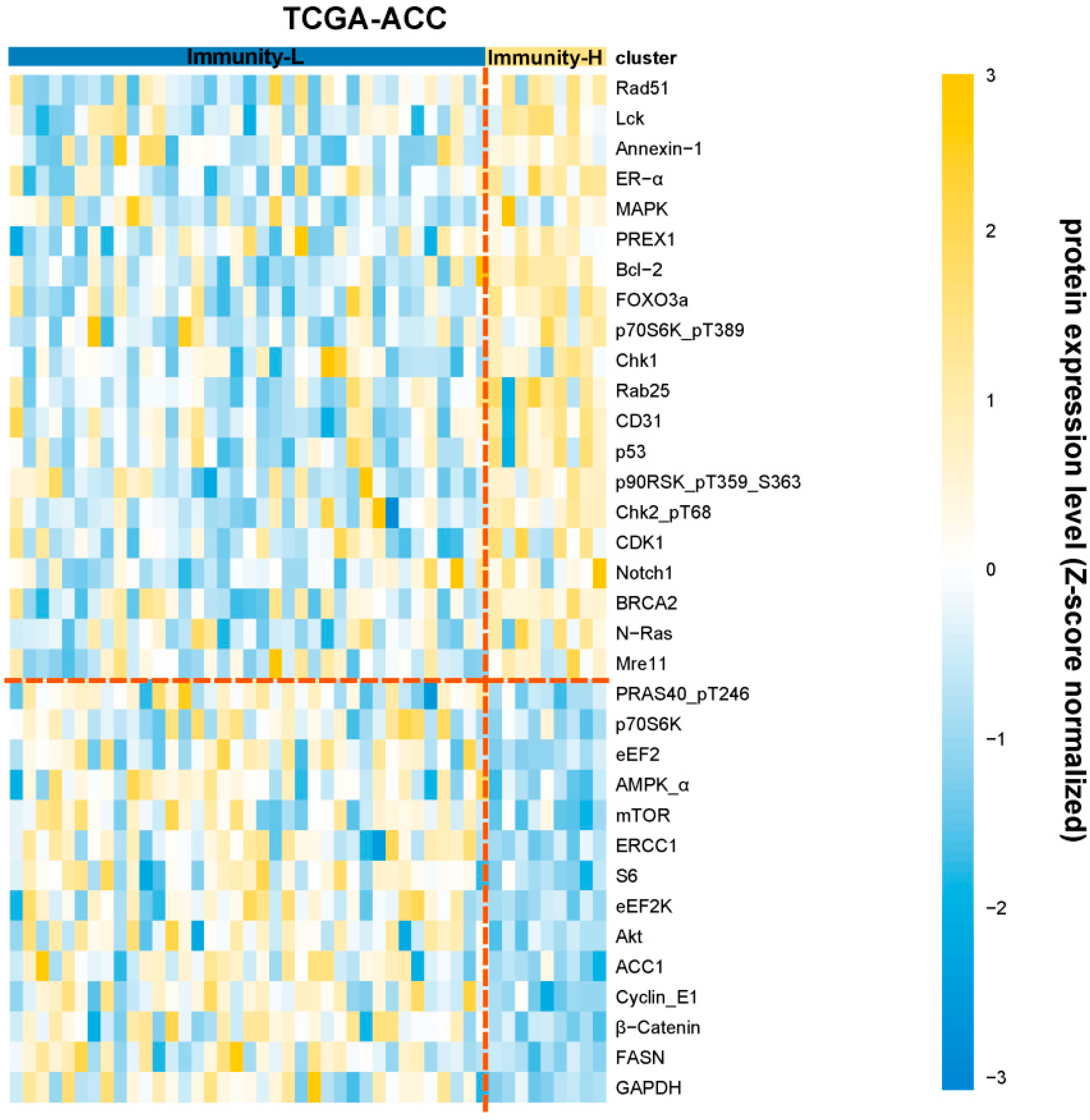
3.5. Identification of Proteins Differentially Expressed between the Immune Subtypes of ACC
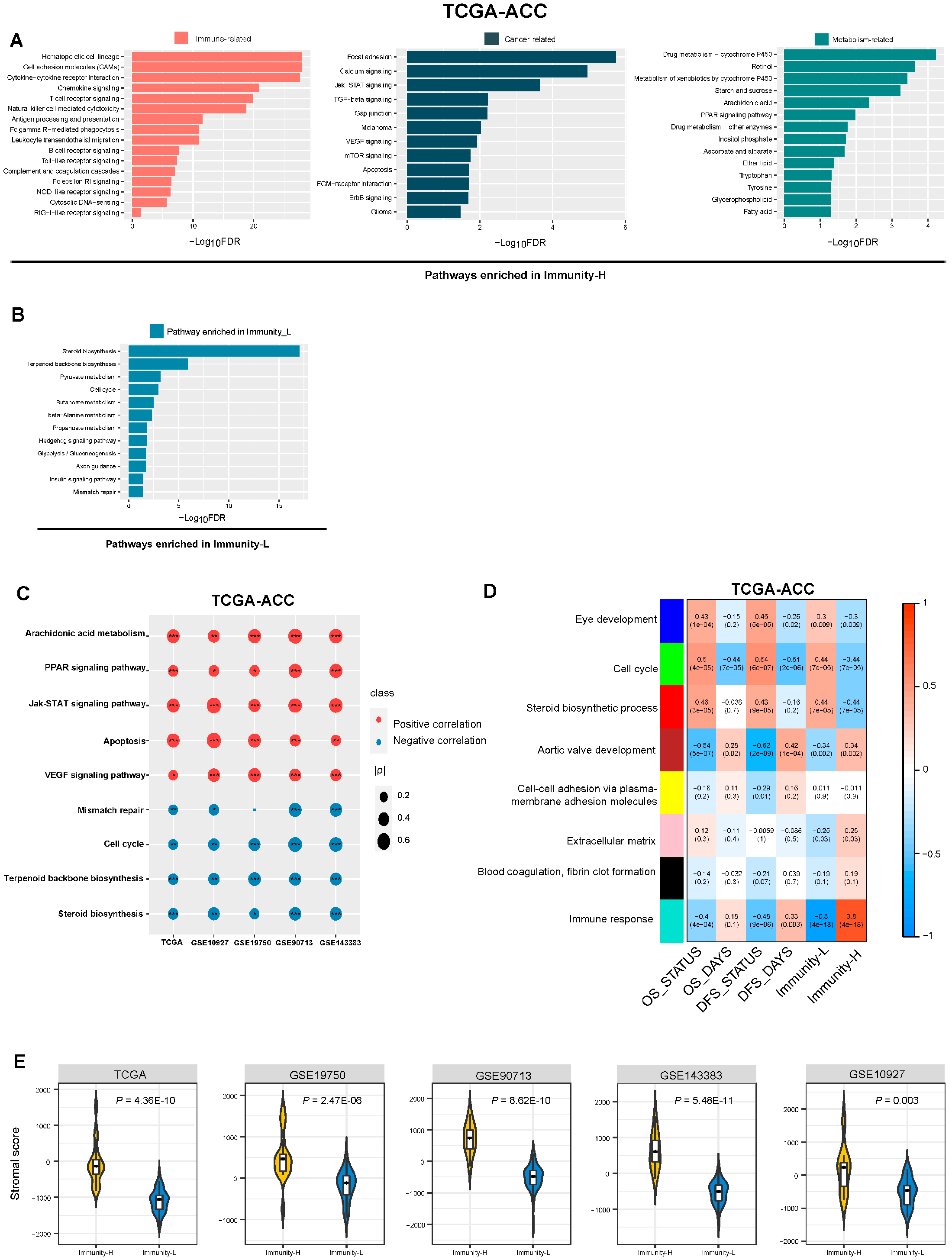
3.6. Identification of Pathways and GO Enriched in the Immune Subtypes of ACC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landwehr, L.S.; Altieri, B.; Schreiner, J.; Sbiera, I.; Weigand, I.; Kroiss, M.; Fassnacht, M.; Sbiera, S. Interplay between glucocorticoids and tumor-infiltrating lymphocytes on the prognosis of adrenocortical carcinoma. J. Immunother. Cancer 2020, 8, e000469. [Google Scholar] [CrossRef]
- Jouinot, A.; Bertherat, J. MANAGEMENT OF ENDOCRINE DISEASE: Adrenocortical carcinoma: Differentiating the good from the poor prognosis tumors. Eur. J. Endocrinol. 2018, 178, R215–R230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassnacht, M.; Johanssen, S.; Quinkler, M.; Bucsky, P.; Willenberg, H.S.; Beuschlein, F.; Terzolo, M.; Mueller, H.H.; Hahner, S.; Allolio, B.; et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: Proposal for a Revised TNM Classification. Cancer 2009, 115, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Abiven, G.; Coste, J.; Groussin, L.; Anract, P.; Tissier, F.; Legmann, P.; Dousset, B.; Bertagna, X.; Bertherat, J. Clinical and Biological Features in the Prognosis of Adrenocortical Cancer: Poor Outcome of Cortisol-Secreting Tumors in a Series of 202 Consecutive Patients. J. Clin. Endocrinol. Metab. 2006, 91, 2650–2655. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- McGrail, D.; Pilié, P.; Rashid, N.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Raj, N.; Zheng, Y.; Kelly, V.; Katz, S.S.; Chou, J.; Do, R.K.G.; Capanu, M.; Zamarin, D.; Saltz, L.B.; Ariyan, C.E.; et al. PD-1 Blockade in Advanced Adrenocortical Carcinoma. J. Clin. Oncol. 2020, 38, 71–80. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch repair deficiency. J. Clin. Oncol. 2015, 33, 2509–2520. [Google Scholar] [CrossRef]
- Lim, A.; Rao, P.; Matin, S. Lynch syndrome and urologic malignancies: A contemporary review. Curr. Opin. Urol. 2019, 29, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I. Data mining in bioinformatics using Weka. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef] [Green Version]
