
In Silico Pleiotropy Analysis in KEGG Signaling Networks Using a Boolean Network Model


Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Boolean Network Model
2.3. Computation of In-Silico Pleiotropic Scores
- For each initial state we obtain two attractors and in the wild-type and the -mutant networks, respectively. For convenience, let and .
- We compute a distance between and defined as follows:where and are the least common multiple and the greatest common divisor, respectively, of and , and is an indicator function where outputs 1 if is true, and 0 otherwise. As a result, represents the minimum ratio of a bitwise difference between the states sequence of in the wild-type and the -mutant attractors over the least common period ( of the two attractors.
- Lastly, we compute the dynamics influence of on denoted by by averaging out over a set of initial states in as follows:
2.4. A Standardized Measure of Degree of Pleiotropy
2.5. Structural Characteristics of Pleiotropic Genes
- A feedback loop (FBL) means a sequence chain of nodes where any node is not repeated except the starting and the end nodes [59,61]. In a given network , an FBL is a closed simple cycle in which all nodes except the starting and ending nodes are not revisited; in other words, a path of a length is represented by a sequence of ordered nodes with no repeated nodes except and . Hence, the is called a feedback loop if . It was known that FBLs play important roles for controlling dynamics behavior of signaling networks [61,62,63,64].
- Centrality properties including the closeness defined as the reciprocal of the average distance from a node to every other node [4], the betweenness defined as the ability of a gene to control communication between genes through the shortest paths [65], the stress based on enumeration of the shortest paths [66], and the eigenvector represented by the principal eigenvector of the adjacency matrix of the network, where each node affects all of its neighbors [52].
2.6. Random Network Generation
2.7. Parallel Computation
2.8. Statistical Analysis
3. Results
3.1. Comparison of with the Observational Pleiotropy
3.2. Relation of and the Functional Importance Genes
3.3. Relation of and the Structural Characteristics
3.4. Biological Evidence of Pleiotropic Genes Based on
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ittisoponpisan, S.; Alhuzimi, E.; Sternberg, M.; David, A. Landscape of Pleiotropic Proteins Causing Human Disease: Structural and System Biology Insights. Hum. Mutat. 2016, 38, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liao, B.-Y.; Zhang, J. Genomic patterns of pleiotropy and the evolution of complexity. Proc. Natl. Acad. Sci. USA 2010, 107, 18034–18039. [Google Scholar] [CrossRef]
- Dudley, A.M.; Janse, D.M.; Tanay, A.; Shamir, R.; Church, G.M. A global view of pleiotropy and phenotypically derived gene function in yeast. Mol. Syst. Biol. 2005, 1, 2005.0001. [Google Scholar] [CrossRef] [PubMed]
