
Nanoscopic Spatial Association between Ras and Phosphatidylserine on the Cell Membrane Studied with Multicolor Super Resolution Microscopy



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmids
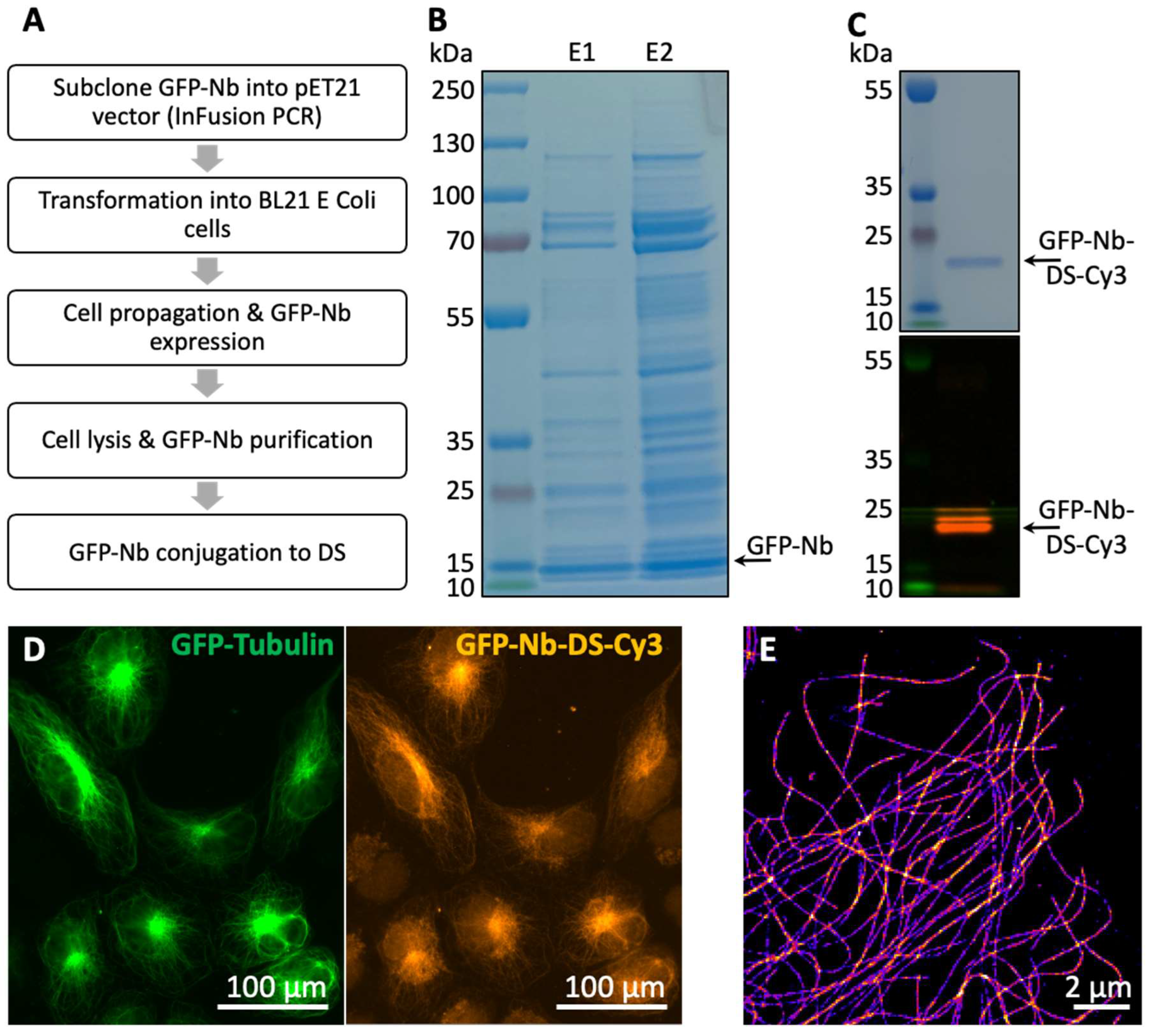
2.3. Nanobody Purification and Conjugation with DNA Oligos
2.4. Cell Culture and Transfection
2.5. Immunostaining
2.6. Microscopy
2.7. Data Acquisition and Analysis
3. Results
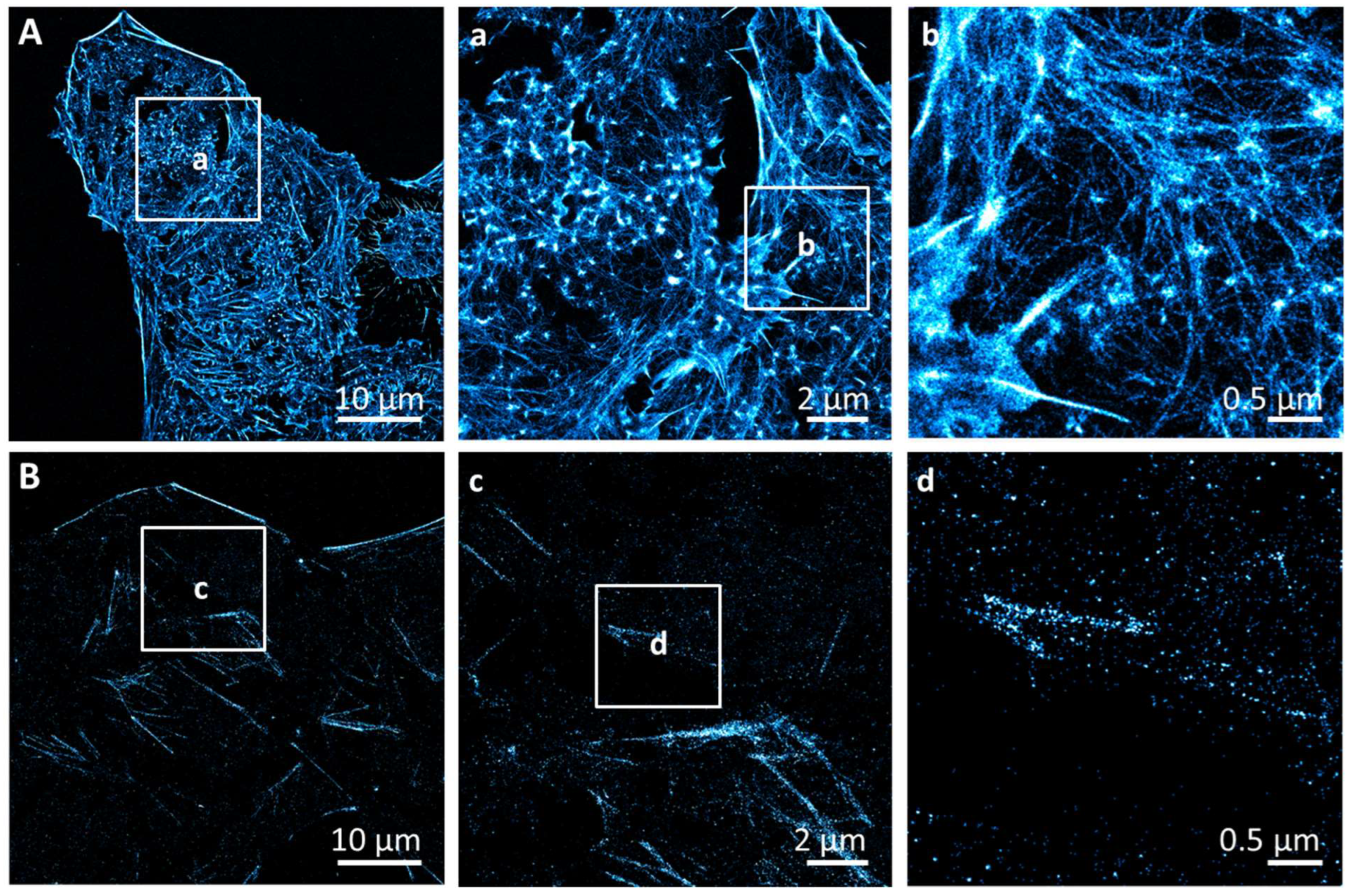
3.1. Phosphatidylserine Plasma Membrane Distribution at the Nanoscale
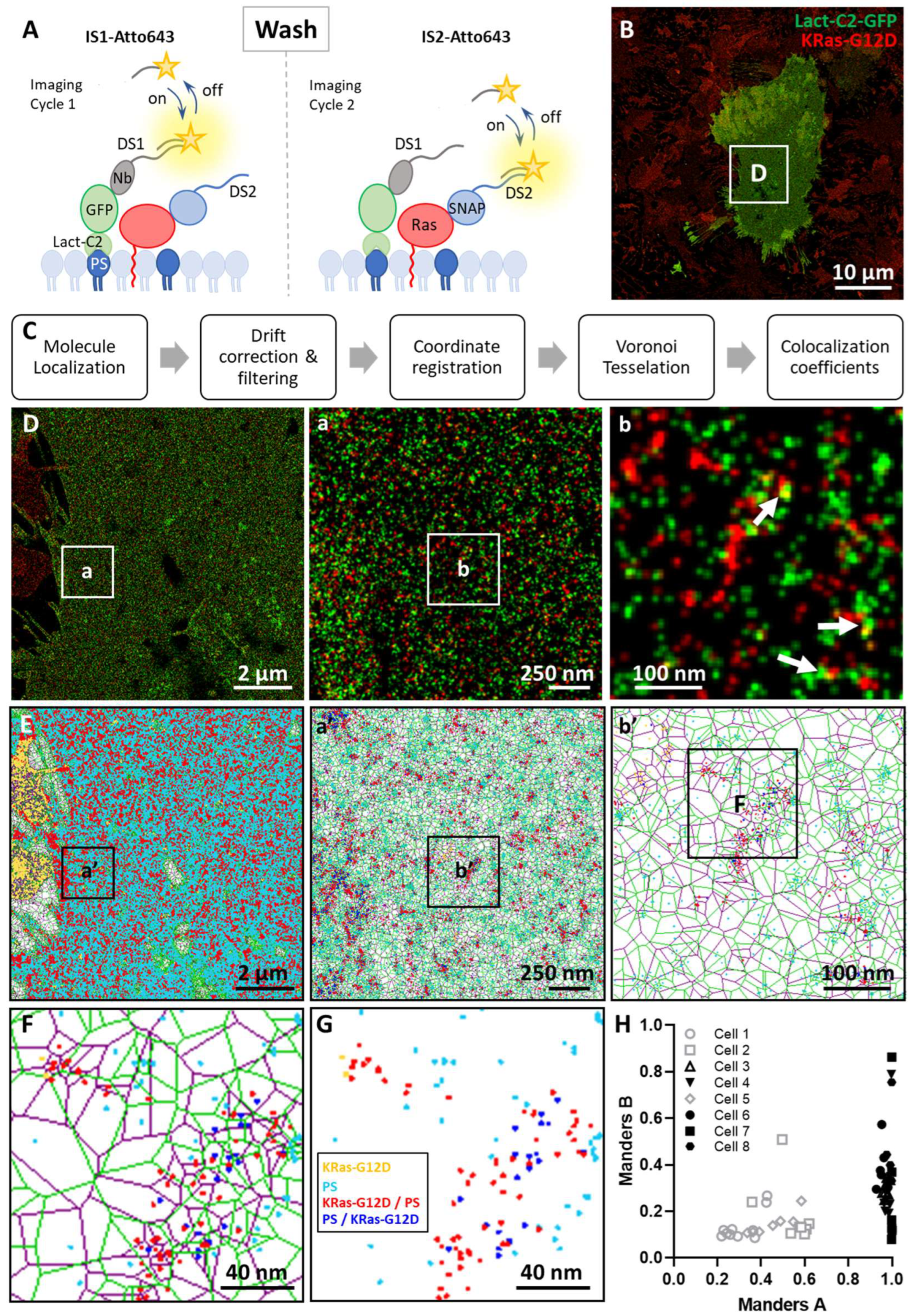
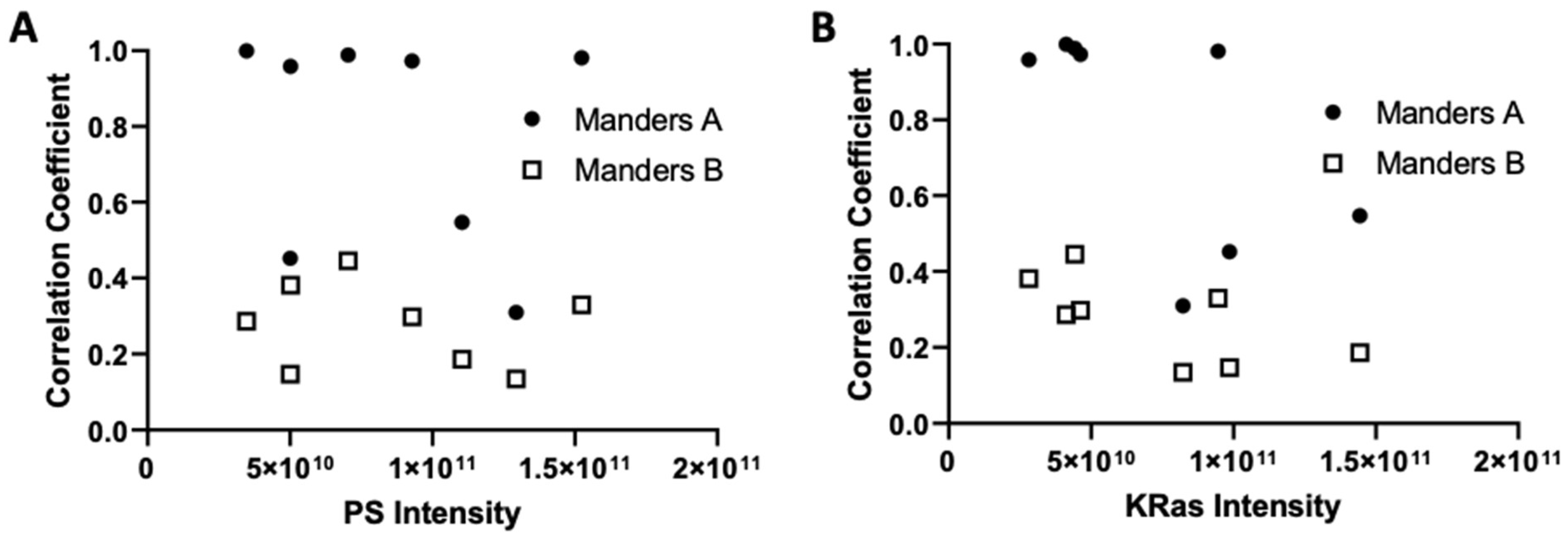
3.2. Imaging the Spatial Association between KRas-G12D and PS at the Nanoscale
3.3. Imaging and Quantitating Interaction between HRas-G12V and PS
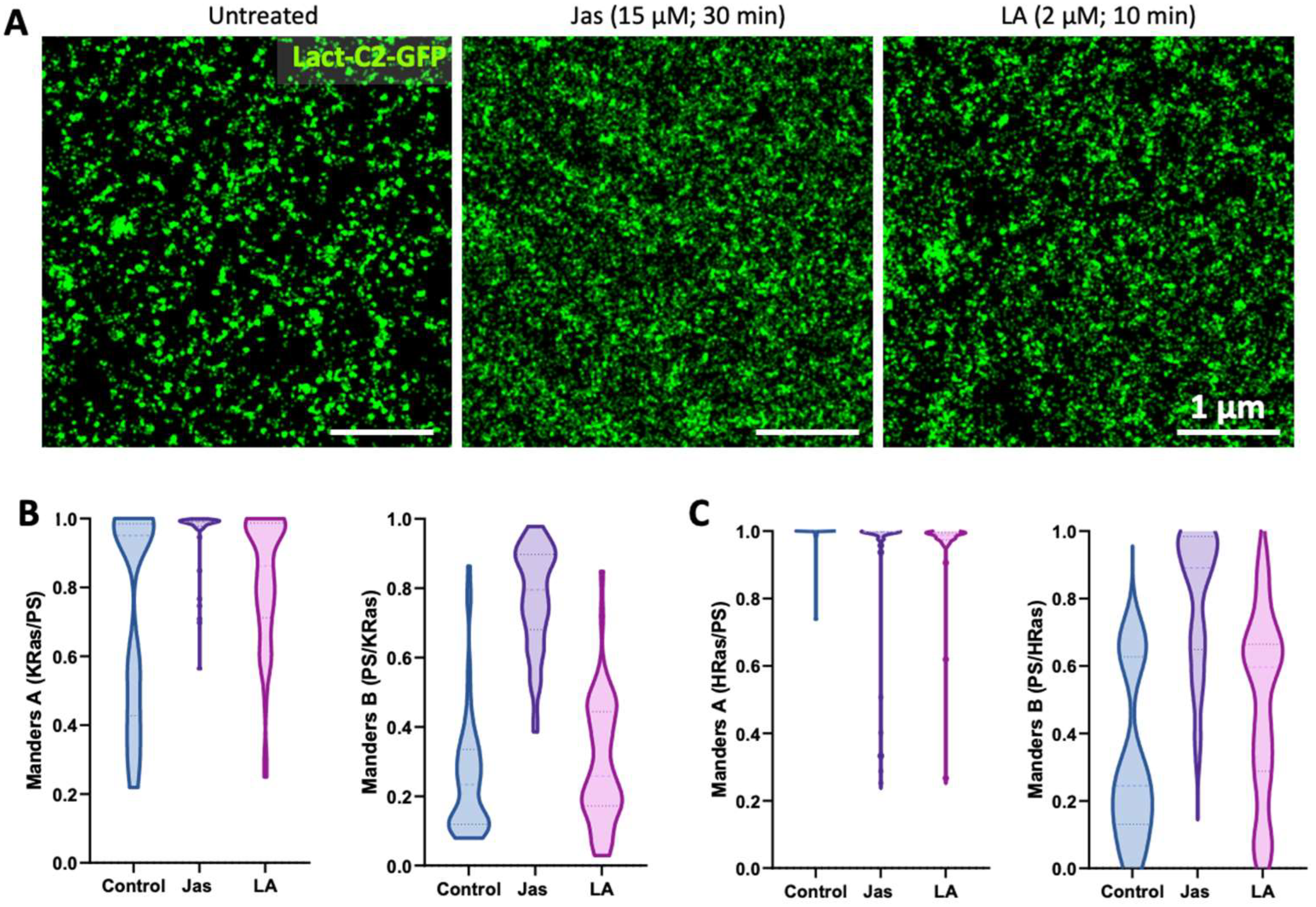
3.4. Dependence of Ras-PS Association on the Actin Cytoskeleton
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




References
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasper, R.; Wittinghofer, F. The Ras switch in structural and historical perspective. Biol. Chem. 2019, 401, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Medarde, A.; De Las Rivas, J.; Santos, E. 40 Years of RAS—A Historic Overview. Genes 2021, 12, 681. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.H.; Cochrane, C.G.; Bourne, J.R.; Solski, A.P.; Buss, E.J.; Der, C.J. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc. Natl. Acad. Sci. USA 1990, 87, 3042–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willumsen, B.M.; Christensen, A.; Hubbert, N.L.; Papageorge, A.G.; Lowy, D.R. The p21 ras C-terminus is required for transformation and membrane association. Nature 1984, 310, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Schafer, W.R.; Trueblood, C.E.; Yang, C.