- Bland, J.M.; Altman, D.G. Statistics Notes: Survival probabilities (the Kaplan-Meier method). BMJ 1998, 317, 1572–1580. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Chen, C.; Wang, X. DITHER: An algorithm for Defining IntraTumor Heterogeneity based on EntRopy. Briefings Bioinform. 2021, 22, bbab202. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Libe, R. Adrenocortical carcinoma (ACC): Diagnosis, prognosis, and treatment. Front. Cell Dev. Biol. 2015, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 30, 363. [Google Scholar] [CrossRef]
- de Reyniès, A.; Assié, G.; Rickman, D.S.; Tissier, F.; Groussin, L.; René-Corail, F.; Dousset, B.; Bertagna, X.; Clauser, E.; Bertherat, J. Gene Expression Profiling Reveals a New Classification of Adrenocortical Tumors and Identifies Molecular Predictors of Malignancy and Survival. J. Clin. Oncol. 2009, 27, 1108–1115. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Ding, G.; Zhou, Z.; Feng, C. beta-Catenin-driven adrenocortical carcinoma is characterized with immune exclusion. Oncotargets Ther. 2018, 11, 2029–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragazzon, B.; Libé, R.; Gaujoux, S.; Assié, G.; Fratticci, A.; Launay, P.; Clauser, E.; Bertagna, X.; Tissier, F.; de Reyniès, A.; et al. Transcriptome Analysis Reveals that p53 and beta-Catenin Alterations Occur in a Group of Aggressive Adrenocortical Cancers. Cancer Res. 2010, 70, 8276–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duell, E.J.; Wiencke, J.K.; Cheng, T.J.; Varkonyi, A.; Zuo, Z.F.; Ashok, T.D.S.; Mark, E.J.; Wain, J.C.; Christiani, D.C.; Kelsey, K.T. Polymorphisms in the DNA repair genes XRCC1 and ERCC2 and biomarkers of DNA damage in human blood mononuclear cells. Carcinogenesis 2000, 21, 965–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Sang, Y.-H.; Cai, S.; Xu, C.; Shi, M.-H. The requirement of mitochondrial RNA polymerase for non-small cell lung cancer cell growth. Cell Death Dis. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018, 11, 1171–1187. [Google Scholar] [CrossRef]
- Baechle, J.J.; Hanna, D.N.; Sekhar, K.R.; Rathmell, J.C.; Rathmell, W.K.; Baregamian, N. Integrative computational immunogenomic profiling of cortisol-secreting adrenocortical carcinoma. J. Cell. Mol. Med. 2021, 25, 10061–10072. [Google Scholar] [CrossRef]
