
STIM Proteins and Regulation of SOCE in ER-PM Junctions

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Properties and Function of the STIM Polybasic Domains
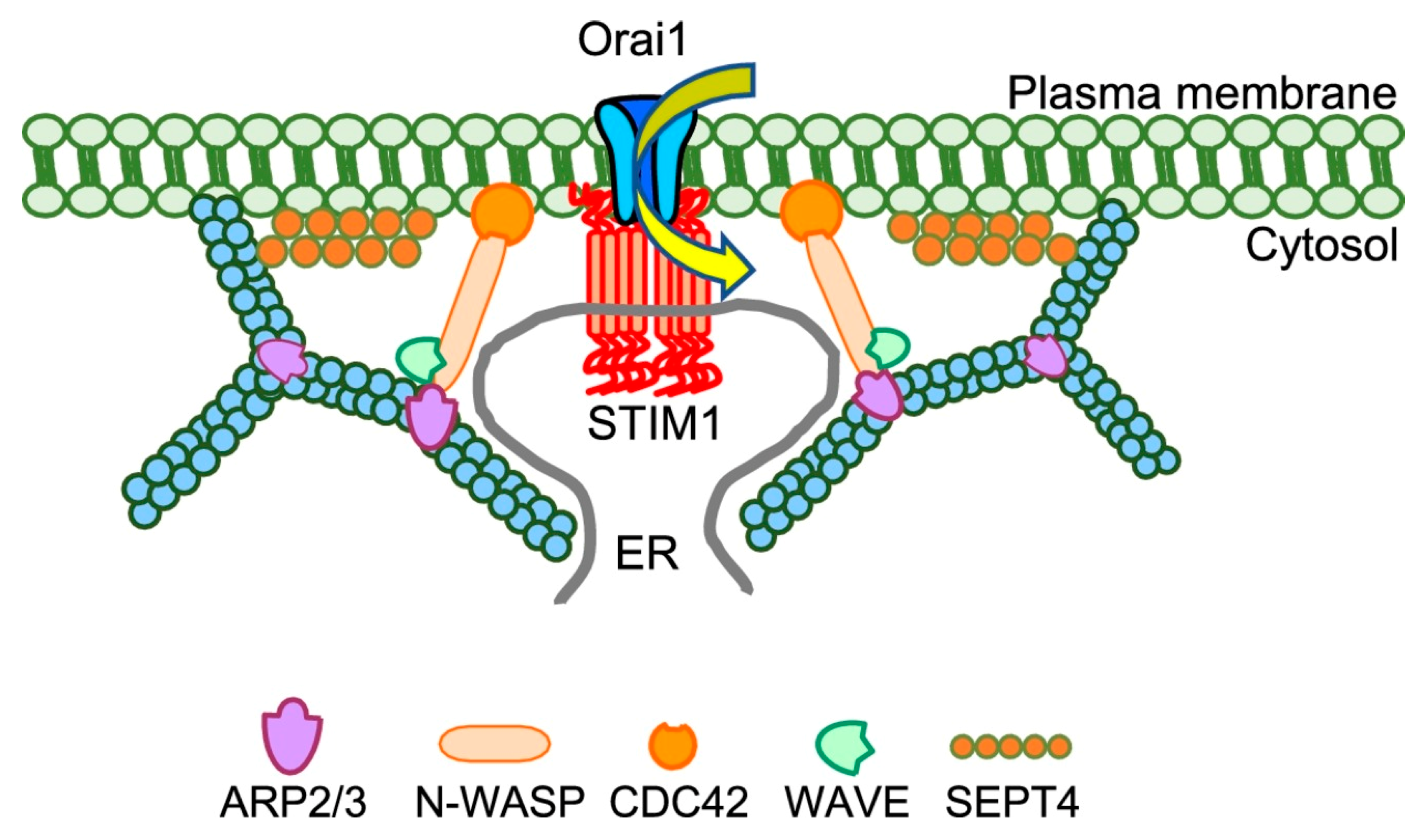
3. Modulation of SOCE by Plasma Membrane PIP2, Actin, and Septins
4. Accessory Proteins Modulating STIMs and SOCE
5. STIM2 Response to ER-Ca2+ Depletion Is a Checkpoint for SOCE Activation
6. Assembly of Ca2+-Signaling Proteins in ER-PM Junctions and Regulation of Cell Function
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Y.-J.; Quintanilla, C.G.; Liou, J. Recent insights into mammalian ER–PM junctions. Curr. Opin. Cell Biol. 2019, 57, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; Ambudkar, I.S. The Endoplasmic Reticulum–Plasma Membrane Junction: A Hub for Agonist Regulation of Ca2+ Entry. Cold Spring Harb. Perspect. Biol. 2020, 12, a035253. [Google Scholar] [CrossRef] [PubMed]
- Berlansky, S.; Humer, C.; Sallinger, M.; Frischauf, I. More Than Just Simple Interaction between STIM and Orai Proteins: CRAC Channel Function Enabled by a Network of Interactions with Regulatory Proteins. Int. J. Mol. Sci. 2021, 22, 471. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qian, T.; He, R.; Wan, C.; Liu, Y.; Yu, H. Endoplasmic Reticulum–Plasma Membrane Contact Sites: Regulators, Mechanisms, and Physiological Functions. Front. Cell Dev. Biol. 2021, 9, 627700. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Ong, H.L.; Saadi, H.; Son, G.-Y.; Shokatian, Z.; Terry, L.E.; Trebak, M.; Yule, D.I.; Ambudkar, I. Functional communication between IP3R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE. Proc. Natl. Acad. Sci. USA 2022, 119, e2114928118. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, C.G.; Lee, W.-R.; Liou, J. Nir1 constitutively localizes at ER–PM junctions and promotes Nir2 recruitment for PIP2 homeostasis. Mol. Biol. Cell 2022, 33, br2. [Google Scholar] [CrossRef]
- Chang, C.-L.; Hsieh, T.-S.; Yang, T.T.; Rothberg, K.G.; Azizoglu, D.B.; Volk, E.; Liao, J.-C.; Liou, J. Feedback Regulation of Receptor-Induced Ca2+ Signaling Mediated by E-Syt1 and Nir2 at Endoplasmic Reticulum-Plasma Membrane Junctions. Cell Rep. 2013, 5, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Balla, T.; Kim, Y.J.; Alvarez-Prats, A.; Pemberton, J. Lipid Dynamics at Contact Sites between the Endoplasmic Reticulum and Other Organelles. Annu. Rev. Cell Dev. Biol. 2019, 35, 85–109. [Google Scholar] [CrossRef]
- Pemberton, J.G.; Kim, Y.J.; Balla, T. Integrated regulation of the phosphatidylinositol cycle and phosphoinositide-driven lipid transport at ER-PM contact sites. Traffic 2020, 21, 200–219. [Google Scholar] [CrossRef]
- Giordano, F.; Saheki, Y.; Idevall-Hagren, O.; Colombo, S.F.; Pirruccello, M.; Milosevic, I.; Gracheva, E.O.; Bagriantsev, S.N.; Borgese, N.; De Camilli, P. PI(4,5)P2-Dependent and Ca2+-Regulated ER-PM Interactions Mediated by the Extended Synaptotagmins. Cell 2013, 153, 1494–1509. [Google Scholar] [CrossRef] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 Is a Feedback Regulator that Stabilizes Basal Cytosolic and Endoplasmic Reticulum Ca2+ Levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Meraner, P.; Kwon, H.T.; Machnes, D.; Oh-Hora, M.; Zimmer, J.; Huang, Y.; Stura, A.; Rao, A.; Hogan, P.G. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat. Struct. Mol. Biol. 2010, 17, 112–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees, J.A.; Messa, M.; Sun, E.W.; Wheeler, H.; Torta, F.; Wenk, M.R.; De Camilli, P.; Reinisch, K.M. Lipid transport by TMEM24 at ER–plasma membrane contacts regulates pulsatile insulin secretion. Science 2017, 355, eaah6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besprozvannaya, M.; Dickson, E.; Li, H.; Ginburg, K.S.; Bers, D.M.; Auwerx, J.; Nunnari, J. GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. eLife 2018, 7, e31019. [Google Scholar] [CrossRef]