-C.; Mayer, M.P.; Rosenberg, S.; Poulter, C.D.; Kim, S.-H.; Rine, J. Enzymatic Coupling of Cholesterol Intermediates to a Mating Pheromone Precursor and to the Ras Protein. Science 1990, 249, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Schafer, W.R.; Kim, R.; Sterne, R.; Thorner, J.; Kim, S.-H.; Rine, J. Genetic and Pharmacological Suppression of Oncogenic Mutations in RAS Genes of Yeast and Humans. Science 1989, 245, 379–385. [Google Scholar] [CrossRef]
- Prior, I.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Plowman, S.J.; Muncke, C.; Parton, R.G.; Hancock, J.F.; Simons, K. H-Ras, K-Ras, and Inner Plasma Membrane Raft Proteins Operate in Nanoclusters with Differential Dependence on the Actin Cytoskeleton. Proc. Natl. Acad. Sci. USA 2005, 25, 15500–15505. [Google Scholar] [CrossRef] [Green Version]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Phelps, C.; Huang, T.; Mostofian, B.; Wu, L.; Zhang, Y.; Tao, K.; Chang, Y.H.; Stork, P.J.; Gray, J.W.; et al. High-throughput, single-particle tracking reveals nested membrane domains that dictate KRasG12D diffusion and trafficking. eLife 2019, 8, e46393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hancock, J. Deciphering lipid codes: K-Ras as a paradigm. Traffic 2017, 19, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Chung, S.; Kim, S.; Li, Z.; Manor, D.; Buck, M. K-Ras G-domain binding with signaling lipid phosphatidylinositol (4,5)-phosphate (PIP2): Membrane association, protein orientation, and function. J. Biol. Chem. 2019, 294, 7068–7084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, M.A.; Stephen, A.G.; Sligar, S.G. PIP2 Influences the Conformational Dynamics of Membrane-Bound KRAS4b. Biochemistry 2019, 58, 3537–3545. [Google Scholar] [CrossRef]