- Vanbrabant, T.; Fassnacht, M.; Assie, G.; Dekkers, O.M. Influence of hormonal functional status on survival in adrenocortical carcinoma: Systematic review and meta-analysis. Eur. J. Endocrinol. 2018, 179, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Muzzi, J.C.; Magno, J.M.; Cardoso, M.A.; de Moura, J.; Castro, M.A.; Figueiredo, B.C. Adrenocortical Carcinoma Steroid Profiles: In Silico Pan-Cancer Analysis of TCGA Data Uncovers Immunotherapy Targets for Potential Improved Outcomes. Front. Endocrinol. 2021, 12, 672319. [Google Scholar] [CrossRef]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Taves, M.D.; Ashwell, J.D. Glucocorticoids in T cell development, differentiation and function. Nat. Rev. Immunol. 2020, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Habra, M.A.; Stephen, B.; Campbell, M.; Hess, K.; Tapia, C.; Xu, M.; Rodon Ahnert, J.; Jimenez, C.; Lee, J.E.; Perrier, N.D.; et al. Phase II clinical trial of pembrolizumab efficacy and safety in advanced adrenocortical carcinoma. J. Immunother. Cancer 2019, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Head, L.; Kiseljak-Vassiliades, K.; Clark, T.J.; Somerset, H.; King, J.; Raeburn, C.; Albuja-Cruz, M.; Weyant, M.; Cleveland, J.; Wierman, M.E.; et al. Response to Immunotherapy in Combination With Mitotane in Patients With Metastatic Adrenocortical Cancer. J. Endocr. Soc. 2019, 3, 2295–2304. [Google Scholar] [CrossRef] [PubMed]
- Cosentini, D.; Grisanti, S.; Dalla Volta, A.; Laganà, M.; Fiorentini, C.; Perotti, P.; Sigala, S.; Berruti, A. Immunotherapy failure in adrenocortical cancer: Where next? Endocr. Connect. 2018, 7, E5–E8. [Google Scholar] [CrossRef] [Green Version]
- Krisenko, M.O.; Geahlen, R. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Et Biophys. Acta-Mol. Cell Res. 2015, 1853, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.D.; Wakelin, M.W.; Larbi, K.Y.; Dewar, A.; Asimakopoulos, G.; Horton, M.A.; Nakada, M.T.; Nourshargh, S. Divergent effects of platelet-endothelial cell adhesion molecule-1 and beta 3 integrin blockade on leukocyte transmigration in vivo. J. Immunol. 2000, 165, 426–434. [Google Scholar] [CrossRef] [Green Version]
- Welch, H.C.; Coadwell, W.J.; Ellson, C.D.; Ferguson, G.J.; Andrews, S.R.; Erdjument-Bromage, H.; Tempst, P.; Hawkins, P.T.; Stephens, L.R. P-Rex1, a PtdIns(3,4,5)P-3- and G beta gamma-regulated guanine-nucleotide exchange factor for Rac. Cell 2002, 108, 809–821. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2017, 62, 50–60. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trillo-Tinoco, J.; Sierra, R.A.; Mohamed, E.; Cao, Y.; de Mingo-Pulido, Á.; Gilvary, D.L.; Anadon, C.M.; Costich, T.L.; Wei, S.; Flores, E.R.; et al. AMPK Alpha-1 Intrinsically Regulates the Function and Differentiation of Tumor Myeloid-Derived Suppressor Cells. Cancer Res. 2019, 79, 5034–5047. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, 1–15. [Google Scholar] [CrossRef]
