
Research Progress on the Role of RNA m6A Modification in Glial Cells in the Regulation of Neurological Diseases



Abstract
: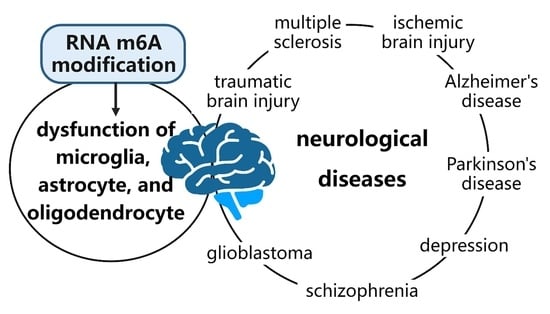
1. Introduction
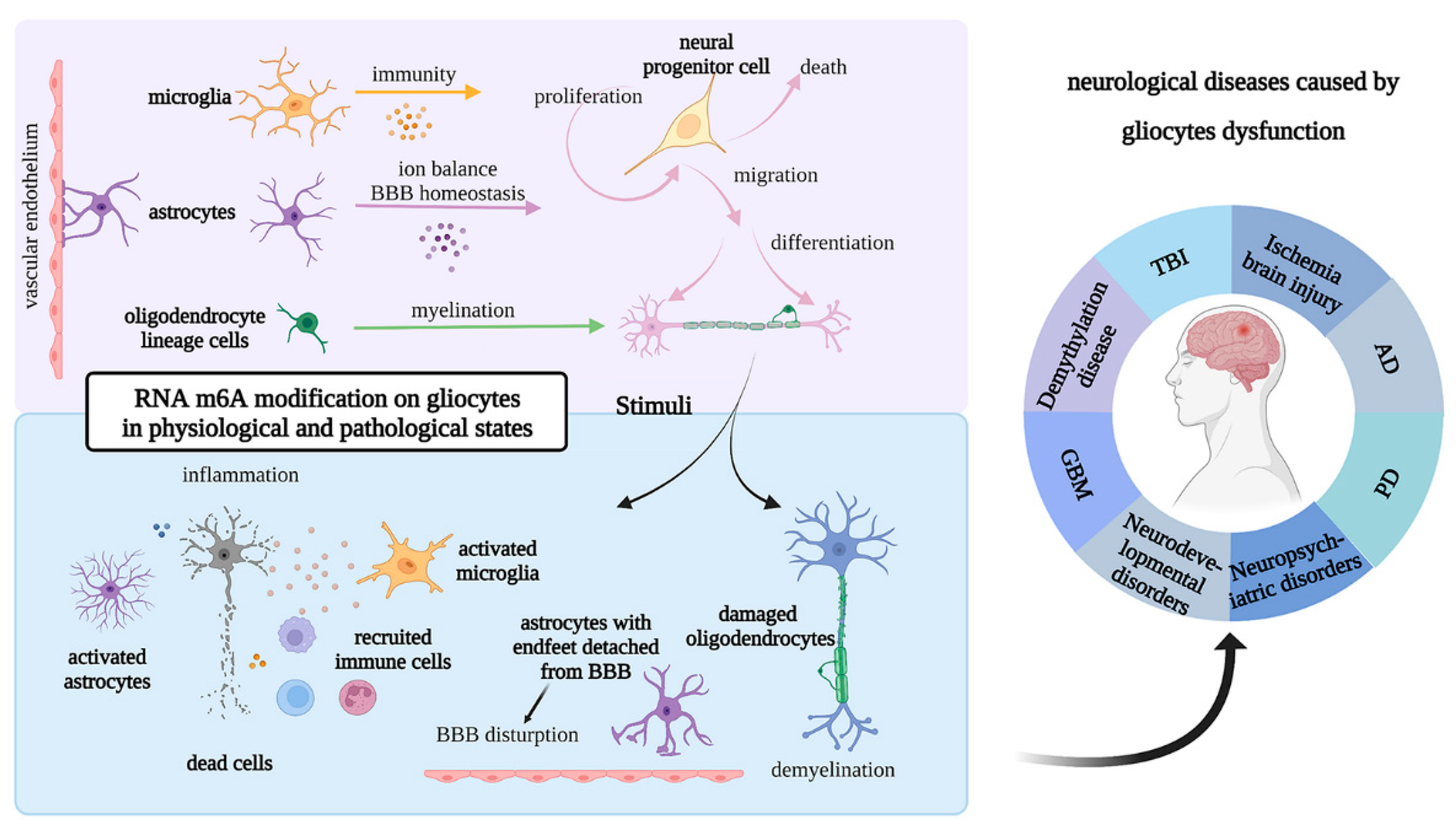
2. Roles of Glial Cells in Physiological and Pathological States
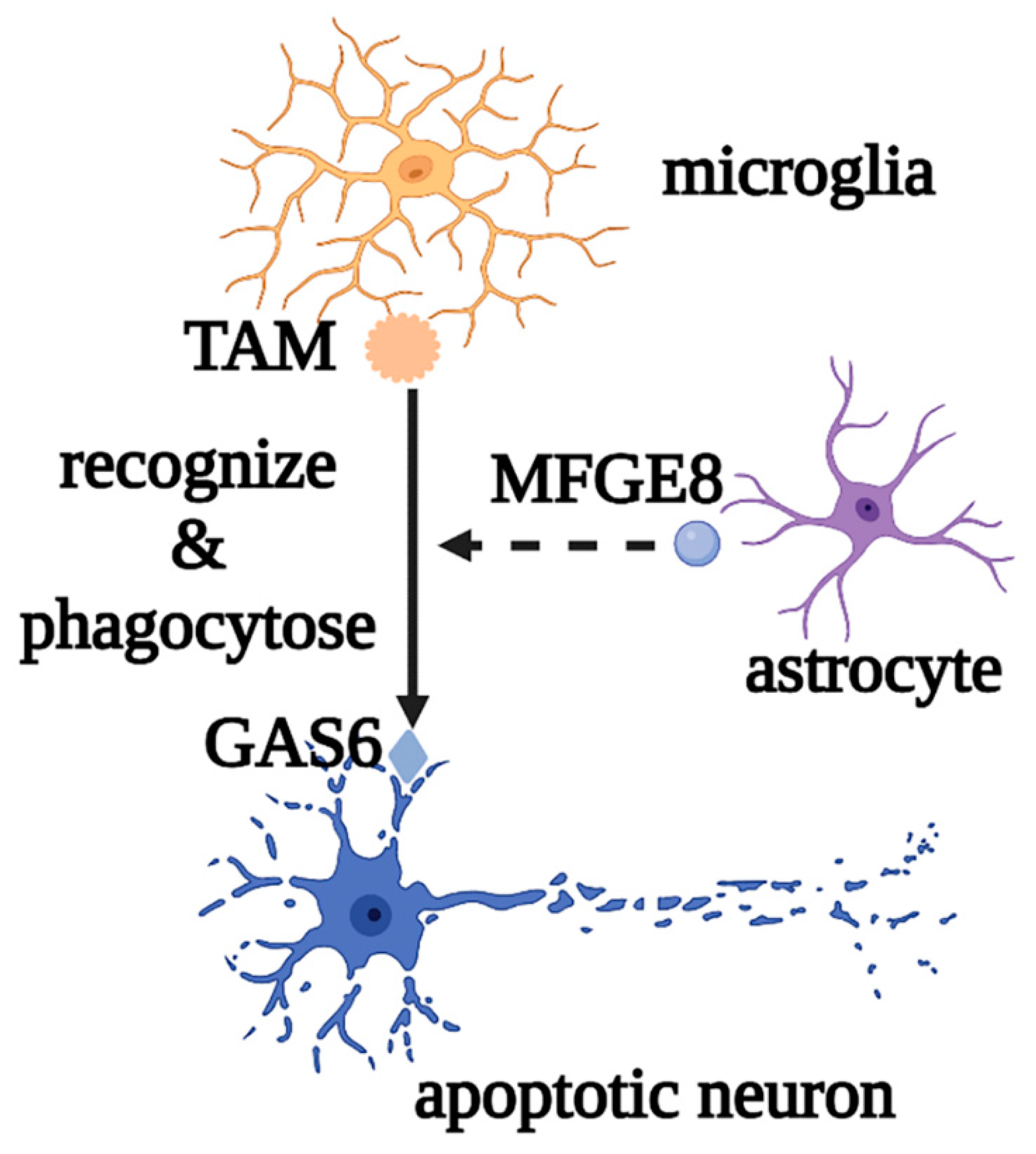
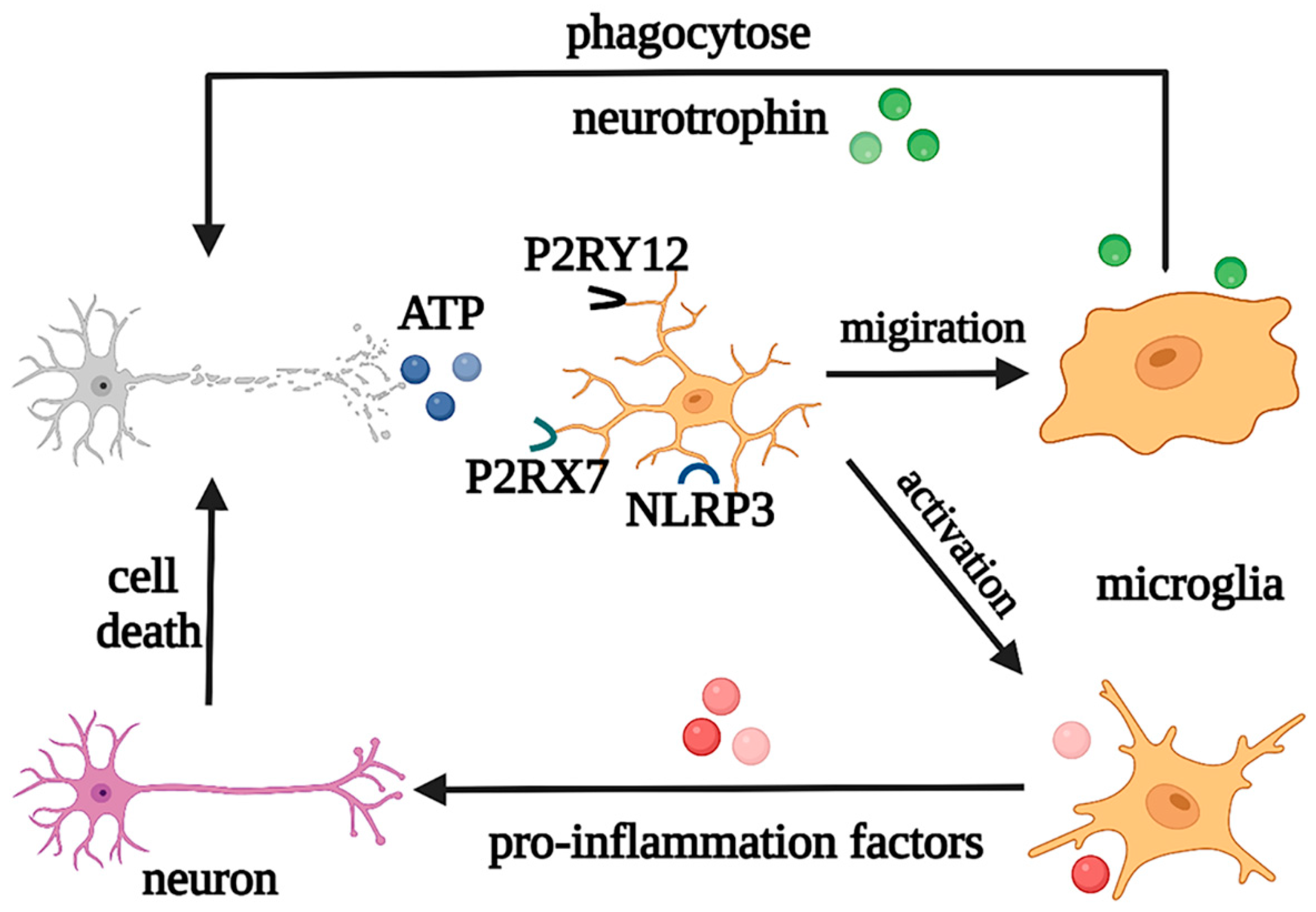
2.1. Roles of Microglia in Physiological and Pathological States
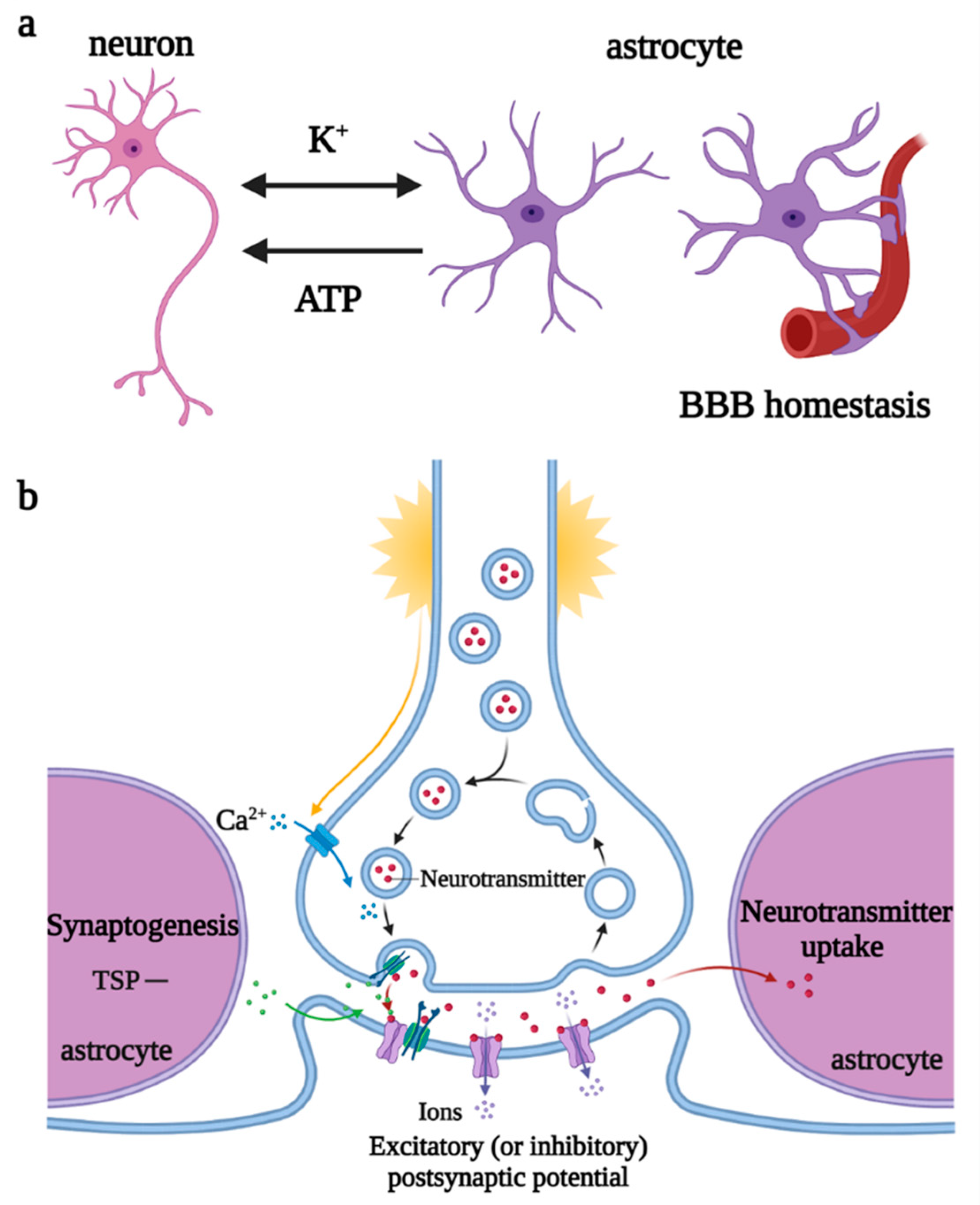
2.2. Roles of Astrocytes in Physiological and Pathological States
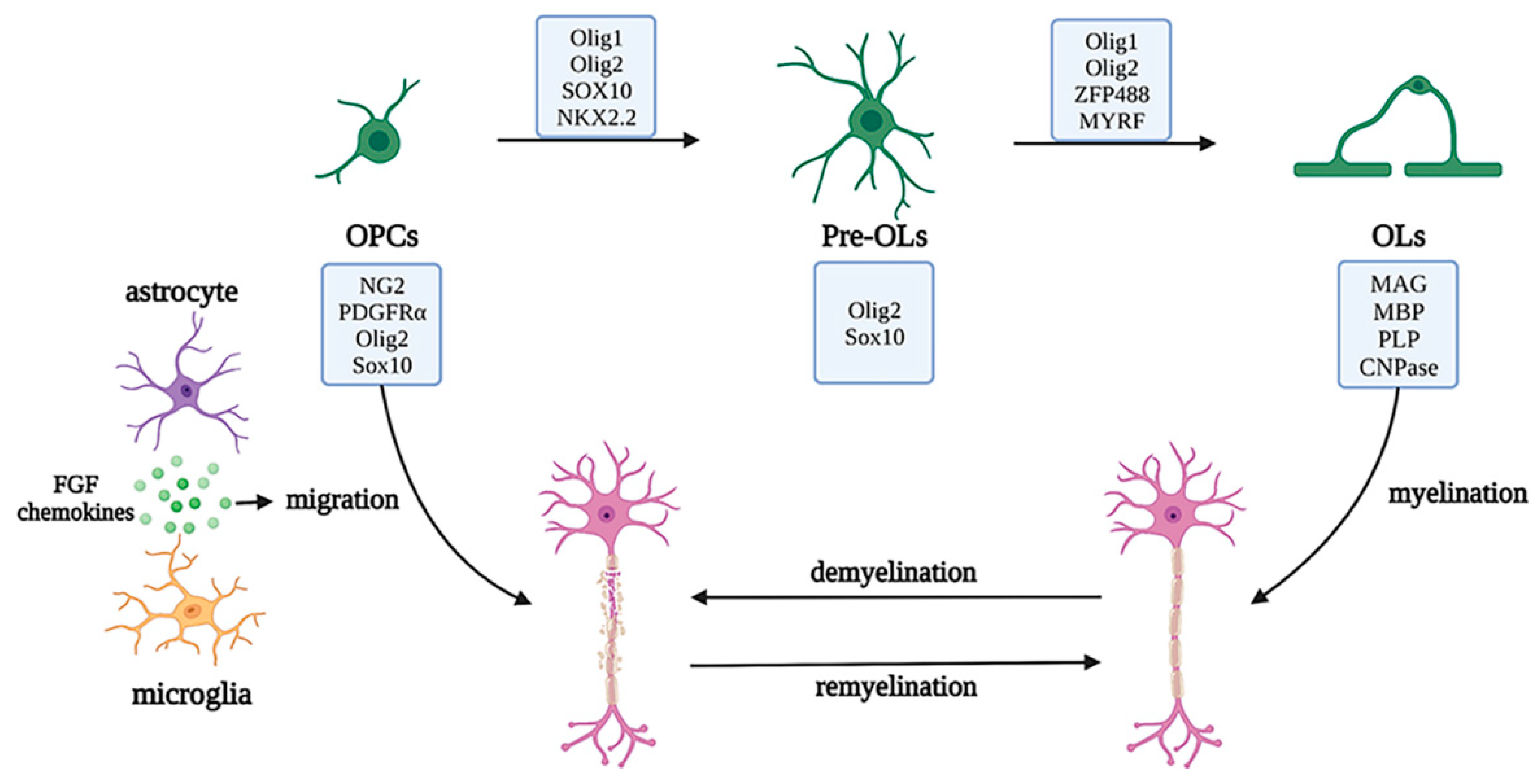
2.3. Roles of Oligodendrocytes in Physiological and Pathological States
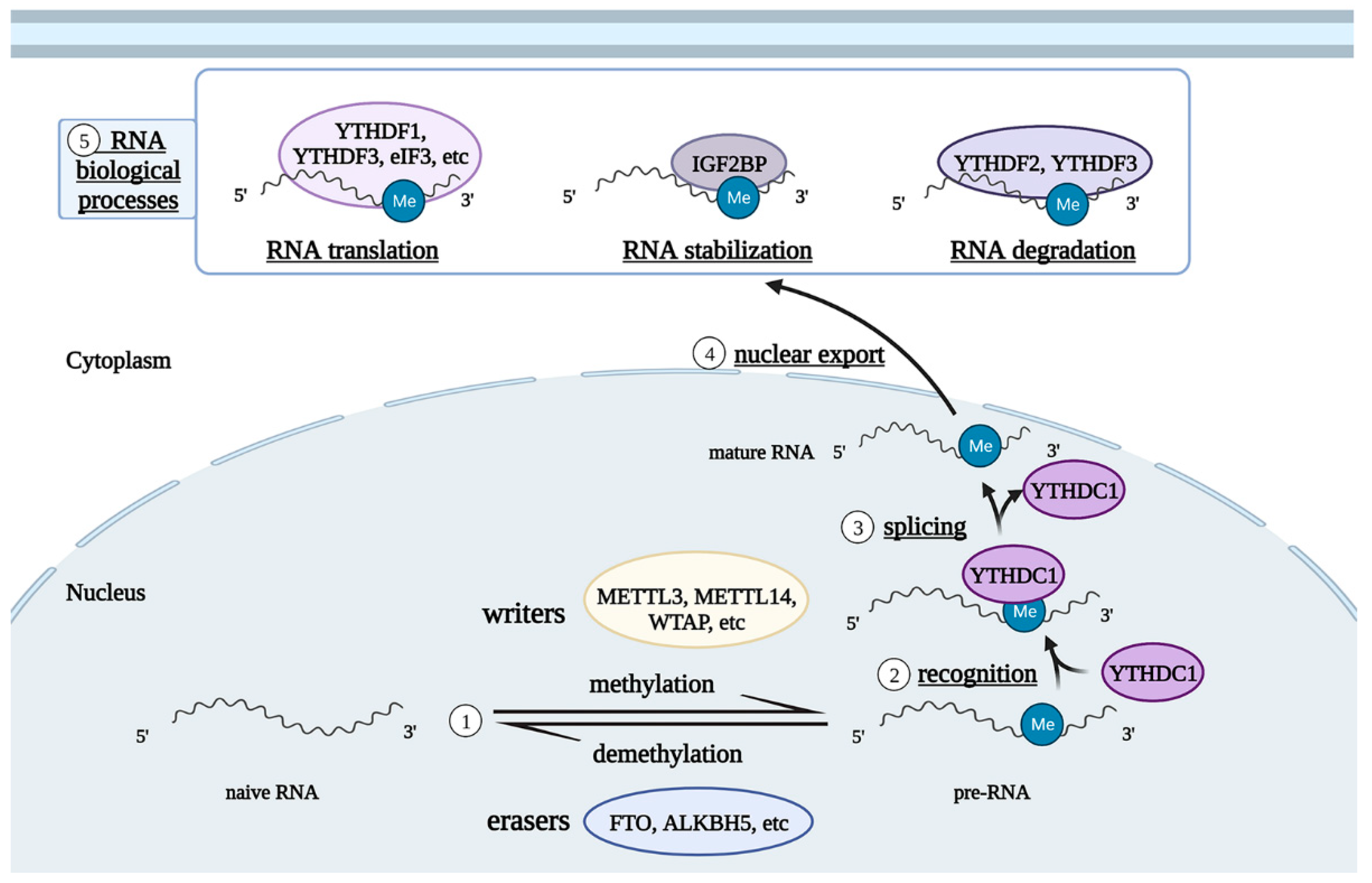
3. Mechanism of RNA m6A-Modified Glial Cells in the Occurrence and Development of Neurological Diseases
3.1. TBI Mediated by RNA m6A-Modified Glial Cells
3.2. Demyelination Diseases Mediated by RNA m6A-Modified Glial Cells
3.3. Ischemic Brain Injury Mediated by RNA m6A-Modified Glial Cells
3.4. AD Mediated by RNA m6A-Modified Glial Cells
3.5. PD Mediated by RNA m6A-Modified Glial Cells
3.6. Neuropsychiatric Disorders Mediated by RNA m6A-Modified Glial Cells
3.7. Neurodevelopmental Disorders Mediated by RNA m6A-Modified Glial Cells
3.8. GBM Mediated by RNA m6A-Modified Glial Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jessen, K.R. Glial cells. Int. J. Biochem. Cell Biol. 2004, 36, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Zuchero, J.B.; Barres, B.A. Glia in mammalian development and disease. Development 2015, 142, 3805–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Schöller, E.; Weichmann, F.; Treiber, T.; Ringle, S.; Treiber, N.; Flatley, A.; Feederle, R.; Bruckmann, A.; Meister, G. Interactions, localization, and phosphorylation of the m6A generating METTL3–METTL14–WTAP complex. RNA 2018, 24, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Bawankar, P.; Lence, T.; Paolantoni, C.; Haussmann, I.U.; Kazlauskiene, M.; Jacob, D.; Heidelberger, J.B.; Richter, F.M.; Nallasivan, M.P.; Morin, V.; et al. Hakai is required for stabilization of core components of the m(6)A mRNA methylation machinery. Nat. Commun. 2021, 12, 3778. [Google Scholar] [CrossRef]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villasenor, R.; Hess, D.; et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Y.; Zhang, J.; Zhu, J.S. The role of m(6)A RNA methylation in human cancer. Mol. Cancer 2019, 18, 103. [Google Scholar] [CrossRef] [Green Version]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m(6)A in the Transcriptome: M(6)A-Binding Proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edens, B.M.; Vissers, C.; Su, J.; Arumugam, S.; Xu, Z.; Shi, H.; Miller, N.; Rojas Ringeling, F.; Ming, G.L.; He, C.; et al. FMRP Modulates Neural Differentiation through m(6)A-Dependent mRNA Nuclear Export. Cell Rep. 2019, 28, 845–854.e5. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Ann. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Baxter, P.S.; Dando, O.; Emelianova, K.; He, X.; McKay, S.; Hardingham, G.E.; Qiu, J. Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 2021, 34, 108882. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Lemke, G. How macrophages deal with death. Nat. Rev. Immunol. 2019, 19, 539–549. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Kanmogne, M.; Klein, R.S. Neuroprotective versus Neuroinflammatory Roles of Complement: From Development to Disease. Trends Neurosci. 2021, 44, 97–109. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Zhou, R.; Ying, J.; Qiu, X.; Yu, L.; Yue, Y.; Liu, Q.; Shi, J.; Li, X.; Qu, Y.; Mu, D. A new cell death program regulated by toll-like receptor 9 through p38 mitogen-activated protein kinase signaling pathway in a neonatal rat model with sepsis associated encephalopathy. Chin. Med. J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Masuch, A.; Shieh, C.H.; van Rooijen, N.; van Calker, D.; Biber, K. Mechanism of microglia neuroprotection: Involvement of P2X7, TNFalpha, and valproic acid. Glia 2016, 64, 76–89. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Gomez Morillas, A.; Besson, V.C.; Lerouet, D. Microglia and Neuroinflammation: What Place for P2RY12? Int. J. Mol. Sci. 2021, 22, 1636. [Google Scholar] [CrossRef]