- Ngo, V.; Sarkar, S.; Neale, C.; Garcia, A.E. How Anionic Lipids Affect Spatiotemporal Properties of KRAS4B on Model Membranes. J. Phys. Chem. B 2020, 124, 5434–5453. [Google Scholar] [CrossRef]
- Travers, T.; López, C.A.; Agamasu, C.; Hettige, J.J.; Messing, S.; García, A.E.; Stephen, A.G.; Gnanakaran, S. Anionic Lipids Impact RAS-Binding Site Accessibility and Membrane Binding Affinity of CRAF RBD-CRD. Biophys. J. 2020, 119, 525–538. [Google Scholar] [CrossRef]
- Gorfe, A.A.; Hanzal-Bayer, M.; Abankwa, D.; Hancock, J.; McCammon, J.A. Structure and Dynamics of the Full-Length Lipid-Modified H-Ras Protein in a 1,2-Dimyristoylglycero-3-phosphocholine Bilayer. J. Med. Chem. 2007, 50, 674–684. [Google Scholar] [CrossRef]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Freed, R.N.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-Y.; Enomoto, M.; Gebregiworgis, T.; Gasmi-Seabrook, G.M.C.; Ikura, M.; Marshall, C.B. Oncogenic KRAS G12D mutation promotes dimerization through a second, phosphatidylserine–dependent interface: A model for KRAS oligomerization. Chem. Sci. 2021, 12, 12827–12837. [Google Scholar] [CrossRef]
- Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R. Membrane-associated Ras dimers are isoform-specific: K-Ras dimers differ from H-Ras dimers. Biochem. J. 2016, 473, 1719–1732. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Neale, C.; Carpenter, T.S.; Shrestha, R.; López, C.A.; Tran, T.H.; Oppelstrup, T.; Bhatia, H.; Stanton, L.G.; Zhang, X.; et al. Machine learning–driven multiscale modeling reveals lipid-dependent dynamics of RAS signaling proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e113297119. