



The Ni(II)-Binding Activity of the Intrinsically Disordered Region of Human NDRG1, a Protein Involved in Cancer Development

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Cloning
2.2. Protein Expression and Purification
2.3. Isothermal Titration Calorimetry
2.4. Circular Dichroism Spectroscopy
2.5. NMR Spectroscopy
2.6. Light Scattering
2.7. Cultures and Cellular Treatments
2.8. Preparation of Protein Extracts
3. Results
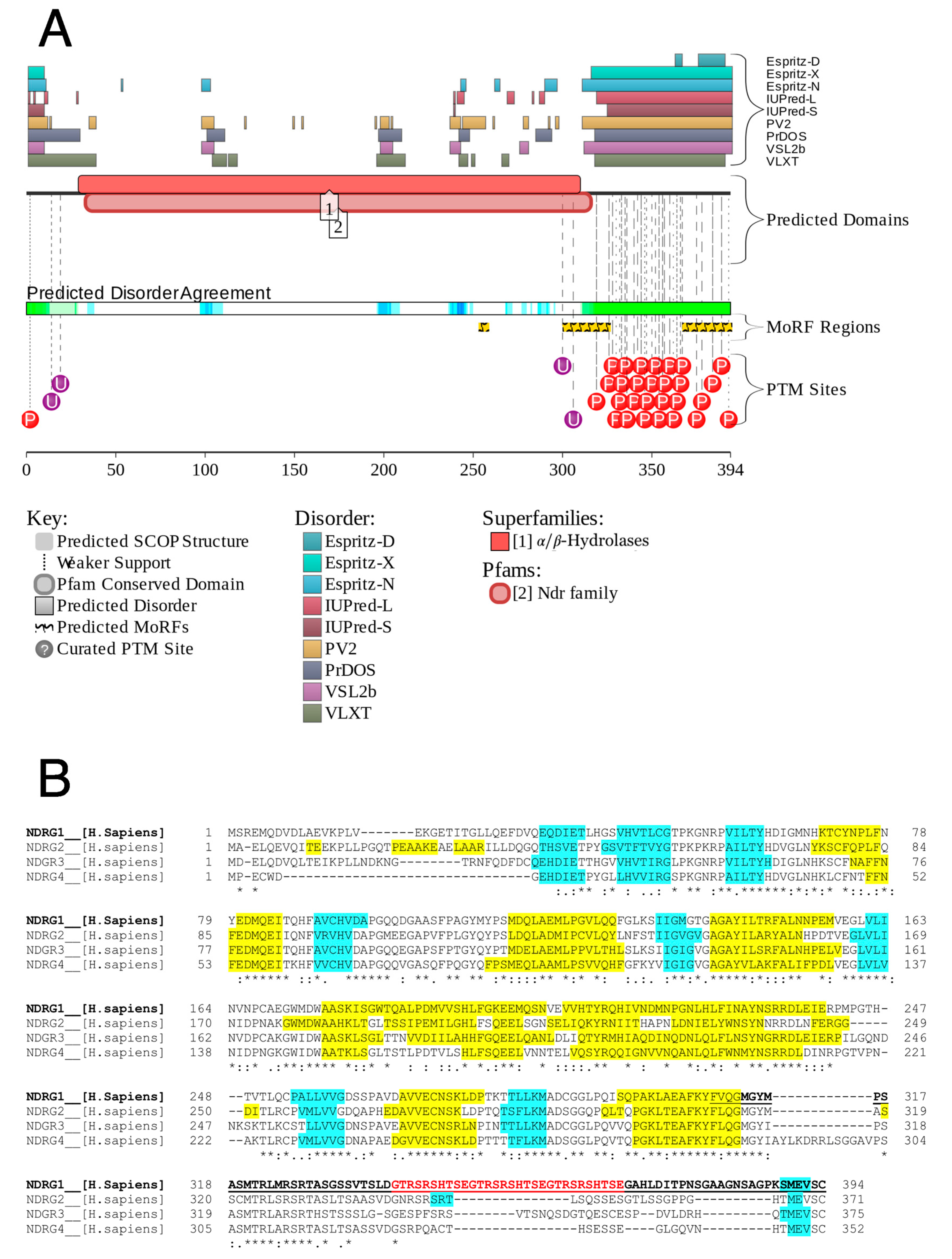
3.1. Analysis of Protein Disorder
3.2. Protein Expression and Purification
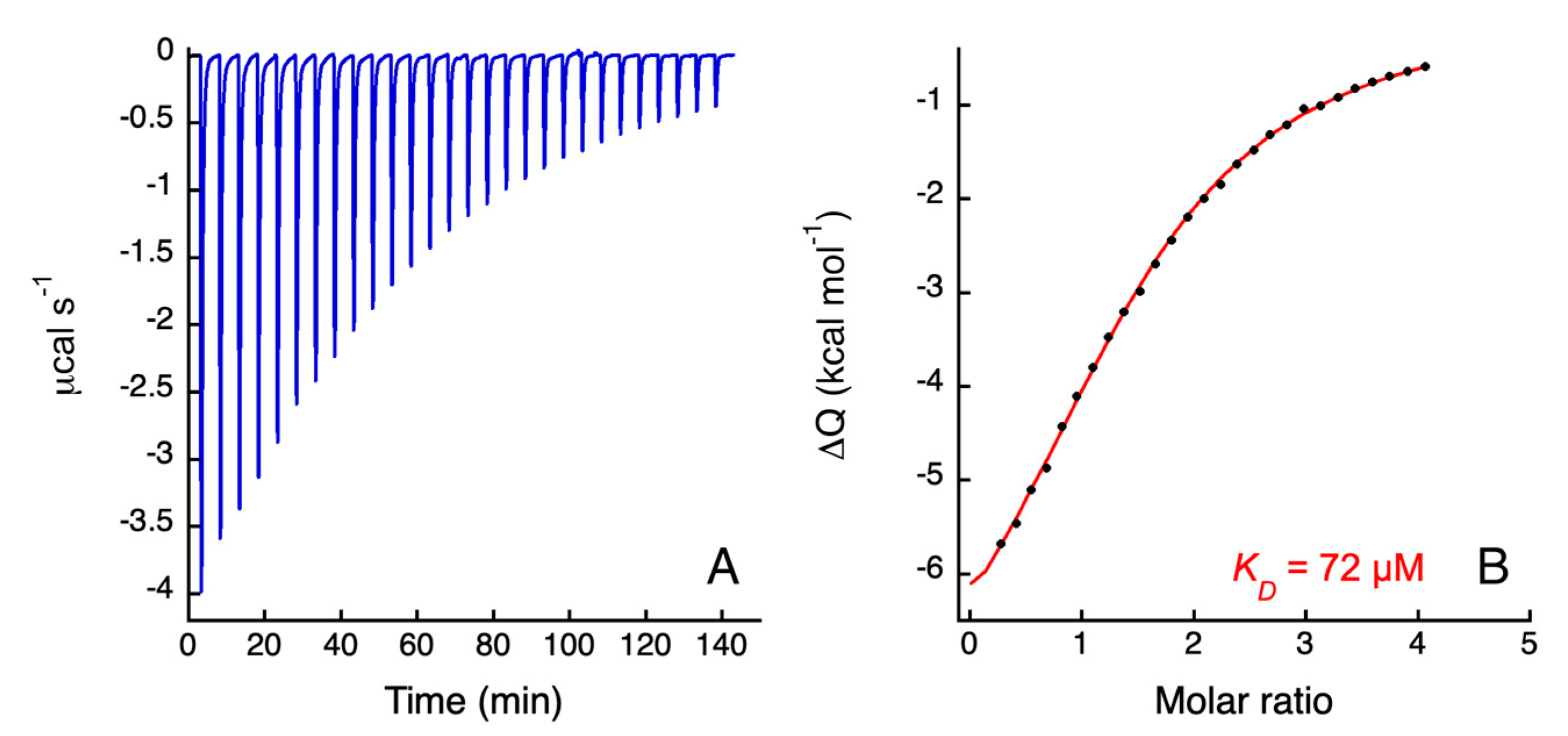
3.3. Isothermal Titration Calorimetry