- Chavali, S.; Barrenas, F.; Kanduri, K.; Benson, M. Network properties of human disease genes with pleiotropic effects. BMC Syst. Biol. 2010, 4, 78. [Google Scholar] [CrossRef]
- Sivakumaran, S.; Agakov, F.; Theodoratou, E.; Prendergast, J.G.; Zgaga, L.; Manolio, T.; Rudan, I.; McKeigue, P.; Wilson, J.F.; Campbell, H. Abundant Pleiotropy in Human Complex Diseases and Traits. Am. J. Hum. Genet. 2011, 89, 607–618. [Google Scholar] [CrossRef]
- Liu, Y.; Koyutürk, M.; Barnholtz-Sloan, J.S.; Chance, M.R. Gene interaction enrichment and network analysis to identify dysregulated pathways and their interactions in complex diseases. BMC Syst. Biol. 2012, 6, 65. [Google Scholar] [CrossRef]
- Polster, R.; Petropoulos, C.J.; Bonhoeffer, S.; Guillaume, F. Epistasis and Pleiotropy Affect the Modularity of the Genotype–Phenotype Map of Cross-Resistance in HIV-1. Mol. Biol. Evol. 2016, 33, 3213–3225. [Google Scholar] [CrossRef]
- Mi, Z.; Guo, B.; Yin, Z.; Li, J.; Zheng, Z. Disease classification via gene network integrating modules and pathways. R. Soc. Open Sci. 2019, 6, 190214. [Google Scholar] [CrossRef]
- Yan, J.; Risacher, S.L.; Shen, L.; Saykin, A.J. Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Briefings Bioinform. 2017, 19, 1370–1381. [Google Scholar] [CrossRef]
- Duveau, F.; Félix, M.-A. Role of Pleiotropy in the Evolution of a Cryptic Developmental Variation in Caenorhabditis elegans. PLoS Biol. 2012, 10, e1001230. [Google Scholar] [CrossRef]
- Seoane, J.A.; Campbell, C.; Day, I.N.M.; Casas, J.P.; Gaunt, T.R. Canonical Correlation Analysis for Gene-Based Pleiotropy Discovery. PLoS Comput. Biol. 2014, 10, e1003876. [Google Scholar] [CrossRef]
- Chung, J.; Alzheimer’s Disease Genetics Consortium; Zhang, X.; Allen, M.; Wang, X.; Ma, Y.; Beecham, G.; Montine, T.J.; Younkin, S.G.; Dickson, D.W.; et al. Genome-wide pleiotropy analysis of neuropathological traits related to Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 22. [Google Scholar] [CrossRef]
- Collet, J.M.; McGuigan, K.; Allen, S.; Chenoweth, S.F.; Blows, M.W. Mutational Pleiotropy and the Strength of Stabilizing Selection within and between Functional Modules of Gene Expression. Genetics 2018, 208, 1601–1616. [Google Scholar] [CrossRef]
- Alzoubi, D.; Desouki, A.A.; Lercher, M.J. Alleles of a gene differ in pleiotropy, often mediated through currency metabolite production, in E. coli and yeast metabolic simulations. Sci. Rep. 2018, 8, 17252. [Google Scholar] [CrossRef]
- A Stoney, R.; Ames, R.M.; Nenadic, G.; Robertson, D.L.; Schwartz, J.-M. Disentangling the multigenic and pleiotropic nature of molecular function. BMC Syst. Biol. 2015, 9, S3. [Google Scholar] [CrossRef]
- Yazdani, A.; Yazdani, A.; Elsea, S.H.; Schaid, D.J.; Kosorok, M.R.; Dangol, G.; Samiei, A. Genome analysis and pleiotropy assessment using causal networks with loss of function mutation and metabolomics. BMC Genom. 2019, 20, 395. [Google Scholar] [CrossRef]