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, H.; Rodkey, T.; Ariotti, N.; Parton, R.G.; Hancock, J.F. Signal Integration by Lipid-Mediated Spatial Cross Talk between Ras Nanoclusters. Mol. Cell. Biol. 2014, 34, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.-J.; van der Hoeven, D.; Zhou, Y.; Maekawa, M.; Ma, X.; Chen, W.; Fairn, G.D.; Hancock, J.F. Inhibition of Acid Sphingomyelinase Depletes Cellular Phosphatidylserine and Mislocalizes K-Ras from the Plasma Membrane. Mol. Cell. Biol. 2016, 36, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Hancock, J. Ras Proteolipid Nanoassemblies on the Plasma Membrane Sort Lipids With High Selectivity. Adv. Biomembr. Lipid Self-Assem. 2017, 25, 41–62. [Google Scholar] [CrossRef]
- Liang, H.; Mu, H.; Jean-Francois, F.; Lakshman, B.; Sarkar-Banerjee, S.; Zhuang, Y.; Zeng, Y.; Gao, W.; Zaske, A.M.; Nissley, D.V.; et al. Membrane curvature sensing of the lipid-anchored K-Ras small GTPase. Life Sci. Alliance 2019, 2, e201900343. [Google Scholar] [CrossRef]
- Kay, J.G.; Koivusalo, M.; Ma, X.; Wohland, T.; Grinstein, S. Phosphatidylserine dynamics in cellular membranes. Mol. Biol. Cell 2012, 23, 2198–2212. [Google Scholar] [CrossRef]
- Yeung, T.; Gilbert, G.E.; Shi, J.; Silvius, J.; Kapus, A.; Grinstein, S. Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science 2008, 319, 210–213. [Google Scholar] [CrossRef]
- Zhou, Y.; Prakash, P.S.; Liang, H.; Gorfe, A.A.; Hancock, J.F. The KRAS and other prenylated polybasic domain membrane anchors recognize phosphatidylserine acyl chain structure. Proc. Natl. Acad. Sci. USA 2021, 118, e2014605118. [Google Scholar] [CrossRef]