- Ong, H.L.; Subedi, K.P.; Son, G.-Y.; Liu, X.; Ambudkar, I.S. Tuning store-operated calcium entry to modulate Ca2+-dependent physiological processes. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Albert, A.P.; Large, W.A. Store-operated Ca2+-permeable non-selective cation channels in smooth muscle cells. Cell Calcium. 2003, 33, 345–356. [Google Scholar] [CrossRef]
- Park, Y.-J.; Yoo, S.-A.; Kim, M.; Kim, W.-U. The Role of Calcium–Calcineurin–NFAT Signaling Pathway in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 195. [Google Scholar] [CrossRef]
- Son, G.-Y.; Subedi, K.P.; Ong, H.L.; Noyer, L.; Saadi, H.; Zheng, C.; Bhardwaj, R.; Feske, S.; Ambudkar, I.S. STIM2 targets Orai1/STIM1 to the AKAP79 signaling complex and confers coupling of Ca2+ entry with NFAT1 activation. Proc. Natl. Acad. Sci. USA 2020, 117, 16638–16648. [Google Scholar] [CrossRef]
- Novello, M.J.; Zhu, J.; Feng, Q.; Ikura, M.; Stathopulos, P.B. Structural elements of stromal interaction molecule function. Cell Calcium 2018, 73, 88–94. [Google Scholar] [CrossRef]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardín, I. STIM and Orai1 Variants in Store-Operated Calcium Entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum–plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subedi, K.P.; Ong, H.L.; Son, G.-Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavez, M.; Thompson, A.C.; Arnott, H.J.; Mitchell, C.B.; D’Atri, I.; Don, E.K.; Chilton, J.K.; Scott, E.K.; Lin, J.Y.; Young, K.M.; et al. STIM1 Is Required for Remodeling of the Endoplasmic Reticulum and Microtubule Cytoskeleton in Steering Growth Cones. J. Neurosci. 2019, 39, 5095–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Hogan, P.G. STIM calcium sensing and conformational change. J. Physiol. 2020, 598, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, R.; Müller, H.-M.; Nickel, W.; Seedorf, M. Oligomerization and Ca2+/calmodulin control binding of the ER Ca2+-sensors STIM1 and STIM2 to plasma membrane lipids. Biosci. Rep. 2013, 33, 833–845. [Google Scholar] [CrossRef]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A Conserved, Lipid-Mediated Sorting Mechanism of Yeast Ist2 and Mammalian STIM Proteins to the Peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 Clusters and Activates CRAC Channels via Direct Binding of a Cytosolic Domain to Orai1. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Crocker, E.; McLaughlin, S.; Smith, S.O. Binding of Peptides with Basic and Aromatic Residues to Bilayer Membranes: Phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J. Biol. Chem. 2003, 278, 21459–21466. [Google Scholar] [CrossRef] [Green Version]
- Gambhir, A.; Hangyás-Mihályné, G.; Zaitseva, I.; Cafiso, D.S.; Wang, J.; Murray, D.; Pentyala, S.N.; Smith, S.O.; McLaughlin, S. Electrostatic Sequestration of PIP2 on Phospholipid Membranes by Basic/Aromatic Regions of Proteins. Biophys. J. 2004, 86, 2188–2207. [Google Scholar] [CrossRef] [Green Version]