- Nguyen, T.-P.; Liu, W.-C.; Jordán, F. Inferring pleiotropy by network analysis: Linked diseases in the human PPI network. BMC Syst. Biol. 2011, 5, 179. [Google Scholar] [CrossRef]
- Li, S.; Assmann, S.M.; Albert, R. Predicting Essential Components of Signal Transduction Networks: A Dynamic Model of Guard Cell Abscisic Acid Signaling. PLoS Biol. 2006, 4, e312. [Google Scholar] [CrossRef]
- Kwon, Y.-K.; Kim, J.; Cho, K.-H. Dynamical Robustness against Multiple Mutations in Signaling Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 13, 996–1002. [Google Scholar] [CrossRef]
- Shastry, B. Overexpression of genes in health and sickness. A bird’s eye view. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1995, 112, 1–13. [Google Scholar] [CrossRef]
- Kwon, Y.-K.; Cho, K.-H. Analysis of feedback loops and robustness in network evolution based on Boolean models. BMC Bioinform. 2007, 8, 430. [Google Scholar] [CrossRef]
- Raeymaekers, L. Dynamics of Boolean Networks Controlled by Biologically Meaningful Functions. J. Theor. Biol. 2002, 218, 331–341. [Google Scholar] [CrossRef]
- Schwab, J.D.; Kühlwein, S.D.; Ikonomi, N.; Kühl, M.; Kestler, H.A. Concepts in Boolean network modeling: What do they all mean? Comput. Struct. Biotechnol. J. 2020, 18, 571–582. [Google Scholar] [CrossRef]
- Wang, R.-S.; Saadatpour, A.; Albert, R. Boolean modeling in systems biology: An overview of methodology and applications. Phys. Biol. 2012, 9, 055001. [Google Scholar] [CrossRef]
- Gupta, S.; Hashimoto, R.F. Dynamical Analysis of a Boolean Network Model of the Oncogene Role of lncRNA ANRIL and lncRNA UFC1 in Non-Small Cell Lung Cancer. Biomolecules 2022, 12, 420. [Google Scholar] [CrossRef]
- Wooten, D.J.; Zañudo, J.G.T.; Murrugarra, D.; Perry, A.M.; Dongari-Bagtzoglou, A.; Laubenbacher, R.; Nobile, C.J.; Albert, R. Mathematical modeling of the Candida albicans yeast to hyphal transition reveals novel control strategies. PLoS Comput. Biol. 2021, 17, e1008690. [Google Scholar] [CrossRef]
- Guberman, E.; Sherief, H.; Regan, E.R. Boolean model of anchorage dependence and contact inhibition points to coordinated inhibition but semi-independent induction of proliferation and migration. Comput. Struct. Biotechnol. J. 2020, 18, 2145–2165. [Google Scholar] [CrossRef]
- Gupta, S.; Panda, P.K.; Hashimoto, R.F.; Samal, S.K.; Mishra, S.; Verma, S.K.; Mishra, Y.K.; Ahuja, R. Dynamical modeling of miR-34a, miR-449a, and miR-16 reveals numerous DDR signaling pathways regulating senescence, autophagy, and apoptosis in HeLa cells. Sci. Rep. 2022, 12, 4911. [Google Scholar] [CrossRef]
- Mazaya, M.; Trinh, H.-C.; Kwon, Y.-K. Construction and analysis of gene-gene dynamics influence networks based on a Boolean model. BMC Syst. Biol. 2017, 11, 133. [Google Scholar] [CrossRef]
- Robinson, P.N.; Köhler, S.; Bauer, S.; Seelow, D.; Horn, D.; Mundlos, S. The Human Phenotype Ontology: A Tool for Annotating and Analyzing Human Hereditary Disease. Am. J. Hum. Genet. 2008, 83, 610–615. [Google Scholar] [CrossRef]