- Yue, N.; Huang, H.; Zhu, X.; Han, Q.; Wang, Y.; Li, B.; Liu, Q.; Wu, G.; Zhang, Y.; Yu, J. Activation of P2X7 receptor and NLRP3 inflammasome assembly in hippocampal glial cells mediates chronic stress-induced depressive-like behaviors. J. Neuroinflamm. 2017, 14, 102. [Google Scholar] [CrossRef] [Green Version]
- Menaceur, C.; Gosselet, F.; Fenart, L.; Saint-Pol, J. The Blood-Brain Barrier, an Evolving Concept Based on Technological Advances and Cell-Cell Communications. Cells 2021, 11, 133. [Google Scholar] [CrossRef]
- Allen, N.J. Astrocyte regulation of synaptic behavior. Ann. Rev. Cell Dev. Biol. 2014, 30, 439–463. [Google Scholar] [CrossRef]
- Lezmy, J.; Arancibia-Cárcamo, I.L.; Quintela-López, T.; Sherman, D.L.; Brophy, P.J.; Attwell, D. Astrocyte Ca(2+)-evoked ATP release regulates myelinated axon excitability and conduction speed. Science 2021, 374, eabh2858. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Huo, J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem. Int. 2021, 148, 105080. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Sanmarco, L.M.; Polonio, C.M.; Wheeler, M.A.; Quintana, F.J. Functional immune cell-astrocyte interactions. J. Exp. Med. 2021, 218, e20202715. [Google Scholar] [CrossRef]
- Jo, M.; Kim, J.H.; Song, G.J.; Seo, M.; Hwang, E.M.; Suk, K. Astrocytic Orosomucoid-2 Modulates Microglial Activation and Neuroinflammation. J. Neurosci. 2017, 37, 2878–2894. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.; Kim, J.H.; Kim, J.H.; Lee, W.H.; Lee, M.S.; Suk, K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J. Neuroinflamm. 2012, 9, 149. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Li, Z.; Zhang, G.X.; Guan, Y. Wnt signaling in remyelination in multiple sclerosis: Friend or foe? Mol. Neurobiol. 2014, 49, 1117–1125. [Google Scholar] [CrossRef]
- Clayton, B.L.L.; Tesar, P.J. Oligodendrocyte progenitor cell fate and function in development and disease. Curr. Opin. Cell Biol. 2021, 73, 35–40. [Google Scholar] [CrossRef]
- De Castro, F.; Bribian, A.; Ortega, M.C. Regulation of oligodendrocyte precursor migration during development, in adulthood and in pathology. Cell Mol. Life Sci. 2013, 70, 4355–4368. [Google Scholar] [CrossRef] [PubMed]
- Rosko, L.; Smith, V.N.; Yamazaki, R.; Huang, J.K. Oligodendrocyte Bioenergetics in Health and Disease. Neuroscientist 2019, 25, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019, 42, 263–277. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat 2015, 11, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Khellaf, A.; Khan, D.Z.; Helmy, A. Recent advances in traumatic brain injury. J. Neurol. 2019, 266, 2878–2889. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, Y.; Ma, H.; Zeng, R.; Liu, R.; Wang, P.; Jin, X.; Zhao, Y. Epitranscriptomic profiling of N6-methyladenosine-related RNA methylation in rat cerebral cortex following traumatic brain injury. Mol. Brain 2020, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mao, J.; Wang, X.; Lin, Y.; Hou, G.; Zhu, J.; Xie, B. Genome-wide screening of altered m6A-tagged transcript profiles in the hippocampus after traumatic brain injury in mice. Epigenomics 2019, 11, 805–819. [Google Scholar] [CrossRef]
- Xu, H.; Dzhashiashvili, Y.; Shah, A.; Kunjamma, R.B.; Weng, Y.L.; Elbaz, B.; Fei, Q.; Jones, J.S.; Li, Y.I.; Zhuang, X.; et al. m(6)A mRNA Methylation Is Essential for Oligodendrocyte Maturation and CNS Myelination. Neuron 2020, 105, 293–309.e295. [Google Scholar] [CrossRef]
- Maggipinto, M.; Rabiner, C.; Kidd, G.J.; Hawkins, A.J.; Smith, R.; Barbarese, E. Increased expression of the MBP mRNA binding protein HnRNP A2 during oligodendrocyte differentiation. J. Neurosci. Res. 2004, 75, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, A.; Sun, B.; Sun, J.G.; Zhang, J.; Zhang, T.; Chen, Y.; Xiao, Y.; Gao, Y.; Zhang, Q.; et al. A novel m(6)A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019, 29, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, M.; Huang, H.; Zhu, J.; Song, H.; Zhu, J.; Park, J.; Ji, S.J. Dynamic m6A modification regulates local translation of mRNA in axons. Nucleic Acids Res. 2018, 46, 1412–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Tang, X.; Lu, M.; Sun, S.; Xie, S.; Cai, J.; Zan, J. microRNA-421-3p prevents inflammatory response in cerebral ischemia/reperfusion injury through targeting m6A Reader YTHDF1 to inhibit p65 mRNA translation. Int. Immunopharmacol. 2020, 88, 106937. [Google Scholar] [CrossRef]