- Sönnichsen, F.; Van Eyk, J.E.; Hodges, R.S.; Sykes, B.D. Effect of trifluoroethanol on protein secondary structure: An NMR and CD study using a synthetic actin peptide. Biochemistry 1992, 31, 8790–8798. [Google Scholar] [CrossRef]
- Manford, A.G.; Stefan, C.J.; Yuan, H.L.; MacGurn, J.A.; Emr, S.D. ER-to-Plasma Membrane Tethering Proteins Regulate Cell Signaling and ER Morphology. Dev. Cell 2012, 23, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maass, K.; Fischer, M.A.; Seiler, M.; Temmerman, K.; Nickel, W.; Seedorf, M. A signal comprising a basic cluster and an amphipathic α-helix interacts with lipids and is required for the transport of Ist2 to the yeast cortical ER. J. Cell Sci. 2009, 122, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, W.; Kilic, A.; Schrul, B.; Lorenz, H.; Schwappach, B.; Seedorf, M. Yeast Ist2 Recruits the Endoplasmic Reticulum to the Plasma Membrane and Creates a Ribosome-Free Membrane Microcompartment. PLoS ONE 2012, 7, e39703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Shi, X.; Guo, X.; Li, H.; Xu, C. Ionic protein–lipid interaction at the plasma membrane: What can the charge do? Trends Biochem. Sci. 2014, 39, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Gagnon, E.; Call, M.E.; Schnell, J.R.; Schwieters, C.D.; Carman, C.V.; Chou, J.J.; Wucherpfennig, K.W. Regulation of T Cell Receptor Activation by Dynamic Membrane Binding of the CD3ɛ Cytoplasmic Tyrosine-Based Motif. Cell 2008, 135, 702–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. PIP2 and septin control STIM1/Orai1 assembly by regulating cytoskeletal remodeling via a CDC42-WASP/WAVE-ARP2/3 protein complex. Cell Calcium 2021, 99, 102475. [Google Scholar] [CrossRef]
- Zheng, L.; Stathopulos, P.B.; Li, G.-Y.; Ikura, M. Biophysical characterization of the EF-hand and SAM domain containing Ca2+ sensory region of STIM1 and STIM2. Biochem. Biophys. Res. Commun. 2008, 369, 240–246. [Google Scholar] [CrossRef]
- Frischauf, I.; Schindl, R.; Derler, I.; Bergsmann, J.; Fahrner, M.; Romanin, C. The STIM/Orai coupling machinery. Channels 2008, 2, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.C.; O’Connell, D.; Cahill, D.J.; Linse, S. Calmodulin Binding to the Polybasic C-Termini of STIM Proteins Involved in Store-Operated Calcium Entry. Biochemistry 2008, 47, 6089–6091. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Ikura, M. Calmodulin in Action: Diversity in Target Recognition and Activation Mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Crivici, A.; Ikura, M. Molecular and Structural Basis of Target Recognition by Calmodulin. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Saarikangas, J.; Zhao, H.; Lappalainen, P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinosi-tides. Physiol. Rev. 2010, 90, 259–289. [Google Scholar] [CrossRef] [Green Version]
- Vasilev, F.; Ezhova, Y.; Chun, J.T. Signaling Enzymes and Ion Channels Being Modulated by the Actin Cytoskeleton at the Plasma Membrane. Int. J. Mol. Sci. 2021, 22, 10366. [Google Scholar] [CrossRef]
- Senju, Y.; Kalimeri, M.; Koskela, E.V.; Somerharju, P.; Zhao, H.; Vattulainen, I.; Lappalainen, P. Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, E8977–E8986. [Google Scholar] [CrossRef] [Green Version]
- Lyubchenko, T.A.; Wurth, G.A.; Zweifach, A. The actin cytoskeleton and cytotoxic T lymphocytes: Evidence for multiple roles that could affect granule exocytosis-dependent target cell killing. J. Physiol. 2003, 547, 835–847. [Google Scholar] [CrossRef]