- Kim, J.-R.; Kim, J.; Kwon, Y.-K.; Lee, H.-Y.; Heslop-Harrison, P.; Cho, K.-H. Reduction of Complex Signaling Networks to a Representative Kernel. Sci. Signal. 2011, 4, ra35. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Groza, T.; Köhler, S.; Moldenhauer, D.; Vasilevsky, N.; Baynam, G.; Zemojtel, T.; Schriml, L.M.; Kibbe, W.A.; Schofield, P.N.; Beck, T.; et al. The Human Phenotype Ontology: Semantic Unification of Common and Rare Disease. Am. J. Hum. Genet. 2015, 97, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘Omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef]
- Zhang, R.; Lin, Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2008, 37, D455–D458. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: An updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2015, 44, D1023–D1031. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, J.; Zhao, Z. TSGene: A web resource for tumor suppressor genes. Nucleic Acids Res. 2012, 41, D970–D976. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J. Genet. Genom. 2017, 44, 119–121. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; I Furlong, L. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef] [PubMed]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Geiler-Samerotte, K.A.; Li, S.; Lazaris, C.; Taylor, A.; Ziv, N.; Ramjeawan, C.; Paaby, A.B.; Siegal, M.L. Extent and context dependence of pleiotropy revealed by high-throughput single-cell phenotyping. PLoS Biol. 2020, 18, e3000836. [Google Scholar] [CrossRef] [PubMed]
- Christodoulaki, E.; Nolte, V.; Lai, W.-Y.; Schlötterer, C. Natural variation in Drosophila shows weak pleiotropic effects. Genome Biol. 2022, 23, 116. [Google Scholar] [CrossRef]
- Campbell, C.; Albert, R. Stabilization of perturbed Boolean network attractors through compensatory interactions. BMC Syst. Biol. 2014, 8, 53. [Google Scholar] [CrossRef]
- Trinh, H.-C.; Le, D.-H.; Kwon, Y.-K. PANET: A GPU-Based Tool for Fast Parallel Analysis of Robustness Dynamics and Feed-Forward/Feedback Loop Structures in Large-Scale Biological Networks. PLoS ONE 2014, 9, e103010. [Google Scholar] [CrossRef]
- Mendes, N.D.; Lang, F.; Le Cornec, Y.-S.; Mateescu, R.; Batt, G.; Chaouiya, C. Composition and abstraction of logical regulatory modules: Application to multicellular systems. Bioinformatics 2013, 29, 749–757. [Google Scholar] [CrossRef]
- Kauffman, S.; Peterson, C.; Samuelsson, B.; Troein, C. Genetic networks with canalyzing Boolean rules are always stable. Proc. Natl. Acad. Sci. USA 2004, 101, 17102–17107. [Google Scholar] [CrossRef]
- Kauffman, S.; Peterson, C.; Samuelsson, B.; Troein, C. Random Boolean network models and the yeast transcriptional network. Proc. Natl. Acad. Sci. USA 2003, 100, 14796–14799. [Google Scholar] [CrossRef]
- Harris, S.E.; Sawhill, B.K.; Wuensche, A.; Kauffman, S. A model of transcriptional regulatory networks based on biases in the observed regulation rules. Complexity 2002, 7, 23–40. [Google Scholar] [CrossRef]