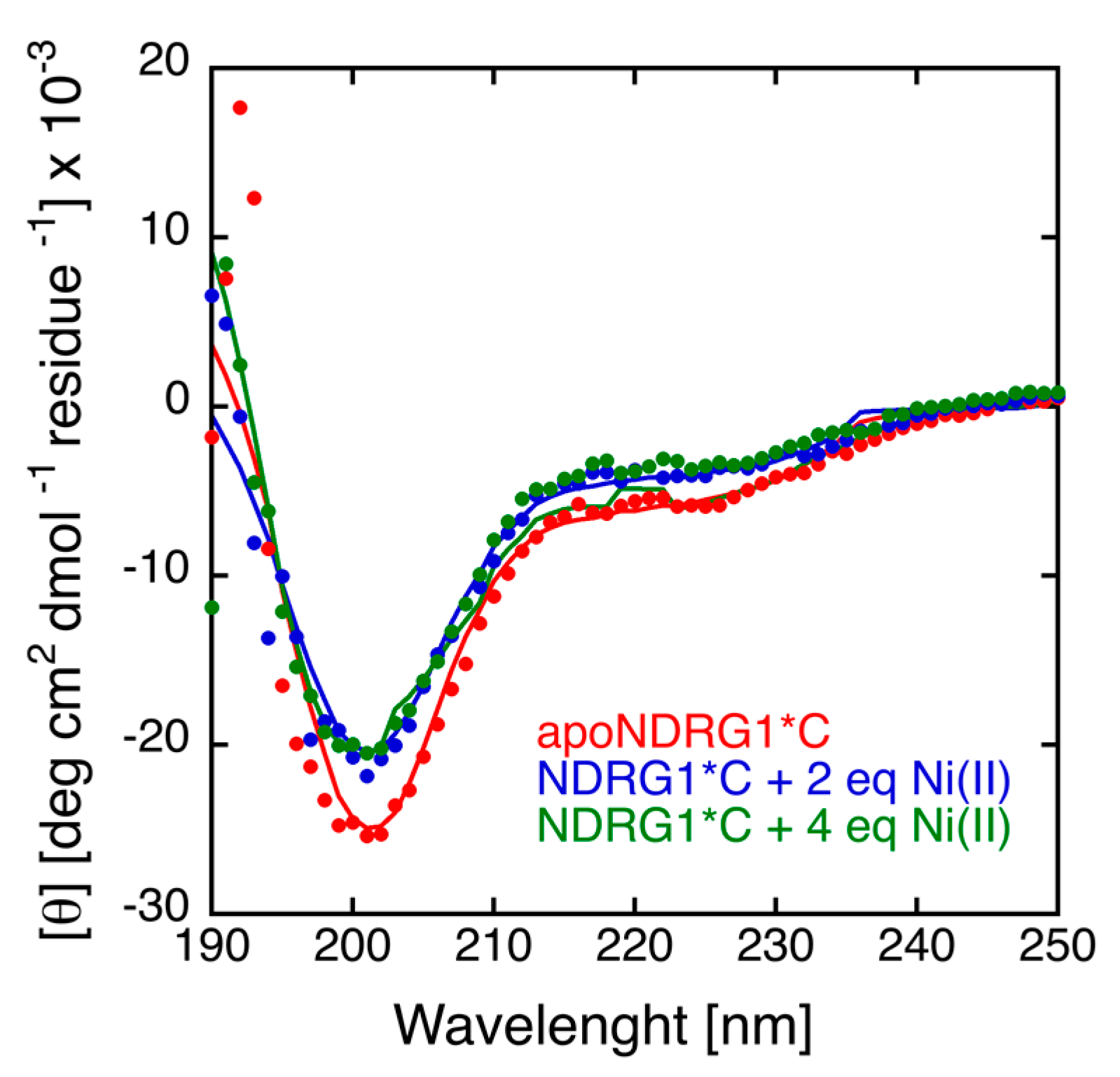
3.4. Circular Dichroism
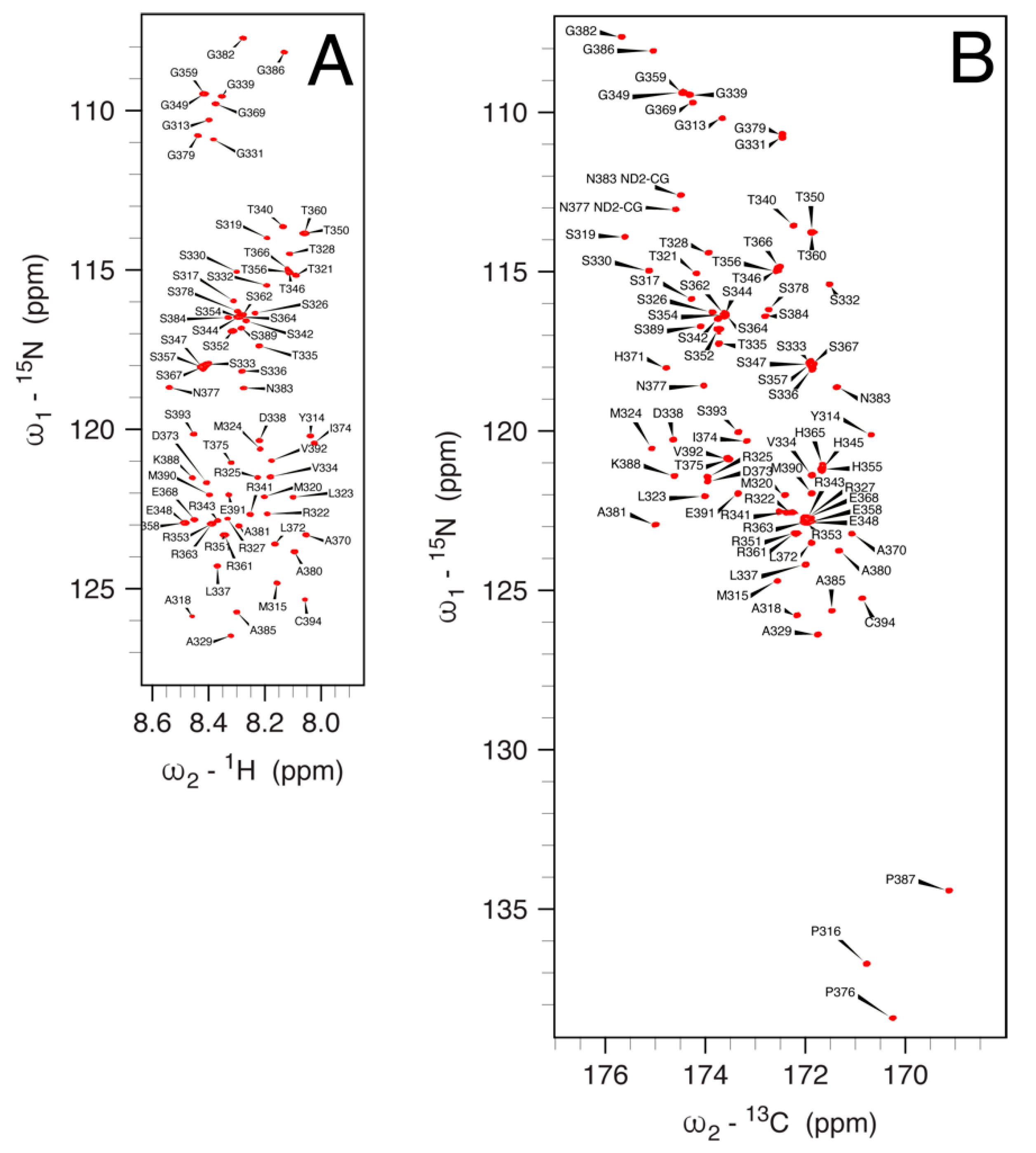
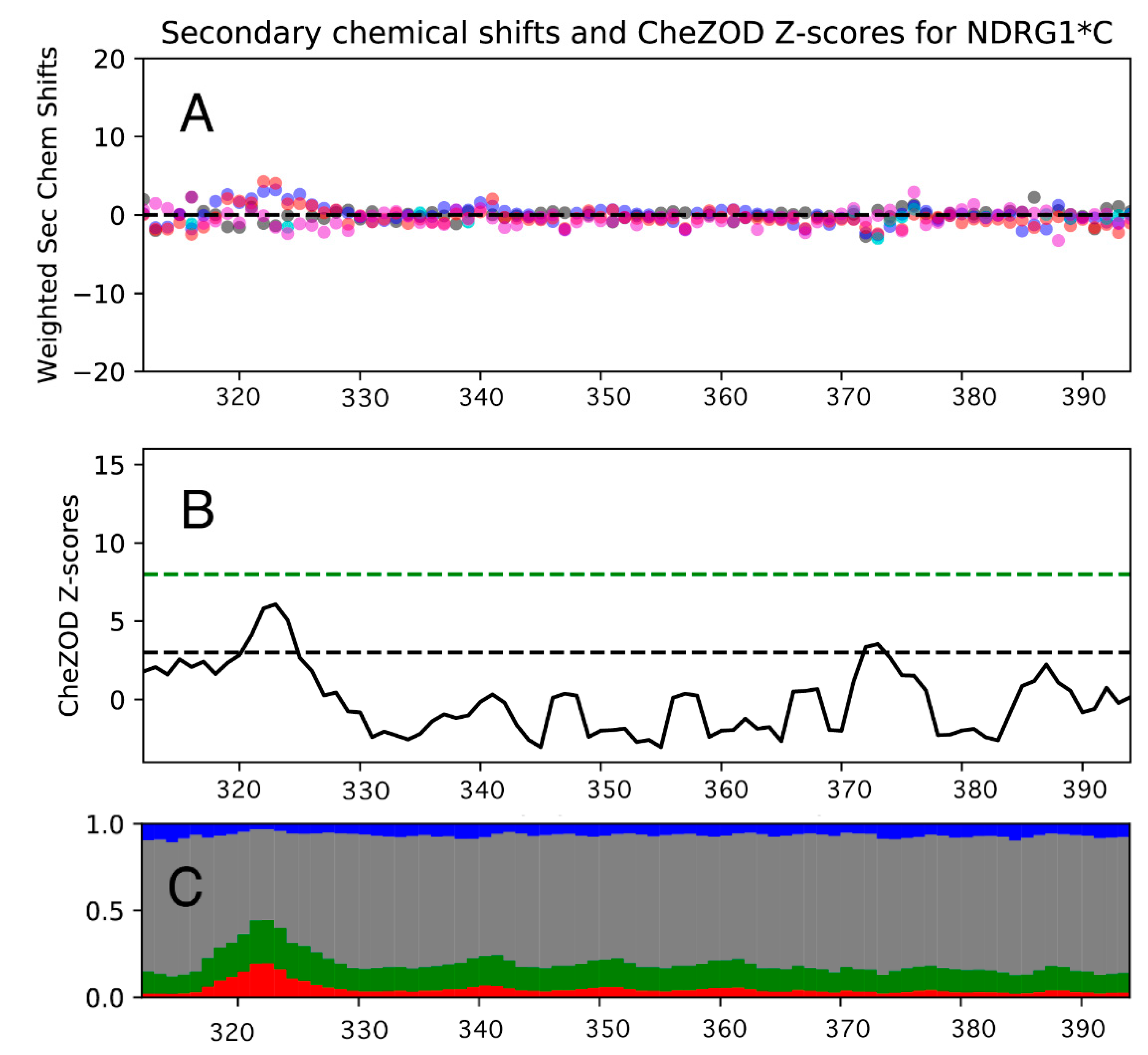
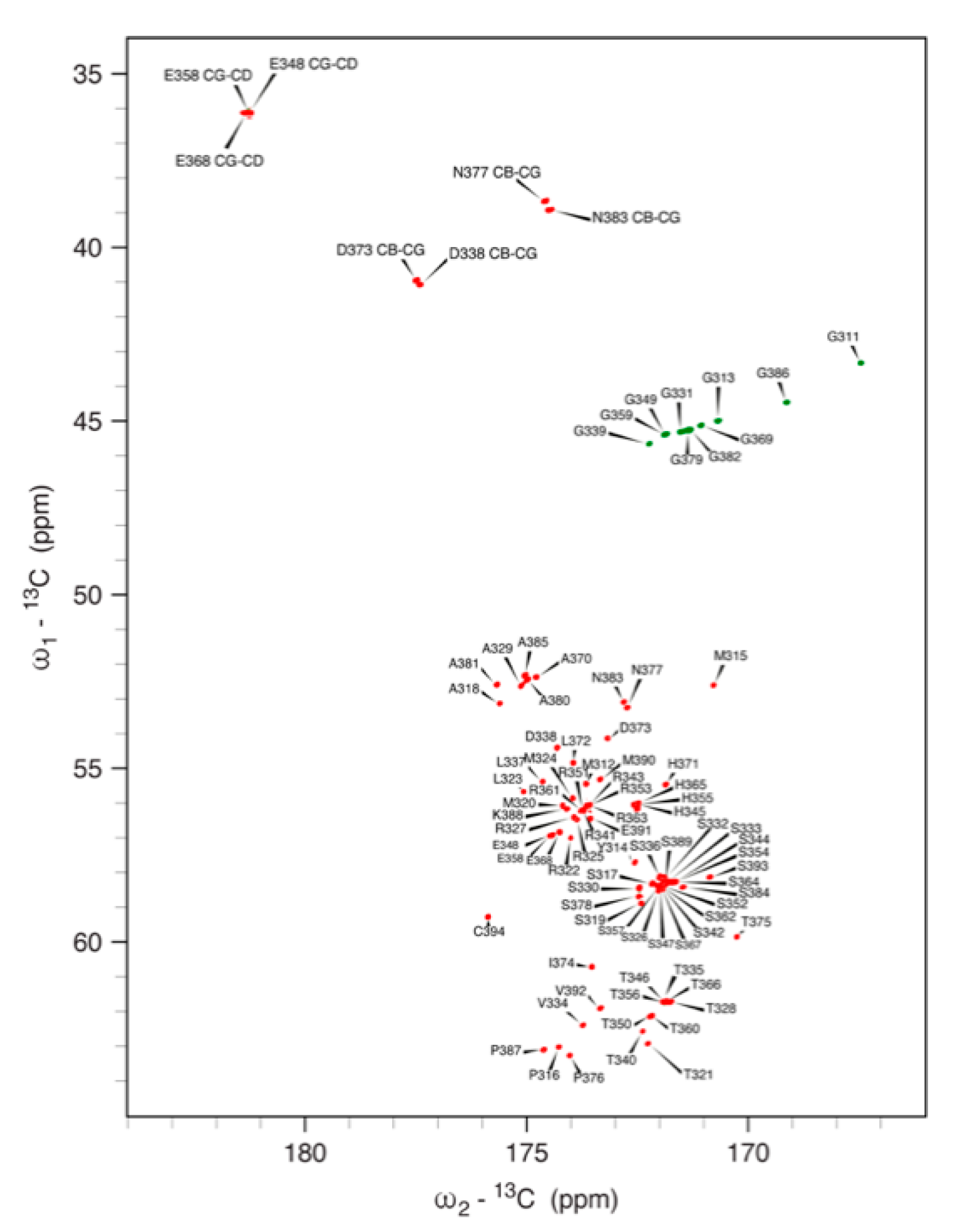
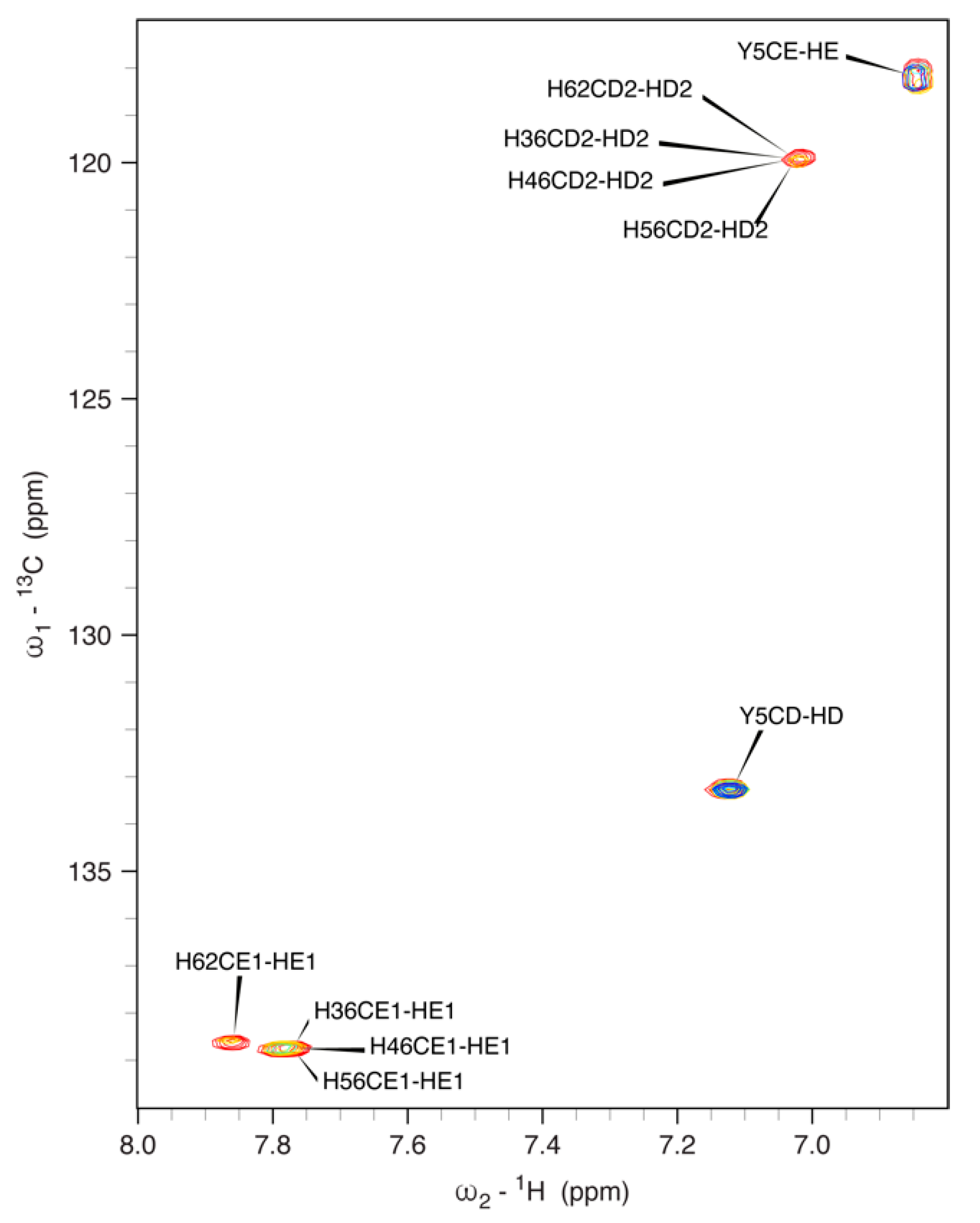
3.5. NMR Spectroscopy on hNDRG1*C
3.5.1. 1H, 13C and 15N Signal Assignment
3.5.2. Effect of pH on the NMR Spectra