- Arrieumerlou, C.; Randriamampita, C.; Bismuth, G.; Trautmann, A. Rac Is Involved in Early TCR Signaling. J. Immunol. 2000, 165, 3182–3189. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Sato, K.; Hori, M.; Ozaki, H.; Karaki, H. FcεRI cross-linking-induced actin assembly mediates calcium signalling in RBL-2H3 mast cells. Br. J. Pharmacol. 2002, 136, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Baeker, T.R.; Simons, E.R.; Rothstein, T.L. Cytochalasin induces an increase in cytosolic free calcium in murine B lymphocytes. J. Immunol. 1987, 138, 2691–2697. [Google Scholar]
- Van Haelst, C.; Rothstein, T.L. Cytochalasin stimulates phosphoinositide metabolism in murine B lymphocytes. J. Immunol. 1988, 140, 1256–1258. [Google Scholar] [PubMed]
- Hartzell, C.A.; Jankowska, K.I.; Burkhardt, J.K.; Lewis, R.S. Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. eLife 2016, 5, e14850. [Google Scholar] [CrossRef]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.K.; Hasan, G. Regulation of Store-Operated Ca2+ Entry by Septins. Front. Cell Dev. Biol. 2016, 4, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, Z.; Zhang, C.; Quintana, A.; Lillemeier, B.F.; Hogan, P.G. Septins organize endoplasmic reticulum-plasma membrane junctions for STIM1-ORAI1 calcium signalling. Sci. Rep. 2019, 9, 10839. [Google Scholar] [CrossRef]
- Takenawa, T.; Miki, H. WASP and WAVE family proteins: Key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809. [Google Scholar] [CrossRef]
- Watson, J.; Owen, D.; Mott, H.R. Cdc42 in actin dynamics: An ordered pathway governed by complex equilibria and directional effector handover. Small GTPases 2017, 8, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Deb, B.K.; Hasan, G. SEPT7-mediated regulation of Ca2+ entry through Orai channels requires other septin subunits. Cytoskeleton 2019, 76, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum–plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef] [Green Version]
- Hodeify, R.; Selvaraj, S.; Wen, J.; Arredouani, A.; Hubrack, S.; Dib, M.; Al-Thani, S.N.; McGraw, T.; Machaca, K. A STIM1-dependent ‘trafficking trap’ mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels. J. Cell Sci. 2015, 128, 3143–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, T.-S.; Chen, Y.-J.; Chang, C.-L.; Lee, W.-R.; Liou, J. Cortical actin contributes to spatial organization of ER–PM junctions. Mol. Biol. Cell 2017, 28, 3171–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Mao, Y.S.; Janmey, P.A.; Yin, H.L. Phosphatidylinositol 4, 5 Bisphosphate and the Actin Cytoskeleton. In Phosphoinositides II: The Diverse Biological Functions; Subcellular Biochemistry; Springer: Berlin, Germany, 2012; Volume 59, pp. 177–215. [Google Scholar] [CrossRef]
- Chichili, G.R.; Rodgers, W. Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol. Life Sci. CMLS 2009, 66, 2319–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, X.; Saheki, Y.; De Camilli, P. Ca2+ releases E-Syt1 autoinhibition to couple ER-plasma membrane tethering with lipid transport. EMBO J. 2018, 37, 219–234. [Google Scholar] [CrossRef]