- Samal, A.; Jain, S. The regulatory network of E. coli metabolism as a Boolean dynamical system exhibits both homeostasis and flexibility of response. BMC Syst. Biol. 2008, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Trinh, H.-C.; Kwon, Y.-K. Effective Boolean dynamics analysis to identify functionally important genes in large-scale signaling networks. Biosystems 2015, 137, 64–72. [Google Scholar] [CrossRef]
- Davidich, M.I.; Bornholdt, S. Boolean Network Model Predicts Knockout Mutant Phenotypes of Fission Yeast. PLoS ONE 2013, 8, e71786. [Google Scholar] [CrossRef]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; A Roth, J.; Tanaka, N. Overexpression of the wild-type p53 gene inhibits NF-κB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, C.; Chan, C.; Kang, W.; Sun, Y.; Emerson, R.; Robins, H.; Levine, A.J. The evolution of thymic lymphomas in p53 knockout mice. Genes Dev. 2014, 28, 2613–2620. [Google Scholar] [CrossRef]
- Prelich, G. Gene Overexpression: Uses, Mechanisms, and Interpretation. Genetics 2012, 190, 841–854. [Google Scholar] [CrossRef]
- Taylor, M.A.; Wilczek, A.M.; Roe, J.L.; Welch, S.M.; Runcie, D.E.; Cooper, M.D.; Schmitt, J. Large-effect flowering time mutations reveal conditionally adaptive paths through fitness landscapes in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2019, 116, 17890–17899. [Google Scholar] [CrossRef]
- Bozic, I.; Nowak, M.A. Timing and heterogeneity of mutations associated with drug resistance in metastatic cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15964–15968. [Google Scholar] [CrossRef]
- Prill, R.J.; Iglesias, P.; Levchenko, A. Dynamic Properties of Network Motifs Contribute to Biological Network Organization. PLoS Biol. 2005, 3, e343. [Google Scholar] [CrossRef]
- Klein, C.; Marino, A.; Sagot, M.-F.; Milreu, P.V.; Brilli, M. Structural and dynamical analysis of biological networks. Briefings Funct. Genom. 2012, 11, 420–433. [Google Scholar] [CrossRef]
- Kwon, Y.-K. Properties of Boolean dynamics by node classification using feedback loops in a network. BMC Syst. Biol. 2016, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Thieffry, D.; Kaufman, M. Dynamical behaviour of biological regulatory networks—I. Biological role of feedback loops and practical use of the concept of the loop-characteristic state. Bull. Math. Biol. 1995, 57, 247–276. [Google Scholar] [CrossRef]
- Hetmanski, J.H.R.; Zindy, E.; Schwartz, J.M.; Caswell, P.T. A MAPK-Driven Feedback Loop Suppresses Rac Activity to Promote RhoA-Driven Cancer Cell Invasion. PLoS Comput. Biol. 2016, 12, e1004909. [Google Scholar] [CrossRef] [PubMed]
- Yeger-Lotem, E.; Sattath, S.; Kashtan, N.; Itzkovitz, S.; Milo, R.; Pinter, R.Y.; Alon, U.; Margalit, H. Network motifs in integrated cellular networks of transcription–regulation and protein–protein interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5934–5939. [Google Scholar] [CrossRef]