- Chokkalla, A.K.; Mehta, S.L.; Kim, T.; Chelluboina, B.; Kim, J.; Vemuganti, R. Transient Focal Ischemia Significantly Alters the m(6)A Epitranscriptomic Tagging of RNAs in the Brain. Stroke 2019, 50, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Mo, Y.; Li, D.; Yu, Q.; Wang, L.; Lin, F.; Kong, C.; Balelang, M.F.; Zhang, A.; Chen, S.; et al. N(6)-methyladenosine demethylases Alkbh5/Fto regulate cerebral ischemia-reperfusion injury. Ther. Adv. Chronic Dis. 2020, 11, 2040622320916024. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Zhang, Y.; Liang, M.; Du, Z.; Li, H.; Fan, C.; Zhang, H.; Jiang, Y.; Bi, X. N6-methyladenosine (m6A) modification and its clinical relevance in cognitive dysfunctions. Aging 2021, 13, 20716–20737. [Google Scholar] [CrossRef]
- Huang, H.; Camats-Perna, J.; Medeiros, R.; Anggono, V.; Widagdo, J. Altered Expression of the m6A Methyltransferase METTL3 in Alzheimer’s Disease. eNeuro 2020, 7, ENEURO.0125-20.2020. [Google Scholar] [CrossRef]
- Deng, Y.; Zhu, H.; Xiao, L.; Liu, C.; Liu, Y.L.; Gao, W. Identification of the function and mechanism of m6A reader IGF2BP2 in Alzheimer’s disease. Aging 2021, 13, 24086–24100. [Google Scholar] [CrossRef]
- Zhao, F.; Xu, Y.; Gao, S.; Qin, L.; Austria, Q.; Siedlak, S.L.; Pajdzik, K.; Dai, Q.; He, C.; Wang, W.; et al. METTL3-dependent RNA m(6)A dysregulation contributes to neurodegeneration in Alzheimer’s disease through aberrant cell cycle events. Mol. Neurodegener. 2021, 16, 70. [Google Scholar] [CrossRef]
- Cockova, Z.; Honc, O.; Telensky, P.; Olsen, M.J.; Novotny, J. Streptozotocin-Induced Astrocyte Mitochondrial Dysfunction Is Ameliorated by FTO Inhibitor MO-I-500. ACS Chem. Neurosci. 2021, 12, 3818–3828. [Google Scholar] [CrossRef]
- Li, Z.; Moniruzzaman, M.; Dastgheyb, R.M.; Yoo, S.W.; Wang, M.; Hao, H.; Liu, J.; Casaccia, P.; Nogueras-Ortiz, C.; Kapogiannis, D.; et al. Astrocytes deliver CK1 to neurons via extracellular vesicles in response to inflammation promoting the translation and amyloidogenic processing of APP. J. Extracell. Vesicles 2020, 10, e12035. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Guo, M.; Zheng, X.; Ali, S.; Huang, H.; Zhang, L.; Wang, S.; Huang, Y.; Qie, S.; et al. Down-Regulation of m6A mRNA Methylation Is Involved in Dopaminergic Neuronal Death. ACS Chem. Neurosci. 2019, 10, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Wang, S.; Li, J.; Wen, Y.; Cui, R.; Zhang, K.; Liu, Y.; Yang, X.; Zhang, L.; Xu, B.; et al. Protective role of mRNA demethylase FTO on axon guidance molecules of nigro-striatal projection system in manganese-induced parkinsonism. J. Hazard. Mater. 2022, 426, 128099. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhang, Y.; Bai, Y.; Han, B.; Ju, M.; Chen, B.; Yang, L.; Wang, Y.; Zhang, H.; Zhang, H.; et al. N(6)-Methyladenosine Modification of Fatty Acid Amide Hydrolase Messenger RNA in Circular RNA STAG1-Regulated Astrocyte Dysfunction and Depressive-like Behaviors. Biol. Psychiatry 2020, 88, 392–404. [Google Scholar] [CrossRef] [Green Version]
- Engel, M.; Eggert, C.; Kaplick, P.M.; Eder, M.; Roh, S.; Tietze, L.; Namendorf, C.; Arloth, J.; Weber, P.; Rex-Haffner, M.; et al. The Role of m(6)A/m-RNA Methylation in Stress Response Regulation. Neuron 2018, 99, 389–403.e389. [Google Scholar] [CrossRef]
- Shen, J.; Yang, L.; Wei, W. Role of Fto on CaMKII/CREB signaling pathway of hippocampus in depressive-like behaviors induced by chronic restraint stress mice. Behav. Brain Res. 2021, 406, 113227. [Google Scholar] [CrossRef]
- Liu, S.; Xiu, J.; Zhu, C.; Meng, K.; Li, C.; Han, R.; Du, T.; Li, L.; Xu, L.; Liu, R.; et al. Fat mass and obesity-associated protein regulates RNA methylation associated with depression-like behavior in mice. Nat. Commun. 2021, 12, 6937. [Google Scholar] [CrossRef]
- Wu, P.F.; Han, Q.Q.; Chen, F.F.; Shen, T.T.; Li, Y.H.; Cao, Y.; Chen, J.G.; Wang, F. Erasing m(6)A-dependent transcription signature of stress-sensitive genes triggers antidepressant actions. Neurobiol. Stress 2021, 15, 100390. [Google Scholar] [CrossRef]
- Cui, Q.; Shi, H.; Ye, P.; Li, L.; Qu, Q.; Sun, G.; Sun, G.; Lu, Z.; Huang, Y.; Yang, C.G.; et al. m(6)A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017, 18, 2622–2634. [Google Scholar] [CrossRef]