- Kattan, W.; Chen, W.; Ma, X.; Lan, T.H.; Van Der Hoeven, D.; van der Hoeven, R.; Hancock, J.F. Targeting plasma membrane phosphatidylserine content to inhibit oncogenic KRAS function. Life Sci. Alliance 2019, 2, e201900431. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wong, C.-O.; Cho, K.-J.; van der Hoeven, D.; Liang, H.; Thakur, D.P.; Luo, J.; Babic, M.; Zinsmaier, K.E.; Zhu, M.X.; et al. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science 2015, 349, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Fairn, G.D.; Schieber, N.L.; Ariotti, N.; Murphy, S.; Kuerschner, L.; Webb, R.I.; Grinstein, S.; Parton, R.G. High-resolution mapping reveals topologically distinct cellular pools of phosphatidylserine. J. Cell Biol. 2011, 194, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [Green Version]
- Hess, S.T.; Girirajan, T.P.; Mason, M.D. Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys. J. 2006, 91, 4258–4272. [Google Scholar] [CrossRef] [Green Version]
- Jungmann, R.; Avendaño, M.S.; Woehrstein, J.B.; Dai, M.; Shih, W.M.; Yin, P. Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 2014, 11, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Civitci, F.; Shangguan, J.; Zheng, T.; Tao, K.; Rames, M.; Kenison, J.; Zhang, Y.; Wu, L.; Phelps, C.; Esener, S.; et al. Fast and multiplexed superresolution imaging with DNA-PAINT-ERS. Nat. Commun. 2020, 11, 4339. [Google Scholar] [CrossRef]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef]
- Katoh, Y.; Nozaki, S.; Hartanto, D.; Miyano, R.; Nakayama, K. Architectures of multisubunit complexes revealed by a visible immunoprecipitation assay using fluorescent fusion proteins. J. Cell Sci. 2015, 128, 2351–2362. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, A.; Huang, T.; Lin, L.-J.; Nan, X. Photoactivated Localization Microscopy with Bimolecular Fluorescence Complementation (BiFC-PALM). J. Vis. Exp. 2015, e53154. [Google Scholar] [CrossRef] [Green Version]