- Freeman, L.C. A Set of Measures of Centrality Based on Betweenness. Sociometry 1977, 40, 35–41. [Google Scholar] [CrossRef]
- Shimbel, A. Structural parameters of communication networks. Bull. Math. Biol. 1953, 15, 501–507. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Albert, R. Emergence of Scaling in Random Networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef]
- Schoonjans, F.; Zalata, A.; Depuydt, C.; Comhaire, F. MedCalc: A new computer program for medical statistics. Comput. Methods Programs Biomed. 1995, 48, 257–262. [Google Scholar] [CrossRef]
- Cui, Q.; Ma, Y.; Jaramillo, M.; Bari, H.; Awan, A.; Yang, S.; Zhang, S.; Liu, L.; Lu, M.; O’Connor-McCourt, M.; et al. A map of human cancer signaling. Mol. Syst. Biol. 2007, 3, 152. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Han, Z.-G.; He, K.-Y. Malignancy of Cancers and Synthetic Lethal Interactions Associated with Mutations of Cancer Driver Genes. Medicine 2016, 95, e2697. [Google Scholar] [CrossRef]
- Loeb, K.R.; Loeb, L.A. Significance of multiple mutations in cancer. Carcinogenesis 2000, 21, 379–385. [Google Scholar] [CrossRef]
- Kent, D.G.; Green, A.R. Order Matters: The Order of Somatic Mutations Influences Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a027060. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, M.; Wang, Y.; Fan, J.; Sun, Q.; Ji, M.; Fan, X.; Xie, J.; Dai, J.; Jin, G.; et al. Cross-Cancer Pleiotropic Analysis Reveals Novel Susceptibility Loci for Lung Cancer. Front. Oncol. 2020, 9, 1492. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, M.A.; Goh, K.-I.; Cusick, M.E.; Barabasi, A.L.; Vidal, M. Drug—Target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, M.; Fortney, K.; Jurisica, I. Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods 2012, 57, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Xu, Y.; Guo, Y.; Yu, Z.; Feng, G.; Liu, P.; Luan, M.; Zhu, H.; Liu, G.; Zhang, M.; et al. The drug target genes show higher evolutionary conservation than non-target genes. Oncotarget 2015, 7, 4961–4971. [Google Scholar] [CrossRef]
- Zhu, P.; Aliabadi, H.M.; Uludağ, H.; Han, J. Identification of Potential Drug Targets in Cancer Signaling Pathways using Stochastic Logical Models. Sci. Rep. 2016, 6, 23078. [Google Scholar] [CrossRef]
- Bedi, O.; Dhawan, V.; Sharma, P.L.; Kumar, P. Pleiotropic effects of statins: New therapeutic targets in drug design. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016, 389, 695–712. [Google Scholar] [CrossRef]
- Winzeler, E.A.; Shoemaker, D.D.; Astromoff, A.; Liang, H.; Anderson, K.; Andre, B.; Bangham, R.; Benito, R.; Boeke, J.D.; Bussey, H.; et al. Functional Characterization of the S. cerevisiae Genome by Gene Deletion and Parallel Analysis. Science 1999, 285, 901–906. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef]