- Li, F.; Yi, Y.; Miao, Y.; Long, W.; Long, T.; Chen, S.; Cheng, W.; Zou, C.; Zheng, Y.; Wu, X.; et al. N(6)-Methyladenosine Modulates Nonsense-Mediated mRNA Decay in Human Glioblastoma. Cancer Res. 2019, 79, 5785–5798. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.; Li, X.; He, L.; Rong, X.; Wang, H.; Pan, J.; Lu, Z.; Zhang, X.; Peng, Y. Decreased RNA m(6)A methylation enhances the process of the epithelial mesenchymal transition and vasculogenic mimicry in glioblastoma. Am. J. Cancer Res. 2022, 12, 893–906. [Google Scholar] [PubMed]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Zepecki, J.P.; Karambizi, D.; Fajardo, J.E.; Snyder, K.M.; Guetta-Terrier, C.; Tang, O.Y.; Chen, J.S.; Sarkar, A.; Fiser, A.; Toms, S.A.; et al. miRNA-mediated loss of m6A increases nascent translation in glioblastoma. PLoS Genet. 2021, 17, e1009086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bogler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [Green Version]
- Kowalski-Chauvel, A.; Lacore, M.G.; Arnauduc, F.; Delmas, C.; Toulas, C.; Cohen-Jonathan-Moyal, E.; Seva, C. The m6A RNA Demethylase ALKBH5 Promotes Radioresistance and Invasion Capability of Glioma Stem Cells. Cancers 2020, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Dixit, D.; Prager, B.C.; Gimple, R.C.; Poh, H.X.; Wang, Y.; Wu, Q.; Qiu, Z.; Kidwell, R.L.; Kim, L.J.Y.; Xie, Q.; et al. The RNA m6A Reader YTHDF2 Maintains Oncogene Expression and Is a Targetable Dependency in Glioblastoma Stem Cells. Cancer Discov. 2021, 11, 480–499. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Zhou, Y.; Huang, X.; Jiang, X. Long non-coding RNA OIP5-AS1 inhibition upregulates microRNA-129-5p to repress resistance to temozolomide in glioblastoma cells via downregulating IGF2BP2. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef]
- Liu, H.; Qin, S.; Liu, C.; Jiang, L.; Li, C.; Yang, J.; Zhang, S.; Yan, Z.; Liu, X.; Yang, J.; et al. m(6)A reader IGF2BP2-stabilized CASC9 accelerates glioblastoma aerobic glycolysis by enhancing HK2 mRNA stability. Cell Death Discov. 2021, 7, 292. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, X.; Sun, Y.; Yu, J.; Sheng, M.; Yan, S.; Zhu, Q.; Lan, Q. Identification of IGF2BP3 as an Adverse Prognostic Biomarker of Gliomas. Front Genet. 2021, 12, 743738. [Google Scholar] [CrossRef]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Clemente, D.; Ortega, M.C.; Melero-Jerez, C.; de Castro, F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front. Cell. Neurosci. 2013, 7, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Wang, T.; Wu, X.; Liang, J.; Li, J.; Sheng, W. N6-Methyladenosine RNA modification in cerebrospinal fluid as a novel potential diagnostic biomarker for progressive multiple sclerosis. J. Transl. Med. 2021, 19, 316. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.; Karande, M.; Sarkar, A.; Sahu, S.; Sarmah, D.; Datta, A.; Chaudhary, A.; Kalia, K.; Sharma, A.; Wang, X.; et al. Glial Cells Response in Stroke. Cell. Mol. Neurobiol. 2022. [Google Scholar] [CrossRef]
- Shen, X.Y.; Gao, Z.K.; Han, Y.; Yuan, M.; Guo, Y.S.; Bi, X. Activation and Role of Astrocytes in Ischemic Stroke. Front. Cell Neurosci. 2021, 15, 755955. [Google Scholar] [CrossRef]
- Xu, S.; Li, Y.; Chen, J.P.; Li, D.Z.; Jiang, Q.; Wu, T.; Zhou, X.Z. Oxygen glucose deprivation/re-oxygenation-induced neuronal cell death is associated with Lnc-D63785 m6A methylation and miR-422a accumulation. Cell Death Dis. 2020, 11, 816. [Google Scholar] [CrossRef]
- Si, W.; Li, Y.; Ye, S.; Li, Z.; Liu, Y.; Kuang, W.; Chen, D.; Zhu, M. Methyltransferase 3 Mediated miRNA m6A Methylation Promotes Stress Granule Formation in the Early Stage of Acute Ischemic Stroke. Front. Mol. Neurosci. 2020, 13, 103. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Q.; Zhao, X.; Shao, L.; Liu, G.; Zheng, X.; Xie, L.; Zhang, Y.; Sun, C.; Xu, R. YTHDC1 mitigates ischemic stroke by promoting Akt phosphorylation through destabilizing PTEN mRNA. Cell Death Dis. 2020, 11, 977. [Google Scholar] [CrossRef]