- Levet, F.; Julien, G.; Galland, R.; Butler, C.; Beghin, A.; Chazeau, A.; Hoess, P.; Ries, J.; Giannone, G.; Sibarita, J.-B. A tessellation-based colocalization analysis approach for single-molecule localization microscopy. Nat. Commun. 2019, 10, 2379. [Google Scholar] [CrossRef] [Green Version]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-Delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2009, 17, 133–138. [Google Scholar] [CrossRef]
- Platonova, E.; Winterflood, C.M.; Junemann, A.; Albrecht, D.; Faix, J.; Ewers, H. Single-molecule microscopy of molecules tagged with GFP or RFP derivatives in mammalian cells using nanobody binders. Methods 2015, 88, 89–97. [Google Scholar] [CrossRef]
- Jungmann, R.; Steinhauer, C.; Scheible, M.; Kuzyk, A.; Tinnefeld, P.; Simmel, F.C. Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett. 2010, 10, 4756–4761. [Google Scholar] [CrossRef]
- Schnitzbauer, J.; Wang, Y.; Zhao, S.; Bakalar, M.; Nuwal, T.; Chen, B.; Huang, B. Correlation analysis framework for localization-based superresolution microscopy. Proc. Natl. Acad. Sci. USA 2018, 115, 3219–3224. [Google Scholar] [CrossRef] [Green Version]
- Levet, F.; Hosy, E.; Kechkar, A.; Butler, C.; Beghin, A.; Choquet, D.; Sibarita, J.-B. SR-Tesseler: A method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods 2015, 12, 1065–1071. [Google Scholar] [CrossRef]
- Garg, S.; Tang, J.; Rühe, J.; Naumann, C. Actin-induced perturbation of PS lipid–cholesterol interaction: A possible mechanism of cytoskeleton-based regulation of membrane organization. J. Struct. Biol. 2009, 168, 11–20. [Google Scholar] [CrossRef]