- Bien, S.A.; Peters, U. Moving from one to many: Insights from the growing list of pleiotropic cancer risk genes. Br. J. Cancer 2019, 120, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Shi, N.; Feng, Y.; Li, Y.; Tan, J.; Xu, F.; Wang, W.; Sun, C.; Deng, H.; Yang, Y.; et al. Identification of 67 Pleiotropic Genes Associated With Seven Autoimmune/Autoinflammatory Diseases Using Multivariate Statistical Analysis. Front. Immunol. 2020, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Large, E.E.; Padmanabhan, R.; Watkins, K.L.; Campbell, R.F.; Xu, W.; McGrath, P.T. Modeling of a negative feedback mechanism explains antagonistic pleiotropy in reproduction in domesticated Caenorhabditis elegans strains. PLoS Genet. 2017, 13, e1006769. [Google Scholar] [CrossRef] [PubMed]
- Ananthasubramaniam, B.; Herzel, H. Positive Feedback Promotes Oscillations in Negative Feedback Loops. PLoS ONE 2014, 9, e104761. [Google Scholar] [CrossRef]
- Le, D.-H.; Kwon, Y.-K. The effects of feedback loops on disease comorbidity in human signaling networks. Bioinformatics 2011, 27, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Pei, F.; Li, H.; Liu, B.; Bahar, I. Quantitative Systems Pharmacological Analysis of Drugs of Abuse Reveals the Pleiotropy of Their Targets and the Effector Role of mTORC1. Front. Pharmacol. 2019, 10, 191. [Google Scholar] [CrossRef]
- Gu, J.J.; Ryu, J.R.; Pendergast, A.M. Abl tyrosine kinases in T-cell signaling. Immunol. Rev. 2009, 228, 170–183. [Google Scholar] [CrossRef]
- Graham, S.E.; Nielsen, J.B.; Zawistowski, M.; Zhou, W.; Fritsche, L.G.; Gabrielsen, M.E.; Skogholt, A.H.; Surakka, I.; Hornsby, W.E.; Fermin, D.; et al. Sex-specific and pleiotropic effects underlying kidney function identified from GWAS meta-analysis. Nat. Commun. 2019, 10, 1847. [Google Scholar] [CrossRef]
- Hill, W.G.; Zhang, X.-S. Assessing pleiotropy and its evolutionary consequences: Pleiotropy is not necessarily limited, nor need it hinder the evolution of complexity. Nat. Rev. Genet. 2012, 13, 296. [Google Scholar] [CrossRef]
- Brown, S.D.M.; Lad, H.V. The dark genome and pleiotropy: Challenges for precision medicine. Mamm. Genome 2019, 30, 212–216. [Google Scholar] [CrossRef]
- Naldi, A.; Carneiro, J.; Chaouiya, C.; Thieffry, D. Diversity and Plasticity of Th Cell Types Predicted from Regulatory Network Modelling. PLoS Comput. Biol. 2010, 6, e1000912. [Google Scholar] [CrossRef] [PubMed]
- Trinh, H.C.; Kwon, Y.K. Edge-based sensitivity analysis of signaling networks by using Boolean dynamics. Bioinformatics 2016, 32, i763–i771. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
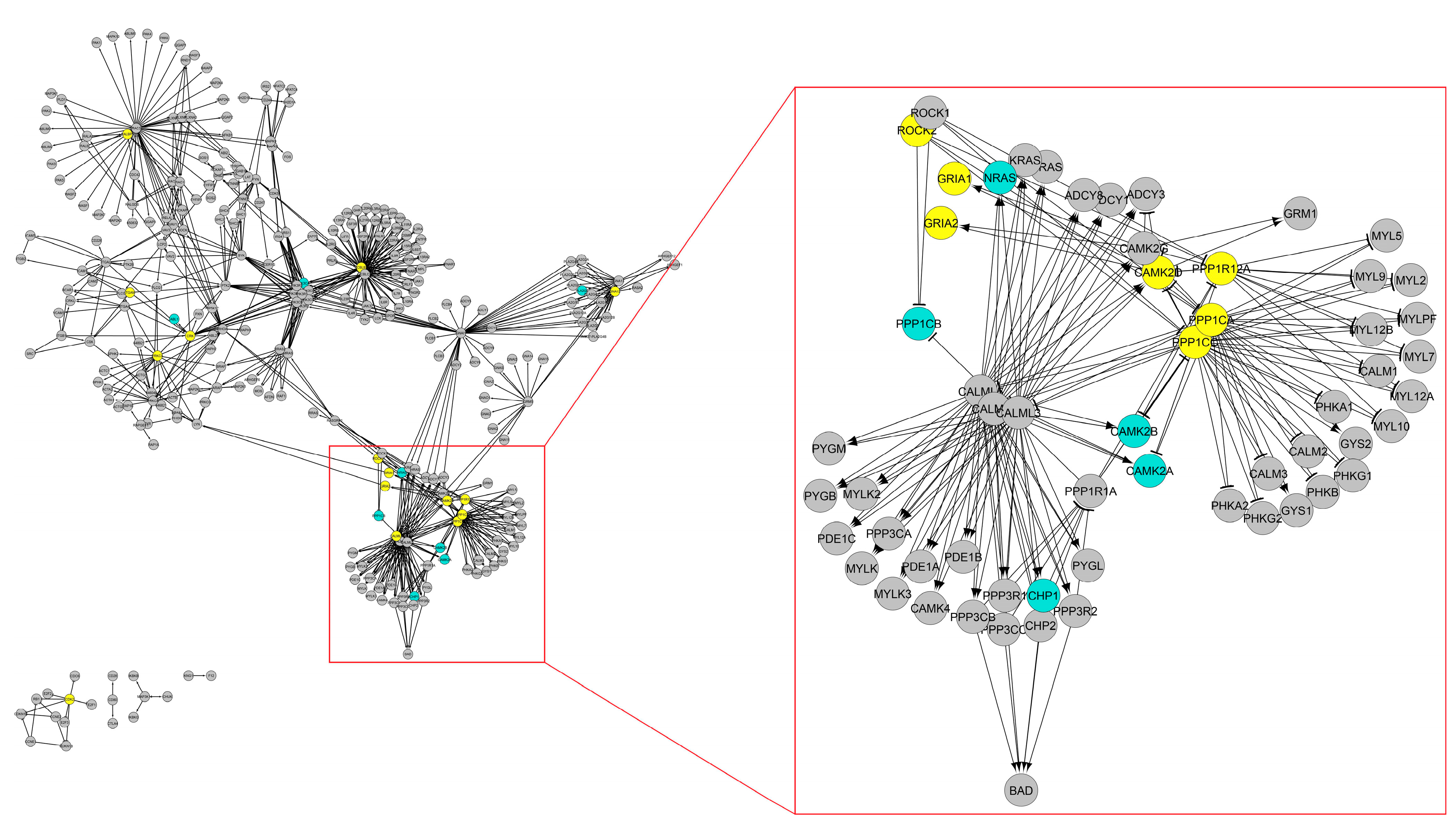{kind=link}
| No. | Gene Name | HPO-Associated | |
|---|---|---|---|
| 1 | COL2A1 | 842 | 22 |
| 2 | FGFR1 | 1045 | 7 |
| 3 | FGFR2 | 1137 | 3 |
| 4 | FGFR3 | 1006 | 5 |
| 5 | LIMK1 | 733 | 2 |
| 6 | NRAS | 728 | 1 |
| 7 | PIK3CA | 751 | 9 |
| 8 | PRKAR1A | 697 | 1 |
| 9 | PTEN | 838 | 107 |
| 10 | TGFBR2 | 586 | 78 |
| Total | 8363 | 235 | |
| No. | Gene Name | HPO | NuCancer | DT | ES | TSG | OCG | DG | Deg | In-Deg | Out-Deg | NuFBL | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ABL1 | 1 | 0.6 | −0.35 | 23 | 1 | 1 | 0 | 1 | 1 | 27 | 15 | 12 | 4588 |