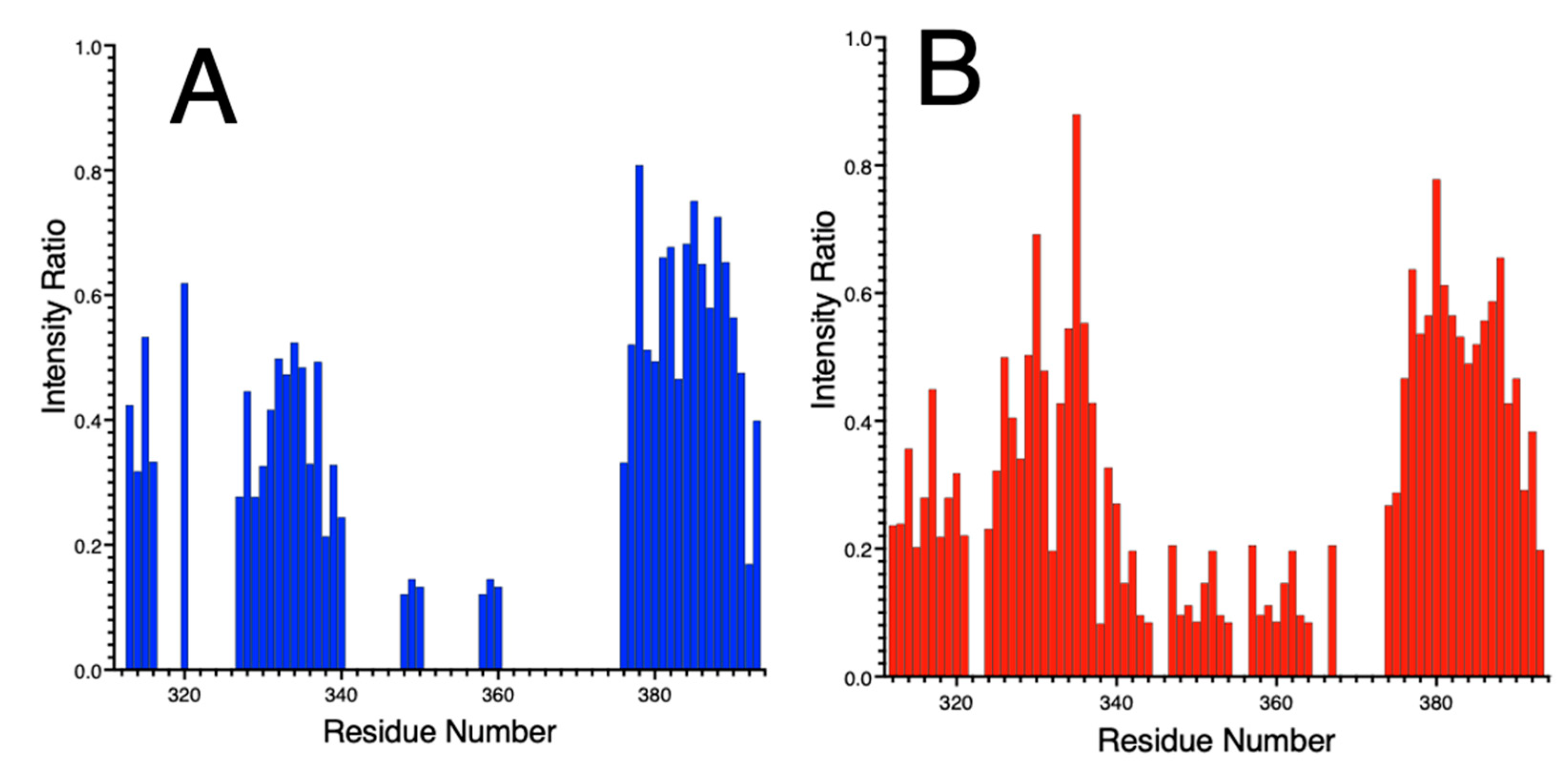
3.5.3. Ni(II) Binding to NDRG1*C by NMR Spectroscopy
3.5.4. Effects of Ni(II) Binding by Paramagnetic NMR
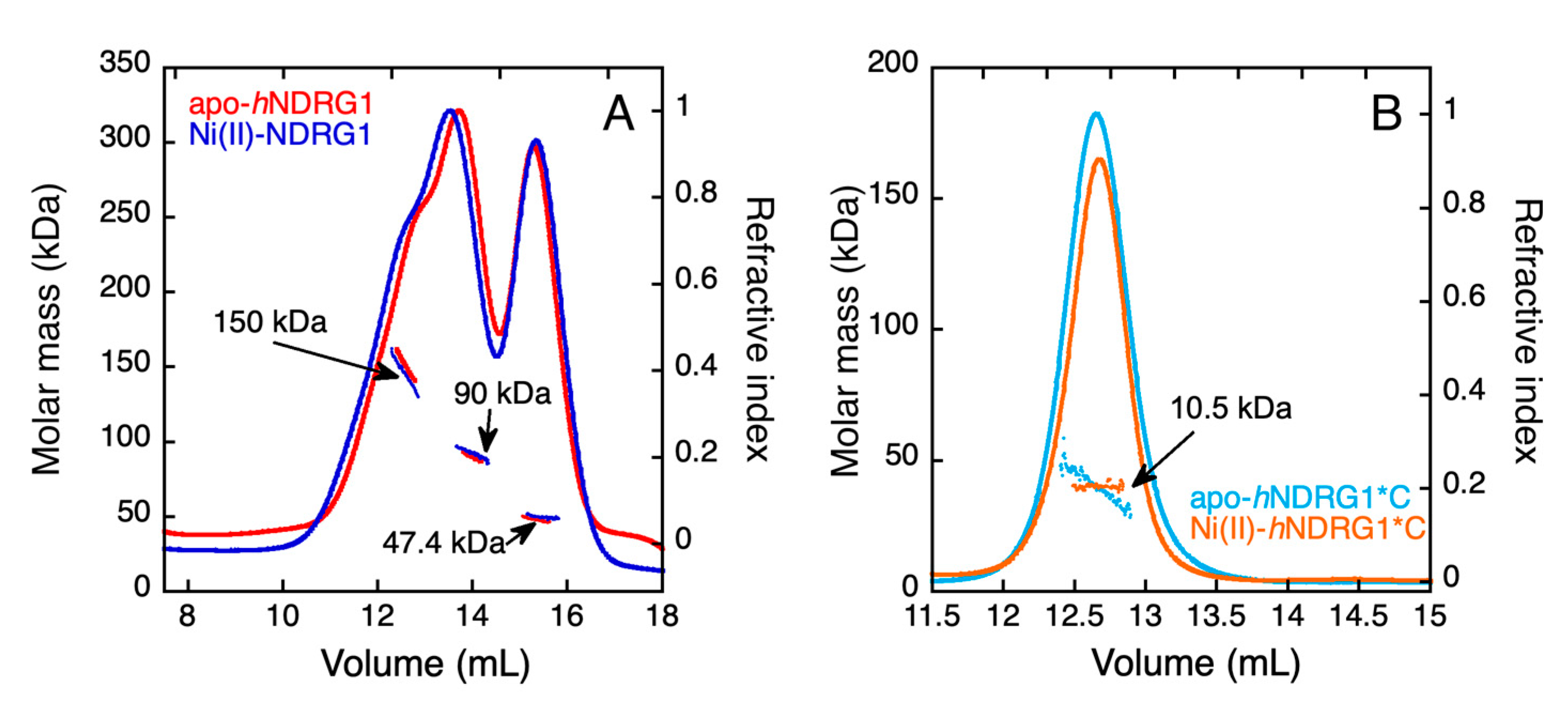
3.6. Light Scattering
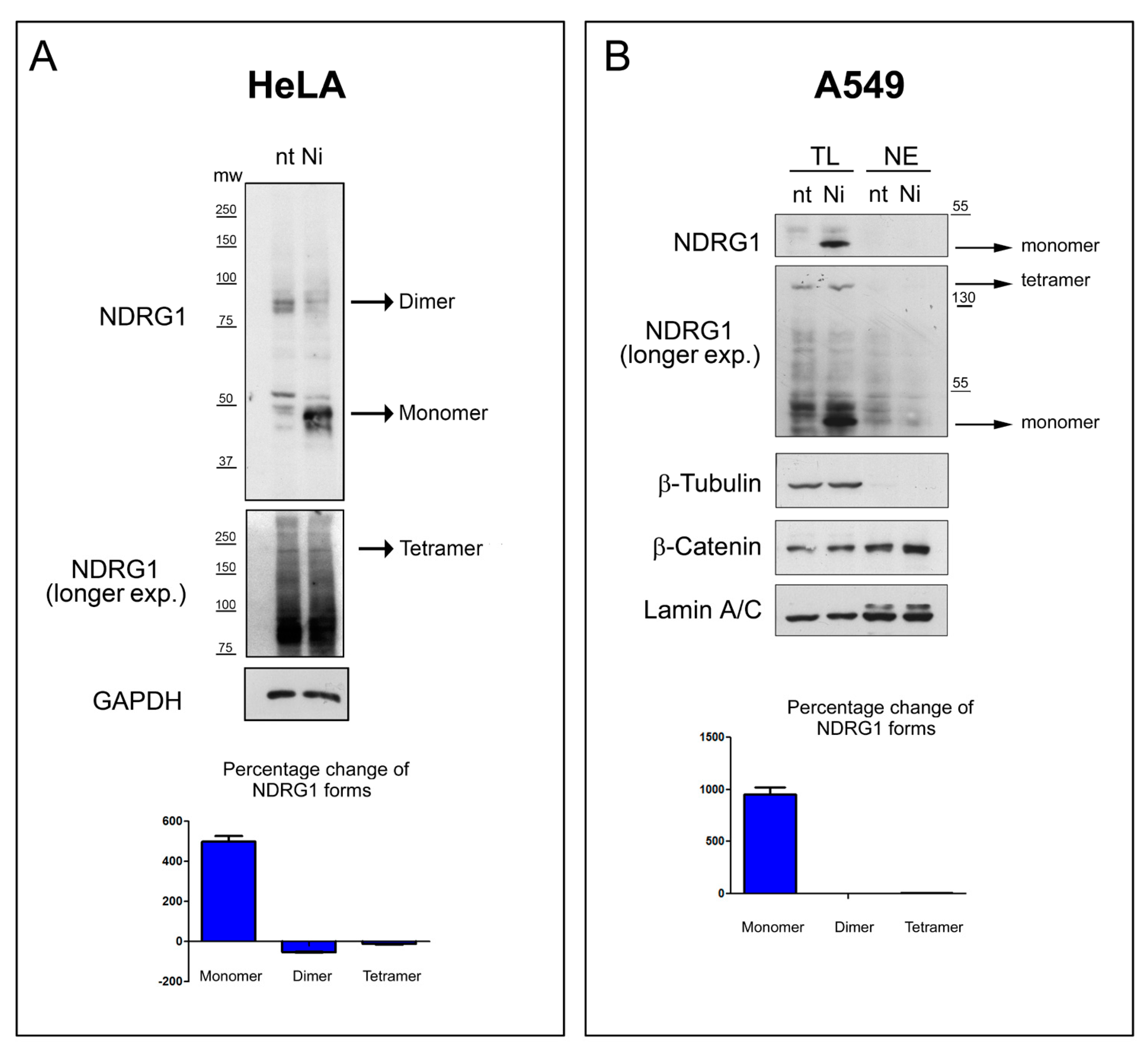
3.7. In-Cell Experiments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- IARC. Outdoor Air Pollution; International Angency for Research on Cancer: Lyon, France, 2016; Volume 109, Available online: https://publications.iarc.fr/Book-And-Report-Series/Iarc-Monographs-On-The-Identification-Of-Carcinogenic-Hazards-To-Humans/Outdoor-Air-Pollution-2015 (accessed on 31 August 2022).