- Zhu, R.; Tian, D.; Zhao, Y.; Zhang, C.; Liu, X. Genome-Wide Detection of m(6)A-Associated Genetic Polymorphisms Associated with Ischemic Stroke. J. Mol. Neurosci. 2021, 71, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Wang, F.; Huang, N.X.; Xiao, L.; Mei, F. Oligodendrocytes and Myelin: Active players in Neurodegenerative brains? Dev. Neurobiol. 2022, 82, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez De La Cruz, B.; Markus, R.; Malla, S.; Haig, M.I.; Gell, C.; Sang, F.; Bellows, E.; Sherif, M.A.; McLean, D.; Lourdusamy, A.; et al. Modifying the m(6)A brain methylome by ALKBH5-mediated demethylation: A new contender for synaptic tagging. Mol. Psychiatry 2021, 26, 7141–7153. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kam, T.I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Koranda, J.L.; Dore, L.; Shi, H.; Patel, M.J.; Vaasjo, L.O.; Rao, M.N.; Chen, K.; Lu, Z.; Yi, Y.; Chi, W.; et al. Mettl14 Is Essential for Epitranscriptomic Regulation of Striatal Function and Learning. Neuron 2018, 99, 283–292.e5. [Google Scholar] [CrossRef] [Green Version]
- Hess, M.E.; Hess, S.; Meyer, K.D.; Verhagen, L.A.; Koch, L.; Bronneke, H.S.; Dietrich, M.O.; Jordan, S.D.; Saletore, Y.; Elemento, O.; et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat. Neurosci. 2013, 16, 1042–1048. [Google Scholar] [CrossRef]
- Selberg, S.; Yu, L.Y.; Bondarenko, O.; Kankuri, E.; Seli, N.; Kovaleva, V.; Herodes, K.; Saarma, M.; Karelson, M. Small-Molecule Inhibitors of the RNA M6A Demethylases FTO Potently Support the Survival of Dopamine Neurons. Int. J. Mol. Sci. 2021, 22, 4537. [Google Scholar] [CrossRef]
- Quan, W.; Li, J.; Liu, L.; Zhang, Q.; Qin, Y.; Pei, X.; Chen, J. Influence of N6-Methyladenosine Modification Gene HNRNPC on Cell Phenotype in Parkinson’s Disease. Parkinson’s Dis. 2021, 2021, 9919129. [Google Scholar] [CrossRef] [PubMed]
- Nierenberg, A.A. Residual symptoms in depression: Prevalence and impact. J. Clin. Psychiatry 2015, 76, e1480. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat. Rev. Drug Discov. 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Ribeiro, D.E.; Roncalho, A.L.; Glaser, T.; Ulrich, H.; Wegener, G.; Joca, S. P2X7 Receptor Signaling in Stress and Depression. Int. J. Mol. Sci. 2019, 20, 2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulen, B.; Serinken, M.; Eken, C.; Karcıoglu, Ö.; Kucukdagli, O.T.; Kilic, E.; Akpinar, G.; Nogay, S.; Kuh, M. Serum S100B as a Surrogate Biomarker in the Diagnoses of Burnout and Depression in Emergency Medicine Residents. Acad. Emerg. Med. 2016, 23, 786–789. [Google Scholar] [CrossRef] [Green Version]
- Du, T.; Rao, S.; Wu, L.; Ye, N.; Liu, Z.; Hu, H.; Xiu, J.; Shen, Y.; Xu, Q. An association study of the m6A genes with major depressive disorder in Chinese Han population. J. Affect. Disord. 2015, 183, 279–286. [Google Scholar] [CrossRef]
- Samaan, Z.; Anand, S.S.; Zhang, X.; Desai, D.; Rivera, M.; Pare, G.; Thabane, L.; Xie, C.; Gerstein, H.; Engert, J.C.; et al. The protective effect of the obesity-associated rs9939609 A variant in fat mass- and obesity-associated gene on depression. Mol. Psychiatry 2013, 18, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Gouvêa-Junqueira, D.; Falvella, A.C.B.; Antunes, A.S.L.M.; Seabra, G.; Brandão-Teles, C.; Martins-de-Souza, D.; Crunfli, F. Novel Treatment Strategies Targeting Myelin and Oligodendrocyte Dysfunction in Schizophrenia. Front. Psychiatry 2020, 11, 379. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Uranova, N.A. Reduced number of satellite oligodendrocytes of pyramidal neurons in layer 5 of the prefrontal cortex in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 947–955. [Google Scholar] [CrossRef]
- Brisch, R.; Wojtylak, S.; Saniotis, A.; Steiner, J.; Gos, T.; Kumaratilake, J.; Henneberg, M.; Wolf, R. The role of microglia in neuropsychiatric disorders and suicide. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 272, 929–945. [Google Scholar] [CrossRef]
- Koskuvi, M.; Lehtonen, S.; Trontti, K.; Keuters, M.; Wu, Y.C.; Koivisto, H.; Ludwig, A.; Plotnikova, L.; Virtanen, P.L.J.; Rasanen, N.; et al. Contribution of astrocytes to familial risk and clinical manifestation of schizophrenia. Glia 2022, 70, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, T.; Tsurugizawa, T. Potential of Multiscale Astrocyte Imaging for Revealing Mechanisms Underlying Neurodevelopmental Disorders. Int. J. Mol. Sci. 2021, 22, 10312. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zang, L.; Zhang, F.; Chen, J.; Shen, H.; Shu, L.; Liang, F.; Feng, C.; Chen, D.; Tao, H.; et al. Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum. Mol. Genet. 