| 2 | PIK3CA | 1 | 0.7 | −0.06 | 19 | 1 | 1 | 0 | 1 | 1 | 51 | 45 | 6 | 40,101 |
| 3 | EGFR | 1 | 0.45 | 0.11 | 21 | 1 | 1 | 0 | 1 | 1 | 73 | 41 | 32 | 225 |
| 4 | SERPINA1 | 1 | 0.21 | −0.34 | 60 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 |
| 5 | CAMK2B | 1 | 0.44 | −0.04612 | 0 | 1 | 0 | 0 | 0 | 1 | 15 | 10 | 5 | 19,612 |
| 6 | PPP1CB | 1 | 0.55 | −0.06861 | 1 | 0 | 1 | 0 | 0 | 1 | 28 | 3 | 25 | 30,526 |
| 7 | CAMK2A | 1 | 0.63 | −0.30841 | 0 | 1 | 1 | 0 | 0 | 1 | 15 | 10 | 5 | 19,612 |
| 8 | NRAS | 1 | 0. 78 | −0.40583 | 1 | 0 | 1 | 0 | 1 | 1 | 44 | 28 | 16 | 20,154 |
| 9 | CHP1 | 1 | 0.45 | −0.08359 | 0 | 1 | 1 | 0 | 0 | 1 | 10 | 8 | 2 | 2970 |
| 10 | PLA2G6 | 1 | 1 | −0.42082 | 303 | 1 | 1 | 0 | 0 | 1 | 10 | 10 | 0 | 0 |
| 11 | IGFBP3 | 0 | 0.45 | 0.08 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 0 |
| 12 | PRKCA | 0 | 0.48 | 0.06 | 0 | 1 | 1 | 0 | 1 | 1 | 24 | 7 | 17 | 10,054 |
| 13 | ITGAM | 0 | 0.86 | −0.4 | 0 | 0 | 0 | 0 | 0 | 1 | 9 | 3 | 6 | 260 |
| 14 | ROCK2 | 0 | 0.59 | −0.12 | 0 | 1 | 1 | 0 | 0 | 1 | 7 | 3 | 4 | 39,206 |
| 15 | PPP1CC | 0 | 0.42 | 0.09 | 0 | 1 | 1 | 0 | 0 | 1 | 28 | 3 | 28 | 30,526 |
| 16 | PPP1CA | 0 | 0.52 | −0.02 | 0 | 1 | 1 | 1 | 0 | 1 | 28 | 3 | 25 | 30,526 |
| 17 | PRKAA1 | 0 | 0.40 | −0.2035 | 0 | 1 | 1 | 1 | 0 | 1 | 3 | 0 | 3 | 0 |
| 18 | PPP1R12A | 0 | 0.73 | −0.27844 | 0 | 0 | 1 | 0 | 0 | 1 | 15 | 3 | 12 | 7624 |
| 19 | CDK2 | 0 | 0.61 | −0.36836 | 0 | 1 | 1 | 1 | 0 | 1 | 10 | 3 | 7 | 4 |
| 20 | PPP1CC | 0 | 0.451327 | 0.043803 | 0 | 1 | 1 | 0 | 0 | 1 | 28 | 3 | 25 | 30,526 |
| 21 | RALBP1 | 0 | 0.428571 | −0.00116 | 0 | 0 | 0 | 0 | 0 | 1 | 6 | 2 | 4 | 648 |
| 22 | CBLB | 0 | 0.6045 | −0.02364 | 1 | 0 | 1 | 0 | 1 | 1 | 60 | 4 | 56 | 1202 |
| 23 | WNT11 | 0 | 0.5455 | −0.34588 | 0 | 0 | 1 | 1 | 0 | 1 | 17 | 7 | 10 | 0 |
| 24 | CAMK2D | 0 | 0.75 | −0.27844 | 0 | 1 | 0 | 0 | 0 | 1 | 15 | 10 | 5 | 19,612 |
| 25 | CRK | 0 | 1 | −0.42082 | 0 | 0 | 1 | 0 | 1 | 1 | 45 | 31 | 14 | 5391 |
| 26 | CALML5 | 0 | 0.4455 | 0.036309 | 0 | 0 | 0 | 0 | 0 | 1 | 36 | 9 | 27 | 11,451 |
| 27 | GRIA1 | 0 | 0.90625 | −0.39834 | 0 | 1 | 1 | 0 | 0 | 1 | 10 | 9 | 1 | 13,272 |
| 28 | GRIA2 | 0 | 0.619048 | −0.18102 | 0 | 1 | 1 | 0 | 0 | 1 | 10 | 9 | 1 | 13,272 |
| 29 | GNA12 | 0 | 0.317647 | 0.013827 | 1 | 0 | 1 | 0 | 1 | 1 | 23 | 6 | 17 | 2528 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazaya, M.; Kwon, Y.-K. In Silico Pleiotropy Analysis in KEGG Signaling Networks Using a Boolean Network Model. Biomolecules 2022, 12, 1139. https://doi.org/10.3390/biom12081139
Mazaya M, Kwon Y-K. In Silico Pleiotropy Analysis in KEGG Signaling Networks Using a Boolean Network Model. Biomolecules. 2022; 12(8):1139. https://doi.org/10.3390/biom12081139
Chicago/Turabian StyleMazaya, Maulida, and Yung-Keun Kwon. 2022. "In Silico Pleiotropy Analysis in KEGG Signaling Networks Using a Boolean Network Model" Biomolecules 12, no. 8: 1139. https://doi.org/10.3390/biom12081139