- IARC. Nickel and nickel compounds. In Arsenic, Metals, Fibres, and Dusts; IARC: Lyon, France, 2012; Volume 100C. [Google Scholar]
- Zambelli, B.; Uversky, V.N.; Ciurli, S. Nickel impact on human health: An intrinsic disorder perspective. Biochim. Biophys. Acta 2016, 1864, 1714–1731. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Y.; Brocato, J.; Laulicht, F.; Costa, M. Mechanisms of Nickel Carcinogenesis. Molec. Integr. Toxicol. 2017, 15, 181–197. [Google Scholar] [CrossRef]
- Maxwell, P.; Salnikow, K. HIF-1: An oxygen and metal responsive transcription factor. Cancer Biol. Ther. 2004, 3, 29–35. [Google Scholar] [CrossRef]
- Noguchi, C.T. Is there something fishy about EPO? Blood 2004, 104, 1238. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]
- Wang, Q.; Li, L.H.; Gao, G.D.; Wang, G.; Qu, L.; Li, J.G.; Wang, C.M. HIF-1alpha up-regulates NDRG1 expression through binding to NDRG1 promoter, leading to proliferation of lung cancer A549 cells. Mol. Biol. Rep. 2013, 40, 3723–3729. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, S.; Zhang, W.; Zhang, J.; Liu, X.; Shi, H.; Che, H.; Wang, W.; Li, F.; Yao, L. Human differentiation-related gene NDRG1 is a Myc downstream-regulated gene that is repressed by Myc on the core promoter region. Gene 2008, 417, 5–12. [Google Scholar] [CrossRef]
- Vervoorts, J.; Luscher-Firzlaff, J.; Luscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729. [Google Scholar] [CrossRef]
- Geleta, B.; Makonnen, E. N-myc downstream regulated gene (NDRG):role in cancer metastasis suppression and as drug target in cancer therapeutics. J. Cancer Sci. Ther. 2016, 8, 154–159. [Google Scholar] [CrossRef]
- Lachat, P.; Shaw, P.; Gebhard, S.; van Belzen, N.; Chaubert, P.; Bosman, F.T. Expression of NDRG1, a differentiation-related gene, in human tissues. Histochem. Cell Biol. 2002, 118, 399–408. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, D.; Bae, D.H.; Sahni, S.; Jansson, P.; Zheng, Y.; Zhao, Q.; Yue, F.; Zheng, M.; Kovacevic, Z.; et al. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis 2013, 34, 1943–1954. [Google Scholar] [CrossRef]
- Fang, B.A.; Kovacevic, Z.; Park, K.C.; Kalinowski, D.S.; Jansson, P.J.; Lane, D.J.; Sahni, S.; Richardson, D.R. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim. Biophys. Acta 2014, 1845, 1–19. [Google Scholar] [CrossRef]
- Said, H.M.; Safari, R.; Al-Kafaji, G.; Ernestus, R.I.; Lohr, M.; Katzer, A.; Flentje, M.; Hagemann, C. Time- and oxygen-dependent expression and regulation of NDRG1 in human brain cancer cells. Oncol. Rep. 2017, 37, 3625–3634. [Google Scholar] [CrossRef]
- Ring, B.Z.; Seitz, R.S.; Beck, R.; Shasteen, W.J.; Tarr, S.M.; Cheang, M.C.; Yoder, B.J.; Budd, G.T.; Nielsen, T.O.; Hicks, D.G.; et al. Novel prognostic immunohistochemical biomarker panel for estrogen receptor-positive breast cancer. J. Clin. Oncol. 2006, 24, 3039–3047. [Google Scholar] [CrossRef]
- Strzelczyk, B.; Szulc, A.; Rzepko, R.; Kitowska, A.; Skokowski, J.; Szutowicz, A.; Pawelczyk, T. Identification of high-risk stage II colorectal tumors by combined analysis of the NDRG1 gene expression and the depth of tumor invasion. Ann. Surg Oncol. 2009, 16, 1287–1294. [Google Scholar] [CrossRef]
- Sun, B.; Chu, D.; Li, W.; Chu, X.; Li, Y.; Wei, D.; Li, H. Decreased expression of NDRG1 in glioma is related to tumor progression and survival of patients. J. Neurooncol. 2009, 94, 213–219. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, D.; Yue, F.; Zheng, M.; Kovacevic, Z.; Richardson, D.R. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J. Biol. Chem. 2012, 287, 17016–17028. [Google Scholar] [CrossRef]
- Liu, W.; Xing, F.; Iiizumi-Gairani, M.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Kobayashi, A.; Mo, Y.Y.; et al. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol. Med. 2012, 4, 93–108. [Google Scholar] [CrossRef]
- Nishio, S.; Ushijima, K.; Tsuda, N.; Takemoto, S.; Kawano, K.; Yamaguchi, T.; Nishida, N.; Kakuma, T.; Tsuda, H.; Kasamatsu, T.; et al. Cap43/NDRG1/Drg-1 is a molecular target for angiogenesis and a prognostic indicator in cervical adenocarcinoma. Cancer Lett. 2008, 264, 36–43. [Google Scholar] [CrossRef]
- Chua, M.S.; Sun, H.; Cheung, S.T.; Mason, V.; Higgins, J.; Ross, D.T.; Fan, S.T.; So, S. Overexpression of NDRG1 is an indicator of poor prognosis in hepatocellular carcinoma. Mod. Pathol. 2007, 20, 76–83. [Google Scholar] [CrossRef]
- Masuda, K.; Ono, M.; Okamoto, M.; Morikawa, W.; Otsubo, M.; Migita, T.; Tsuneyoshi, M.; Okuda, H.; Shuin, T.; Naito, S.; et al. Downregulation of Cap43 gene by von Hippel-Lindau tumor suppressor protein in human renal cancer cells. Int. J. Cancer 2003, 105, 803–810. [Google Scholar] [CrossRef]
- Azuma, K.; Kawahara, A.; Hattori, S.; Taira, T.; Tsurutani, J.; Watari, K.; Shibata, T.; Murakami, Y.; Takamori, S.; Ono, M.; et al. NDRG1/Cap43/Drg-1 may predict tumor angiogenesis and poor outcome in patients with lung cancer. J. Thorac. Oncol. 2012, 7, 779–789. [Google Scholar] [CrossRef]
- Dai, T.; Dai, Y.; Murata, Y.; Husni, R.E.; Nakano, N.; Sakashita, S.; Noguchi, M. The prognostic significance of N-myc downregulated gene 1 in lung adenocarcinoma. Pathol. Int. 2018, 68, 224–231. [Google Scholar] [CrossRef]
- Wang, H.; Li, W.; Xu, J.; Zhang, T.; Zuo, D.; Zhou, Z.; Lin, B.; Wang, G.; Wang, Z.; Sun, W.; et al. NDRG1 inhibition sensitizes osteosarcoma cells to combretastatin A-4 through targeting autophagy. Cell Death Dis. 2017, 8, e3048. [Google Scholar] [CrossRef]
- Bae, D.H.; Jansson, P.J.; Huang, M.L.; Kovacevic, Z.; Kalinowski, D.; Lee, C.S.; Sahni, S.; Richardson, D.R. The role of NDRG1 in the pathology and potential treatment of human cancers. J. Clin. Pathol. 2013, 66, 911–917. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Kowalik-Jankowska, T.; Kozlowski, H.; Salnikow, K.; Costa, M. Ni(II) and Cu(II) binding with a 14-aminoacid sequence of Cap43 protein, TRSRSHTSEGTRSR. J. Inorg. Biochem. 2001, 84, 47–54. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Peana, M.; Medici, S.; Anedda, R. An NMR study on nickel binding sites in Cap43 protein fragments. Dalton. Trans. 2009, 28, 5523–5534. [Google Scholar] [CrossRef]
- Shimono, A.; Okuda, T.; Kondoh, H. N-myc-dependent repression of ndr1, a gene identified by direct subtraction of whole mouse embryo cDNAs between wild type and N-myc mutant. Mech. Dev. 1999, 83, 39–52. [Google Scholar] [CrossRef]
- Shaw, E.; McCue, L.A.; Lawrence, C.E.; Dordick, J.S. Identification of a novel class in the alpha/beta hydrolase fold superfamily: The N-myc differentiation-related proteins. Proteins 2 2002, 47, 163–168. [Google Scholar] [CrossRef]
- Hwang, J.; Kim, Y.; Kang, H.B.; Jaroszewski, L.; Deacon, A.M.; Lee, H.; Choi, W.C.; Kim, K.J.; Kim, C.H.; Kang, B.S.; et al. Crystal structure of the human N-Myc downstream-regulated gene 2 protein provides insight into its role as a tumor suppressor. J. Biol. Chem. 2011, 286, 12450–12460. [Google Scholar] [CrossRef]
- Kim, K.R.; Kim, K.A.; Park, J.S.; Jang, J.Y.; Choi, Y.; Lee, H.H.; Lee, D.C.; Park, K.C.; Yeom, Y.I.; Kim, H.J.; et al. Structural and Biophysical Analyses of Human N-Myc Downstream-Regulated Gene 3 (NDRG3) Protein. Biomolecules 2020, 10, 90. [Google Scholar] [CrossRef]
- Mustonen, V.; Muruganandam, G.; Loris, R.; Kursula, P.; Ruskamo, S. Crystal and solution structure of NDRG1, a membrane-binding protein linked to myelination and tumour suppression. FEBS J. 2020, 288, 3507–3529. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, J.H. SUMO modification regulates the protein stability of NDRG1. Biochem. Biophys. Res. Commun. 2015, 459, 161–165. [Google Scholar] [CrossRef]