- Raghupathy, R.; Anilkumar, A.A.; Polley, A.; Singh, P.P.; Yadav, M.; Johnson, C.; Suryawanshi, S.; Saikam, V.; Sawant, S.D.; Panda, A.; et al. Transbilayer Lipid Interactions Mediate Nanoclustering of Lipid-Anchored Proteins. Cell 2015, 161, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Bubb, M.R.; Spector, I.; Beyer, B.B.; Fosen, K.M. Effects of Jasplakinolide on the Kinetics of Actin Polymerization. J. Biol. Chem. 2000, 275, 5163–5170. [Google Scholar] [CrossRef] [Green Version]
- Ayscough, K. [2] Use of latrunculin-A, an actin monomer-binding drug. Methods Enzymol. 1998, 298, 18–25. [Google Scholar] [CrossRef]
- Creech, M.K.; Wang, J.; Nan, X.; Gibbs, S.L. Superresolution Imaging of Clinical Formalin Fixed Paraffin Embedded Breast Cancer with Single Molecule Localization Microscopy. Sci. Rep. 2017, 7, 40766. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC Regulates a Farnesyl-Electrostatic Switch on K-Ras that Promotes its Association with Bcl-Xl on Mitochondria and Induces Apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef]
- Zhou, Y.; Prakash, P.; Liang, H.; Cho, K.-J.; Gorfe, A.A.; Hancock, J.F. Lipid-Sorting Specificity Encoded in K-Ras Membrane Anchor Regulates Signal Output. Cell 2016, 168, 239–251.e16. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Iwasawa, K.; Kalay, Z.; Tsunoyama, T.-A.; Watanabe, Y.; Umemura, Y.M.; Murakoshi, H.; Suzuki, K.G.N.; Nemoto, Y.L.; Morone, N.; et al. Confined diffusion of transmembrane proteins and lipids induced by the same actin meshwork lining the plasma membrane. Mol. Biol. Cell 2016, 27, 1101–1119. [Google Scholar] [CrossRef]
- Muguruma, M.; Nishimuta, S.; Tomisaka, Y.; Ito, T.; Matsumura, S. Organization of the Functional Domains in Membrane Cytoskeletal Protein Talin1. J. Biochem. 1995, 117, 1036–1042. [Google Scholar] [CrossRef]