2017, 26, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, X.; Wang, W.; Shi, H.; Pan, Q.; Lu, Z.; Perez, S.P.; Suganthan, R.; He, C.; Bjoras, M.; et al. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 2018, 19, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, K.J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m(6)A Methylation. Cell 2017, 171, 877–889.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Hou, L.; Park, Y.P.; Molinie, B.; Consortium, G.T.; Gregory, R.I.; Kellis, M. Genetic drivers of m(6)A methylation in human brain, lung, heart and muscle. Nat. Genet. 2021, 53, 1156–1165. [Google Scholar] [CrossRef]
- Qi, X.; Guan, F.; Wen, Y.; Li, P.; Ma, M.; Cheng, S.; Zhang, L.; Liang, C.; Cheng, B.; Zhang, F. Integrating genome-wide association study and methylation functional annotation data identified candidate genes and pathways for schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109736. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, J.; Liu, Z.; Su, J.; Xu, J.; Li, Z.; Meng, H.; Zhang, H.; Huang, M.; Zhao, D.; et al. Identification of an N6-methyladenosine (m6A)-related signature associated with clinical prognosis, immune response, and chemotherapy in primary glioblastomas. Ann. Transl. Med. 2021, 9, 1241. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Hou, K.; Mi, S.; Ji, H.; Ma, S.; Ba, Y.; Hu, S.; Xie, R.; Chen, L. Malignant Evaluation and Clinical Prognostic Values of m6A RNA Methylation Regulators in Glioblastoma. Front. Oncol. 2020, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Patil, V.; Abdulla, S.; Hoheisel, J.D.; Somasundaram, K. N(6)-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes 2019, 10, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassinari, V.; Cesarini, V.; Tomaselli, S.; Ianniello, Z.; Silvestris, D.A.; Ginistrelli, L.C.; Martini, M.; De Angelis, B.; De Luca, G.; Vitiani, L.R.; et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biol. 2021, 22, 51. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Chen, G.; Dong, X.; Li, H.; Li, S.; Cheng, S.; Li, Y.; Wang, L.; Yuan, J.; Qian, Z.; et al. METTL3 Promotes the Resistance of Glioma to Temozolomide via Increasing MGMT and ANPG in a m(6)A Dependent Manner. Front. Oncol. 2021, 11, 702983. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gao, T.; Pang, B.; Su, X.; Guo, C.; Zhang, R.; Pang, Q. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m(6)A-dependent manner. Cell. Death Dis. 2022, 13, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Wang, M.J.; Zheng, J.L.; Wu, Y.H.; Wang, X.; Jiang, X.B. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021, 112, 4543–4552. [Google Scholar] [CrossRef]
- Dong, F.; Qin, X.; Wang, B.; Li, Q.; Hu, J.; Cheng, X.; Guo, D.; Cheng, F.; Fang, C.; Tan, Y.; et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 5876–5888. [Google Scholar] [CrossRef]
- Fang, R.; Chen, X.; Zhang, S.; Shi, H.; Ye, Y.; Shi, H.; Zou, Z.; Li, P.; Guo, Q.; Ma, L.; et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat. Commun. 2021, 12, 177. [Google Scholar] [CrossRef]
- Chai, R.C.; Chang, Y.Z.; Chang, X.; Pang, B.; An, S.Y.; Zhang, K.N.; Chang, Y.H.; Jiang, T.; Wang, Y.Z. YTHDF2 facilitates UBXN1 mRNA decay by recognizing METTL3-mediated m(6)A modification to activate NF-kappaB and promote the malignant progression of glioma. J. Hematol. Oncol. 2021, 14, 109. [Google Scholar] [CrossRef]
- Han, J.; Yu, X.; Wang, S.; Wang, Y.; Liu, Q.; Xu, H.; Wang, X. IGF2BP2 Induces U251 Glioblastoma Cell Chemoresistance by Inhibiting FOXO1-Mediated PID1 Expression Through Stabilizing lncRNA DANCR. Front. Cell Dev. Biol. 2021, 9, 659228. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S. Astrocyte interleukin-3 preps microglia. Trends Immunol. 2021, 42, 937–939. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neurological Diseases | Glial Cells | Changes in RNA m6A Regulators | Downstream RNA or Pathways | Citation | |
|---|---|---|---|---|---|
| TBI | microglia, astrocytes | METTL14, FTO | downregulated | Bcl-2 downregulated, Dll4 and CD14 upregulated | [49] |
| METTL3, WTAP, VIRMA, ALKBH5 | no changes | ||||
| MS | OLs | METTL14 | - | neurofascin 155 | [51] |
| hnRNPA2/B1 | MBP | [52] | |||
| Prrc2a | Olig2 | [53] | |||
| FTO | GAP-43 | [54] | |||