- Idevall-Hagren, O.; Lü, A.; Xie, B.; De Camilli, P. Triggered Ca2+ influx is required for extended synaptotagmin 1-induced ER-plasma membrane tethering. EMBO J. 2015, 34, 2291–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Busnadiego, R.; Saheki, Y.; De Camilli, P. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum–plasma membrane contact sites. Proc. Natl. Acad. Sci. USA 2015, 112, E2004–E2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, F.; Zhou, M.; Huang, X.; Fan, J.; Wei, L.; Boulanger, J.; Liu, Z.; Salamero, J.; Liu, Y.; Chen, L. E-syt1 Re-arranges STIM1 Clusters to Stabilize Ring-shaped ER-PM Contact Sites and Accelerate Ca2+ Store Replenishment. Sci. Rep. 2019, 9, 3975. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; DeHaven, W.I.; Bird, G.S.; Putney, J.W., Jr. Role of the microtubule cytoskeleton in the function of the store-operated Ca2+ channel activator STIM1. J. Cell Sci. 2007, 120, 3762–3771. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.; Gouveia, S.; Van Der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.; Putney, J.W., Jr.; Hoogenraad, C.; et al. STIM1 Is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-L.; Chen, Y.-J.; Quintanilla, C.; Hsieh, T.-S.; Liou, J. EB1 binding restricts STIM1 translocation to ER–PM junctions and regulates store-operated Ca2+ entry. J. Cell Biol. 2018, 217, 2047–2058. [Google Scholar] [CrossRef] [Green Version]
- Pozo-Guisado, E.; Martin-Romero, F.J. The regulation of STIM1 by phosphorylation. Commun. Integr. Biol. 2013, 6, e26283. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Stotz, S.C.; Manasian, Y.; Clapham, D.E. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 19234–19239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, S.; Jung, H.-J.; Kim, K.-D.; Souda, P.; Whitelegge, J.; Gwack, Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikanth, S.; Jew, M.; Kim, K.-D.; Yee, M.-K.; Abramson, J.; Gwack, Y. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duquette, M.; Nadler, M.; Okuhara, D.; Thompson, J.; Shuttleworth, T.; Lawler, J. Members of the thrombospondin gene family bind stromal interaction molecule 1 and regulate calcium channel activity. Matrix Biol. 2014, 37, 15–24. [Google Scholar] [CrossRef]
- Fujii, Y.; Shiota, M.; Ohkawa, Y.; Baba, A.; Wanibuchi, H.; Kinashi, T.; Kurosaki, T.; Baba, Y. Surf4 modulates STIM1-dependent calcium entry. Biochem. Biophys. Res. Commun. 2012, 422, 615–620. [Google Scholar] [CrossRef]
- Kawata, K.; Baba, A.; Shiota, M.; Wanibuchi, H.; Baba, Y. ER membrane protein complex 1 interacts with STIM1 and regulates store-operated Ca2+ entry. J. Biochem. 2021, 170, 483–488. [Google Scholar] [CrossRef]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Rajanikanth, V.; Farber-Katz, S.; Gudlur, A.; Zhang, C.; Jing, J.; Zhou, Y.; Rao, A.; Hogan, P.G. TMEM110 regulates the maintenance and remodeling of mammalian ER–plasma membrane junctions competent for STIM–ORAI signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E7083–E7092. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.S.; Srikanth, S.; Nishi, M.; Ping, P.; Takeshima, H.; Gwack, Y. Junctophilin-4, a component of the endoplasmic reticulum–plasma membrane junctions, regulates Ca2+ dynamics in T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2762–2767. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER–PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammons, J.; Halpage, J.; Mancarella, S. Mapping the Proximity Interaction Network of STIM1 Reveals New Mechanisms of Cytoskeletal Regulation. Cells 2021, 10, 2701. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.S.; Srikanth, S.; Gwack, Y. Modulation of Orai1 and STIM1 by Cellular Factors. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press: Boca Raton, FL, USA, 2018; pp. 73–92. [Google Scholar]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Kweon, Y.C.; Lee, A.R.; Lee, Y.Y.; Park, C.Y. Metastasis enhancer PGRMC1 boosts store-operated Ca2+ entry by uncoiling Ca+ sensor STIM1 for focal adhesion turnover and actomyosin formation. Cell Rep. 2022, 38, 110281. [Google Scholar] [CrossRef] [PubMed]