- Makuch, R.; Zasada, A.; Mabuchi, K.; Krauze, K.; Wang, C.; Dabrowska, R. Phosphatidylserine liposomes can be tethered by caldesmon to actin filaments. Biophys. J. 1997, 73, 1607–1616. [Google Scholar] [CrossRef] [Green Version]
- Buckles, T.C.; Ziemba, B.P.; Masson, G.R.; Williams, R.L.; Falke, J.J. Single-Molecule Study Reveals How Receptor and Ras Synergistically Activate PI3Kα and PIP3 Signaling. Biophys. J. 2017, 113, 2396–2405. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer: | DNA Sequence |
|---|---|
| Anti-GFP-Nb-F | GAAGGAGATATACATATGGCGCAGGTTCAGCTGGTTGAAAGC |
| Anti-GFP-Nb-R | TGCGGCCGCAAGCTTCTAGCTGCTAACGGTAACCTGGGTG |
| pET21b-F | AAGCTTGCGGCCGCACTCGAG |
| pET21-R | ATGTATATCTCCTTCTTAAAGTTAAACAAAATTATTTCTAGAGGGGAATTGTTATCCGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koester, A.M.; Tao, K.; Szczepaniak, M.; Rames, M.J.; Nan, X. Nanoscopic Spatial Association between Ras and Phosphatidylserine on the Cell Membrane Studied with Multicolor Super Resolution Microscopy. Biomolecules 2022, 12, 1033. https://doi.org/10.3390/biom12081033
Koester AM, Tao K, Szczepaniak M, Rames MJ, Nan X. Nanoscopic Spatial Association between Ras and Phosphatidylserine on the Cell Membrane Studied with Multicolor Super Resolution Microscopy. Biomolecules. 2022; 12(8):1033. https://doi.org/10.3390/biom12081033
Chicago/Turabian StyleKoester, Anna M., Kai Tao, Malwina Szczepaniak, Matthew J. Rames, and Xiaolin Nan. 2022. "Nanoscopic Spatial Association between Ras and Phosphatidylserine on the Cell Membrane Studied with Multicolor Super Resolution Microscopy" Biomolecules 12, no. 8: 1033. https://doi.org/10.3390/biom12081033