| Ischemia brain injury | microglia | YTHDF1 | upregulated | p65 | [55] |
| microglia, astrocytes | YTHDF1, YTHDF3 | upregulated | - | [56] | |
| FTO | downregulated | - | [57] | ||
| AD | microglia, astrocytes | METTL3, METTL16 | upregulated | APOE4 | [58] |
| YTHDC2 | downregulated | ||||
| METTL3 | downregulated | tau protein | [59] | ||
| RBM15B | upregulated | - | |||
| IGF2BP2 | upregulated | transcripts related to extracellular matrix receptor interaction, focal adhesion, cytokine-cytokine receptor interaction, and TGF-β signaling pathways | [60] | ||
| METTL3, METTL14, WTAP, FTO, YTHDF1 | no changes | - | [61] | ||
| astrocytes | FTO, YTHDF1 | upregulated | transcripts related to oxidative stress, apoptosis, and mitochondrial functions | [62] | |
| hnRNPC | - | APP | [63] | ||
| PD | microglia, astrocytes | ALKBH5, FTO | upregulated | NMDAR | [64] |
| FTO | downregulated | epinephrine-B2 | [65] | ||
| Depression | astrocytes | ALKBH5 | upregulated | FAAH | [66] |
| microglia, astrocytes | METTL3 | downregulated | transcripts involved in stress response and synaptic plasticity | [67] | |
| FTO, ALKBH5 | upregulated or downregulated | ||||
| FTO | downregulated | CaMKⅡ/CREB | [68] | ||
| β2-adrenergic receptor | [69] | ||||
| upregulated | Cartpt and Ucn | [70] | |||
| GBM | astrocytes, microglia | METTL3 | downregulated | ADAM 19 | [71] |
| upregulated | SRSF | [72] | |||
| downregulated | transcripts related to epithelial–mesenchymal transition and vasculogenic mimicry | [73] | |||
| upregulated | SOX2 | [74] | |||
| METTL14 | downregulated | ADAM 19 | [71] | ||
| FTO | upregulated | CLIP3 | [75] | ||
| ALKBH5 | upregulated | FOXM1 | [76] | ||
| upregulated | transcripts related to epithelial–mesenchymal transition and vasculogenic mimicry | [73] | |||
| upregulated | YAP1 | [77] | |||
| YTHDF2 | upregulated | MYC, VEGF | [78] | ||
| IGF2BP2 | upregulated | OIP5-AS1, miR-129-5p | [79] | ||
| upregulated | lncRNA CASC9 | [80] | |||
| IGF2BP3 | upregulated | - | [81] | ||
| m6A Regulators | Cell Lines or Tissues | Expression Level | m6A Alteration | Functions | Citation |
|---|---|---|---|---|---|
| METTL3 | tumor tissues from GBM patients | ↓ | m6A decreased | METTL3 knockdown enhanced GSC growth and self-renewal, as well as GBM progression probably via ADAM metallopeptidase domain 19 (ADAM, oncogene). | [71] |
| surgical specimens from GBM patients, subcutaneous tumor model and intracranial GBM xenograft model and U251 and U87MG cell lines | ↑ | m6A increased | METTL3/YTHDC1-dependent SRSF mRNA nonsense-mediated decay increased alternative splicing of Bcl-x and nuclear receptor corepressor 2 (maintain cell specificity and tissue homeostasis) and GBM development and proliferation. | [72] | |
| primary tumor GSC, MGG8 | unknown | m6A modification mainly in the 3′ UTR | METTL3-dependent m6A modification regulated RNA editing and Notch pathway stimulation in GSCs. | [125] | |
| U138MG, T98G, A172, U118MG, U87MG, and LN18 | ↑ | m6A increased near the stop codon | Adenosine-to-inosine RNA editing catalyzing protein enhanced by METTL3/YTHDF1 bound with the cyclin-dependent kinase 2 transcript to accelerate cell cycle and control cell proliferation and tumor growth. | [126] | |
| tumor tissues from GBM patients and U87MG cell line | ↓ | m6A decreased | Knockdown of METTL3 regulated epithelial–mesenchymal transition (EMT) and vasculogenic mimicry (VM) processes with decreased survival time. | [73] | |
| U87MG and U251 cell lines exposed to temozolomide (TMZ) and subcutaneous glioma xenograft model | ↑ | m6A increased | METTL3 promoted the TMZ resistance of GBM cells by increasing DNA repair protein MGMT and ANPG levels in an m6A-dependent manner. | [127] | |
| tumor tissues from GBM patients | ↑ | m6A increased | METTL3 methylated SOX2 mRNA, which recruited human antigen R (HuR) to the modified SOX2 and stabilized the mRNA to enhance GSC self-renewal and dedifferentiation and increase DNA repair for radioresistance. | [74] | |
| U87MG and U251 cell lines | ↑ | no change | RNA-binding protein NKAP combined SLC7A11, a ferroptosis defense protein, recruited the splicing factor proline and glutamine-rich to recognize the splice site, and conducted TTS splicing event on SLC7A11 transcript and the retention of the last exon, to protect GBM cells from ferroptosis. | [128] | |