- Erro, A.B.; Jardin, I.; Salido, G.M.; Rosado, J.A. Role of STIM2 in cell function and physiopathology. J. Physiol. 2017, 595, 3111–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 3183. [Google Scholar] [CrossRef]
- Miederer, A.-M.; Alansary, D.; Schwär, G.; Lee, P.-H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [Green Version]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1505. [Google Scholar] [CrossRef]
- Diercks, B.-P.; Werner, R.; Weidemüller, P.; Czarniak, F.; Hernandez, L.; Lehmann, C.; Rosche, A.; Krüger, A.; Kaufmann, U.; Vaeth, M.; et al. ORAI1, STIM1/2, and RYR1 shape subsecond Ca2+ microdomains upon T cell activation. Sci. Signal. 2018, 11, eaat0358. [Google Scholar] [CrossRef] [Green Version]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca2+ signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef]
- Gil, D.; Guse, A.H.; Dupont, G. Three-Dimensional Model of Sub-Plasmalemmal Ca2+ Microdomains Evoked by the Interplay Between ORAI1 and InsP3 Receptors. Front. Immunol. 2021, 12, 659790. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Lin, Y.-P.; Bhardwaj, R.; Tucker, C.J.; Bird, G.S.; Hediger, M.A.; Monico, C.; Amin, N.; Parekh, A.B. The N terminus of Orai1 couples to the AKAP79 signaling complex to drive NFAT1 activation by local Ca2+ entry. Proc. Natl. Acad. Sci. USA 2021, 118, e2012908118. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Kar, P.; Mirams, G.R.; Parekh, A.B. Ca2+ Channel Re-localization to Plasma-Membrane Microdomains Strengthens Activation of Ca2+-Dependent Nuclear Gene Expression. Cell Rep. 2015, 12, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Kar, P.; Barak, P.; Zerio, A.; Lin, Y.-P.; Parekh, A.J.; Watts, V.J.; Cooper, D.M.F.; Zaccolo, M.; Kramer, H.; Parekh, A.B. AKAP79 Orchestrates a Cyclic AMP Signalosome Adjacent to Orai1 Ca2+ Channels. Function 2021, 2, zqab036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun. 2019, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, D.; Everett, K.L.; Halls, M.L.; Pacheco, J.; Skroblin, P.; Vaca, L.; Klussmann, E.; Cooper, D.M.F. Direct Binding Between Orai1 and AC8 Mediates Dynamic Interplay Between Ca2+ and cAMP Signaling. Sci. Signal. 2012, 5, ra29. [Google Scholar] [CrossRef]
- Bruce, J.I.; Shuttleworth, T.J.; Giovannucci, D.R.; Yule, D.I. Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors in Parotid Acinar Cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J. Biol. Chem. 2002, 277, 1340–1348. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.I.; Straub, S.V.; I Yule, D. Crosstalk between cAMP and Ca2+ signaling in non-excitable cells. Cell Calcium 2003, 34, 431–444. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, M.; Narayanasamy, S.; Ong, H.L.; Ambudkar, I. STIM Proteins and Regulation of SOCE in ER-PM Junctions. Biomolecules 2022, 12, 1152. https://doi.org/10.3390/biom12081152
Ahmad M, Narayanasamy S, Ong HL, Ambudkar I. STIM Proteins and Regulation of SOCE in ER-PM Junctions. Biomolecules. 2022; 12(8):1152. https://doi.org/10.3390/biom12081152
Chicago/Turabian StyleAhmad, Moaz, Sasirekha Narayanasamy, Hwei Ling Ong, and Indu Ambudkar. 2022. "STIM Proteins and Regulation of SOCE in ER-PM Junctions" Biomolecules 12, no. 8: 1152. https://doi.org/10.3390/biom12081152