- Ghalayini, M.K.; Dong, Q.; Richardson, D.R.; Assinder, S.J. Proteolytic cleavage and truncation of NDRG1 in human prostate cancer cells, but not normal prostate epithelial cells. Biosci. Rep. 2013, 33, e00042. [Google Scholar] [CrossRef]
- Park, K.C.; Menezes, S.V.; Kalinowski, D.S.; Sahni, S.; Jansson, P.J.; Kovacevic, Z.; Richardson, D.R. Identification of differential phosphorylation and sub-cellular localization of the metastasis suppressor, NDRG1. Biochim. Biophys. Acta Mol. Basis. Dis. 2018, 1864, 2644–2663. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Peana, M.; Kowalik-Jankowska, T.; Kozlowski, H.; Costa, M. Nickel(II) binding to Cap43 protein fragments. J. Inorg. Biochem. 2004, 98, 931–939. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Kowalik-Jankowska, T.; Medici, S.; Peana, M.; Kozlowski, H. Copper(II) binding to Cap43 protein fragments. Dalton Trans. 2008, 6127–6134. [Google Scholar] [CrossRef]
- Sugiki, T.; Murakami, M.; Taketomi, Y.; Kikuchi-Yanoshita, R.; Kudo, I. N-myc downregulated gene 1 is a phosphorylated protein in mast cells. Biol. Pharm. Bull. 2004, 27, 624–627. [Google Scholar] [CrossRef]
- Murray, J.T.; Campbell, D.G.; Morrice, N.; Auld, G.C.; Shpiro, N.; Marquez, R.; Peggie, M.; Bain, J.; Bloomberg, G.B.; Grahammer, F.; et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem. J. 2004, 384, 477–488. [Google Scholar] [CrossRef] [Green Version]
- McCaig, C.; Potter, L.; Abramczyk, O.; Murray, J.T. Phosphorylation of NDRG1 is temporally and spatially controlled during the cell cycle. Biochem. Biophys. Res. Commun. 2011, 411, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Banz, V.M.; Medova, M.; Keogh, A.; Furer, C.; Zimmer, Y.; Candinas, D.; Stroka, D. Hsp90 transcriptionally and post-translationally regulates the expression of NDRG1 and maintains the stability of its modifying kinase GSK3beta. Biochim. Biophys. Acta 2009, 1793, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Hosoi, F.; Izumi, H.; Maruyama, Y.; Ureshino, H.; Watari, K.; Kohno, K.; Kuwano, M.; Ono, M. Identification of sites subjected to serine/threonine phosphorylation by SGK1 affecting N-myc downstream-regulated gene 1 (NDRG1)/Cap43-dependent suppression of angiogenic CXC chemokine expression in human pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 396, 376–381. [Google Scholar] [CrossRef]
- You, G.R.; Chang, J.T.; Li, H.F.; Cheng, A.J. Multifaceted and Intricate Oncogenic Mechanisms of NDRG1 in Head and Neck Cancer Depend on Its C-Terminal 3R-Motif. Cells 2022, 11, 1581. [Google Scholar] [CrossRef]
- Miraula, M.; Ciurli, S.; Zambelli, B. Intrinsic disorder and metal binding in UreG proteins from Archae hyperthermophiles: GTPase enzymes involved in the activation of Ni(II) dependent urease. J. Biol. Inorg. Chem. 2015, 20, 739–755. [Google Scholar] [CrossRef]
- Bogomolovas, J.; Simon, B.; Sattler, M.; Stier, G. Screening of fusion partners for high yield expression and purification of bioactive viscotoxins. Protein Expr. Purif. 2009, 64, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Azatian, S.B.; Kaur, N.; Latham, M.P. Increasing the buffering capacity of minimal media leads to higher protein yield. J. Biomol. NMR 2019, 73, 11–17. [Google Scholar] [CrossRef]
- Marley, J.; Lu, M.; Bracken, C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 2001, 20, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Stola, M.; Musiani, F.; Mangani, S.; Turano, P.; Safarov, N.; Zambelli, B.; Ciurli, S. The nickel site of Bacillus pasteurii UreE, a urease metallo-chaperone, as revealed by metal-binding studies and X-ray absorption spectroscopy. Biochemistry 2006, 45, 6495–6509. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyaki, E.; Kun, J.; Moussong, E.; Lee, Y.H.; Goto, Y.; Refregiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Bermel, W.; Bertini, I.; Csizmok, V.; Felli, I.C.; Pierattelli, R.; Tompa, P. H-start for exclusively heteronuclear NMR spectroscopy: The case of intrinsically disordered proteins. J. Magn. Reson. 2009, 198, 275–281. [Google Scholar] [CrossRef]
- Bermel, W.; Bertini, I.; Felli, I.C.; Pierattelli, R. Speeding up (13)C direct detection biomolecular NMR spectroscopy. J. Am. Chem. Soc. 2009, 131, 15339–15345. [Google Scholar] [CrossRef]
- Felli, I.C.; Pierattelli, R. Novel methods based on (13)C detection to study intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Delaglio, F.; Torchia, D.A.; Bax, A. Sparse multidimensional iterative lineshape-enhanced (SMILE) reconstruction of both non-uniformly sampled and conventional NMR data. J. Biomol. NMR 2017, 68, 101–118. [Google Scholar] [CrossRef]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef]
- Lee, W.; Rahimi, M.; Lee, Y.; Chiu, A. POKY: A software suite for multidimensional NMR and 3D structure calculation of biomolecules. Bioinformatics 2021, 37, 3041–3042. [Google Scholar] [CrossRef]
- Lee, W.; Westler, W.M.; Bahrami, A.; Eghbalnia, H.R.; Markley, J.L. PINE-SPARKY: Graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics 2009, 25, 2085–2087. [Google Scholar] [CrossRef]
- Bahrami, A.; Assadi, A.H.; Markley, J.L.; Eghbalnia, H.R. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput. Biol. 2009, 5, e1000307. [Google Scholar] [CrossRef]
- Wang, L.; Eghbalnia, H.R.; Bahrami, A.; Markley, J.L. Linear analysis of carbon-13 chemical shift differences and its application to the detection and correction of errors in referencing and spin system identifications. J. Biomol. NMR 2005, 32, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Piccioli, M. Cobalt (II)- and Nickel (II)-Substituted Proteins. In Encyclopedia of Magnetic Resonance; John Wiley & Sons, Ltd.: New York, NY, USA, 1996; pp. 1365–1378. [Google Scholar]
- Charlwood, P.A. Partial Specific Volumes of Proteins in Relation to Composition and Environment. J. Am. Chem. Soc. 1957, 79, 776–781. [Google Scholar] [CrossRef]
- Oates, M.E.; Romero, P.; Ishida, T.; Ghalwash, M.; Mizianty, M.J.; Xue, B.; Dosztanyi, Z.; Uversky, V.N.; Obradovic, Z.; Kurgan, L.; et al. D(2)P(2): Database of disordered protein predictions. Nucleic Acids Res. 2013, 41, D508–D516. [Google Scholar] [CrossRef] [PubMed]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein. Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Schubert, M.; Labudde, D.; Oschkinat, H.; Schmieder, P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. J. Biomol. NMR 2002, 24, 149–154. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Prediction of Xaa-Pro peptide bond conformation from sequence and chemical shifts. J. Biomol. NMR 2010, 46, 199–204. [Google Scholar] [CrossRef]
- Sharma, D.; Rajarathnam, K. 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 2000, 18, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Mulder, F.A. There is Diversity in Disorder-“In all Chaos there is a Cosmos, in all Disorder a Secret Order”. Front. Mol. Biosci. 2016, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.T.; Mulder, F.A.A. CheSPI: Chemical shift secondary structure population inference. J. Biomol. NMR 2021, 75, 273–291. [Google Scholar] [CrossRef]