| METTL14 | tumor tissues from GBM patients | ↓ | m6A decreased | METTL14 knockdown enhanced GSC growth and self-renewal and GBM progression probably via ADAM. | [71] |
| FTO | tumor tissues from GBM patients | ↑ | m6A decreased | miR-145 in differentiated glioma cells (DGCs) mediated the formation of FTO/AGO1/ILF3/miR-145 complexes on clinically relevant tumor suppressor gene (CLIP3) and promoted CLIP3 demethylation by FTO and nascent translation to induce the transformation of DGCs and GSCs. | [75] |
| TS576, GBM-GSC-23, and GBM-6 cell lines | ↑ | - | FTO inhibitors impaired self-renewal in GSCs. | [71] | |
| U251, LN229, U87, SHG-44, and LN18 cell lines | ↑ | - | lncRNA just proximal to the X- inactive specific transcript modulated and stabilized PDK1 in an FTO-dependent manner to promote aerobic glycolysis and TMZ chemoresistance. | [129] | |
| ALKBH5 | GBM xenografts and surgical specimen tissue slides | ↑ | m6A decreased in FOXM1 pre-mRNA | ALKBH5 demethylated forkhead box protein M1(FOXM1) and affected HuR association with FOXM1 nascent transcripts in GSCs, leading to cell proliferation and tumor growth. | [76] |
| U87 and GL261 cell lines | ↑ | most m6A-modified transcripts downregulated | ALKBH5 stabilized lncRNA NEAT1 by demethylation; therefore, NEAT1 controlled paraspeckle assembly and SFPQ relocation from CXCL8 promoter and facilitated TAM recruitment and immunosuppression through CXCL8/IL8. | [130] | |
| GBM biopsy specimens | ↑ | - | ALKBH5 regulated GBM invasion through YAP1 expression and increased radioresistance by regulating homologous recombination and DNA-damage repair. | [77] | |
| tumor tissues from GBM patients and U87MG cell line | ↑ | m6A decreased | ALKBH5 overexpression causes a highly scattered pattern of cytoskeleton through the rearrangement of F-actin to enhance the EMT and VM process and growth of GBM cells. | [73] | |
| YTHDF2 | surgical resection samples from patients | ↑ | No change in m6A peaks, but in m6A distribution | YTHDF2 stabilized MYC and VEGF transcripts in an m6A-dependent manner following upregulated expression of IGFBP3 to promote tumor growth. | [78] |
| Hs683, SW1783, T98G, U87MG, LN299 cell lines, and animals injected with GSC or LN299 cells | ↑ | - | Sustained by EGFR/SRC/ERK signaling, YTHDF2 downregulated LXRα and HIVEP2 through m6A-dependent mRNA decay and inhibited LXRα-dependent cholesterol homeostasis, to promote tumorigenesis. | [131] | |
| CGGA website and TCGA database, and H4, LN299, and U87 cell lines | ↑ | m6A decreased in UBXN1 mRNA | YTHDF2 accelerated UBXN1 mRNA decay by recognizing the m6A modification mediated by METTL3 and enhanced NF-κB activation, to promote GBM progression. | [132] | |
| IGF2BP2 | U251 cell line, surgical tissue samples, and mouse xenograft model | ↑ | - | IGF2BP2 induced GBM cell chemoresistance by downregulating forkhead box protein O1-mediated phosphotyrosine interaction domain containing 1 expression via stabilizing lncRNA DANCR. | [133] |
| GBM surgical specimens and U251, U87, A172, and SHG44 cell lines | ↑ | - | IGF2BP2 regulated by the sponge interaction between OIP5-AS1 and miR-129-5p promoted cell chemoresistance to TMZ and cell growth in GBM. | [79] | |
| GBM surgical specimens and U87MG and U251 cell lines | ↑ | m6A peaks in 5′ UTR and 3′ UTR | IGF2BP2 stabilized lncRNA CASC9 to accelerate the aerobic glycosis of GBM by enhancing hexokinase 2 mRNA stability. | [80] | |
| IGF2BP3 | GBM specimens | ↑ | - | Upregulated IGF2BP3 expression was associated with poor OS and prognosis. | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, S.; Su, X.; Ying, J.; Li, S.; Qu, Y.; Mu, D. Research Progress on the Role of RNA m6A Modification in Glial Cells in the Regulation of Neurological Diseases. Biomolecules 2022, 12, 1158. https://doi.org/10.3390/biom12081158
You S, Su X, Ying J, Li S, Qu Y, Mu D. Research Progress on the Role of RNA m6A Modification in Glial Cells in the Regulation of Neurological Diseases. Biomolecules. 2022; 12(8):1158. https://doi.org/10.3390/biom12081158
Chicago/Turabian StyleYou, Siyi, Xiaojuan Su, Junjie Ying, Shiping Li, Yi Qu, and Dezhi Mu. 2022. "Research Progress on the Role of RNA m6A Modification in Glial Cells in the Regulation of Neurological Diseases" Biomolecules 12, no. 8: 1158. https://doi.org/10.3390/biom12081158