- Medina-Arana, V.; Delgado, L.; González, L.; Bravo, A.; Diaz, H.; Salido, E.; Riverol, D.; González-Aguilera, J.J.; Fernández-Peralta, A.M. Adrenocortical carcinoma, an unusual extracolonic tumor associated with Lynch II syndrome. Fam. Cancer 2011, 10, 265–271. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Wang, X.; Li, M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 2019, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Doghman-Bouguerra, M.; Finetti, P.; Durand, N.; Parise, I.Z.S.; Sbiera, S.; Cantini, G.; Canu, L.; Hescot, S.; Figueiredo, M.M.O.; Komechen, H.; et al. Cancer-testis Antigen FATE1 Expression in Adrenocortical Tumors Is Associated with A Pervasive Autoimmune Response and Is A Marker of Malignancy in Adult, but Not Children, ACC. Cancers 2020, 12, 689. [Google Scholar] [CrossRef] [Green Version]
- Sbiera, S.; Schmull, S.; Assié, G.; Voelker, H.-U.; Kraus, L.; Beyer, M.; Ragazzon, B.; Beuschlein, F.; Willenberg, H.S.; Hahner, S.; et al. High Diagnostic and Prognostic Value of Steroidogenic Factor-1 Expression in Adrenal Tumors. J. Clin. Endocrinol. Metab. 2010, 95, E161–E171. [Google Scholar] [CrossRef] [Green Version]
- Poli, G.; Ruggiero, C.; Cantini, G.; Canu, L.; Baroni, G.; Armignacco, R.; Jouinot, A.; Santi, R.; Ercolino, T.; Ragazzon, B.; et al. Fascin-1 Is a Novel Prognostic Biomarker Associated With Tumor Invasiveness in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2018, 104, 1712–1724. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Nie, R.; Luo, J.; Wang, X.; You, L. Identifying Immune-Specific Subtypes of Adrenocortical Carcinoma Based on Immunogenomic Profiling. Biomolecules 2023, 13, 104. https://doi.org/10.3390/biom13010104
Lu Q, Nie R, Luo J, Wang X, You L. Identifying Immune-Specific Subtypes of Adrenocortical Carcinoma Based on Immunogenomic Profiling. Biomolecules. 2023; 13(1):104. https://doi.org/10.3390/biom13010104
Chicago/Turabian StyleLu, Qiqi, Rongfang Nie, Jiangti Luo, Xiaosheng Wang, and Linjun You. 2023. "Identifying Immune-Specific Subtypes of Adrenocortical Carcinoma Based on Immunogenomic Profiling" Biomolecules 13, no. 1: 104. https://doi.org/10.3390/biom13010104