- Molday, R.S.; Englander, S.W.; Kallen, R.G. Primary structure effects on peptide group hydrogen exchange. Biochemistry 1972, 11, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Pelton, J.G.; Torchia, D.A.; Meadow, N.D.; Roseman, S. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 1993, 2, 543–558. [Google Scholar] [CrossRef]
- Li, S.; Hong, M. Protonation, tautomerization, and rotameric structure of histidine: A comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [Google Scholar] [CrossRef]
- Platzer, G.; Okon, M.; McIntosh, L.P. pH-dependent random coil 1H, 13C, and 15N chemical shifts of the ionizable amino acids: A guide for protein pKa measurements. J. Biomol. NMR 2014, 60, 109–129. [Google Scholar] [CrossRef]
- Dass, R.; Corliano, E.; Mulder, F.A.A. The contribut.tion of electrostatics to hydrogen exchange in the unfolded protein state. Biophys. J. 2021, 120, 4107–4114. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 16, 521–655. [Google Scholar] [CrossRef]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Connelly, G.P.; Bai, Y.; Jeng, M.F.; Englander, S.W. Isotope effects in peptide group hydrogen exchange. Proteins 1993, 17, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Spronk, C.; Zerko, S.; Gorka, M.; Kozminski, W.; Bardiaux, B.; Zambelli, B.; Musiani, F.; Piccioli, M.; Basak, P.; Blum, F.C.; et al. Structure and dynamics of Helicobacter pylori nickel-chaperone HypA: An integrated approach using NMR spectroscopy, functional assays and computational tools. J. Biol. Inorg. Chem. 2018, 23, 1309–1330. [Google Scholar] [CrossRef]
- Ming, L.-J.; Banci, L.; Luchinat, C.; Bertini, I.; Valentine, J.S. Characterization of copper-nickel and silvernickel bovine superoxide dismutase by 1H NMR spectroscopy. Inorg. Chem. 1988, 27, 4458–4463. [Google Scholar] [CrossRef]
- Donaire, A.; Salgado, J.; Moratal, J.M. Determination of the magnetic axes of cobalt(II) and nickel(II) azurins from 1H NMR data: Influence of the metal and axial ligands on the origin of magnetic anisotropy in blue copper proteins. Biochemistry 1998, 37, 8659–8673. [Google Scholar] [CrossRef]
- Salgado, J.; Kalverda, A.P.; Diederix, R.E.; Canters, G.W.; Moratal, J.M.; Lawler, A.T.; Dennison, C. Paramagnetic NMR investigations of Co(II) and Ni(II) amicyanin. J. Biol. Inorg. Chem. 1999, 4, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, B.J.; Duarte, I.C.; Macedo, A.L.; Volkman, B.F.; Nunes, S.G.; Moura, I.; Markley, J.L.; Moura, J.J. An NMR structural study of nickel-substituted rubredoxin. J. Biol. Inorg. Chem. 2010, 15, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.C.; Jagannathan, L.; Tanwar, V.S.; Zhang, X.; Zang, C.; Cuddapah, S. Nickel exposure induces persistent mesenchymal phenotype in human lung epithelial cells through epigenetic activation of ZEB1. Mol. Carcinog. 2018, 57, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Maroney, M.J.; Ciurli, S. Bioinorganic Chemistry of Nickel. Inorganics 2019, 7, 131. [Google Scholar] [CrossRef]
- Musiani, F.; Zambelli, B.; Bazzani, M.; Mazzei, L.; Ciurli, S. Nickel-responsive transcriptional regulators. Metallomics 2015, 7, 1305–1318. [Google Scholar] [CrossRef]
- Zambelli, B.; Ciurli, S. Nickel and human health. Met. Ions Life Sci. 2013, 13, 321–357. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Costa, M. Metals and molecular carcinogenesis. Carcinogenesis 2020, 41, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, H.; Wu, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Nickel Carcinogenesis Mechanism: DNA Damage. Int. J. Mol. Sci. 2019, 20, 4690. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef]
- Choudhary, S.; Lopus, M.; Hosur, R.V. Targeting disorders in unstructured and structured proteins in various diseases. Biophys. Chem. 2022, 281, 106742. [Google Scholar] [CrossRef]
- Zou, H.; Pan, T.; Gao, Y.; Chen, R.; Li, S.; Guo, J.; Tian, Z.; Xu, G.; Xu, J.; Ma, Y.; et al. Pan-cancer assessment of mutational landscape in intrinsically disordered hotspots reveals potential driver genes. Nucleic Acids Res. 2022, 50, e49. [Google Scholar] [CrossRef]
- Basak, P.; Zambelli, B.; Cabelli, D.E.; Ciurli, S.; Maroney, M.J. Pro5 is not essential for the formation of ‘Ni-hook’ in nickel superoxide dismutase. J. Inorg. Biochem. 2022, 234, 111858. [Google Scholar] [CrossRef]
- Rezuke, W.N.; Knight, J.A.; Sunderman, F.W., Jr. Reference values for nickel concentrations in human tissues and bile. Am. J. Ind. Med. 1987, 11, 419–426. [Google Scholar] [CrossRef]
- Dudek-Adamska, D.; Lech, T.; Konopka, T.; Koscielniak, P. Nickel Content in Human Internal Organs. Biol. Trace Elem. Res. 2021, 199, 2138–2144. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; La Penna, G. Metal ions and intrinsically disordered proteins and peptides: From Cu/Zn amyloid-beta to general principles. Acc. Chem. Res. 2014, 47, 2252–2259. [Google Scholar] [CrossRef]
- Pontoriero, L.; Schiavina, M.; Murrali, M.G.; Pierattelli, R.; Felli, I.C. Monitoring the Interaction of alpha-Synuclein with Calcium Ions through Exclusively Heteronuclear Nuclear Magnetic Resonance Experiments. Angew. Chem. Int. Ed. Engl. 2020, 59, 18537–18545. [Google Scholar] [CrossRef] [PubMed]
- Zoroddu, M.A.; Medici, S.; Peana, M.; Anedda, R. NMR studies of zinc binding in a multi-histidinic peptide fragment. Dalton Trans. 2010, 39, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Peana, M.; Medici, S.; Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; Zoroddu, M.A. Manganese and cobalt binding in a multi-histidinic fragment. Dalton Trans. 2013, 42, 16293–16301. [Google Scholar] [CrossRef]
- Salnikow, K.; Su, W.; Blagosklonny, M.V.; Costa, M. Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res. 2000, 60, 3375–3378. [Google Scholar] [PubMed]
- Le, N.T.; Richardson, D.R. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: A link between iron metabolism and proliferation. Blood 2004, 104, 2967–2975. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| His335 | His345 | His355 | His371 | |||||
|---|---|---|---|---|---|---|---|---|
| pH 6.5 | pH 7.5 | pH 6.5 | pH 7.5 | pH 6.5 | pH 7.5 | pH 6.5 | pH 7.5 | |
| (H)N | 121.16 | 121.89 | 121.29 | 121.89 | 121.29 | 121.78 | 118.11 | 118.43 |
| C | 175.24 | 175.69 | 175.24 | 175.69 | 175.20 | 175.60 | 174.54 | 175.14 |
| CA | 56.13 | 56.67 | 56.13 | 56.67 | 56.26 | 56.65 | 55.56 | 56.20 |
| CB | 30.14 | 31.03 | 30.14 | 31.03 | 30.07 | 30.95 | 29.42 | 30.40 |
| HD2 | 7.13 | 6.99 | 7.13 | 6.99 | 7.13 | 6.99 | 7.16 | 6.99 |
| CD2 | 119.97 | 119.86 | 119.97 | 119.86 | 119.97 | 119.86 | 120.0 | 119.86 |
| HE1 | 8.05 | 7.75 | 8.05 | 7.75 | 8.08 | 7.75 | 8.23 | 7.83 |
| CE1 | 137.97 | 138.66 | 137.97 | 138.66 | 137.90 | 138.66 | 137.5 | 138.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beniamino, Y.; Cenni, V.; Piccioli, M.; Ciurli, S.; Zambelli, B. The Ni(II)-Binding Activity of the Intrinsically Disordered Region of Human NDRG1, a Protein Involved in Cancer Development. Biomolecules 2022, 12, 1272. https://doi.org/10.3390/biom12091272
Beniamino Y, Cenni V, Piccioli M, Ciurli S, Zambelli B. The Ni(II)-Binding Activity of the Intrinsically Disordered Region of Human NDRG1, a Protein Involved in Cancer Development. Biomolecules. 2022; 12(9):1272. https://doi.org/10.3390/biom12091272
Chicago/Turabian StyleBeniamino, Ylenia, Vittoria Cenni, Mario Piccioli, Stefano Ciurli, and Barbara Zambelli. 2022. "The Ni(II)-Binding Activity of the Intrinsically Disordered Region of Human NDRG1, a Protein Involved in Cancer Development" Biomolecules 12, no. 9: 1272. https://doi.org/10.3390/biom12091272