

l-Ornithine-N5-monooxygenase (PvdA) Substrate Analogue Inhibitors for Pseudomonas aeruginosa Infections Treatment: Drug Repurposing Computational Studies
 , , , and
, , , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Collection of Substrate Analogues
2.2. Computational Details
2.3. Preparation of Protein and Ligand Structures
2.4. Molecular Docking Studies
2.5. Prediction of Drug Properties
2.6. Drug-Likeness
2.7. Molecular Dynamics Simulation
3. Results
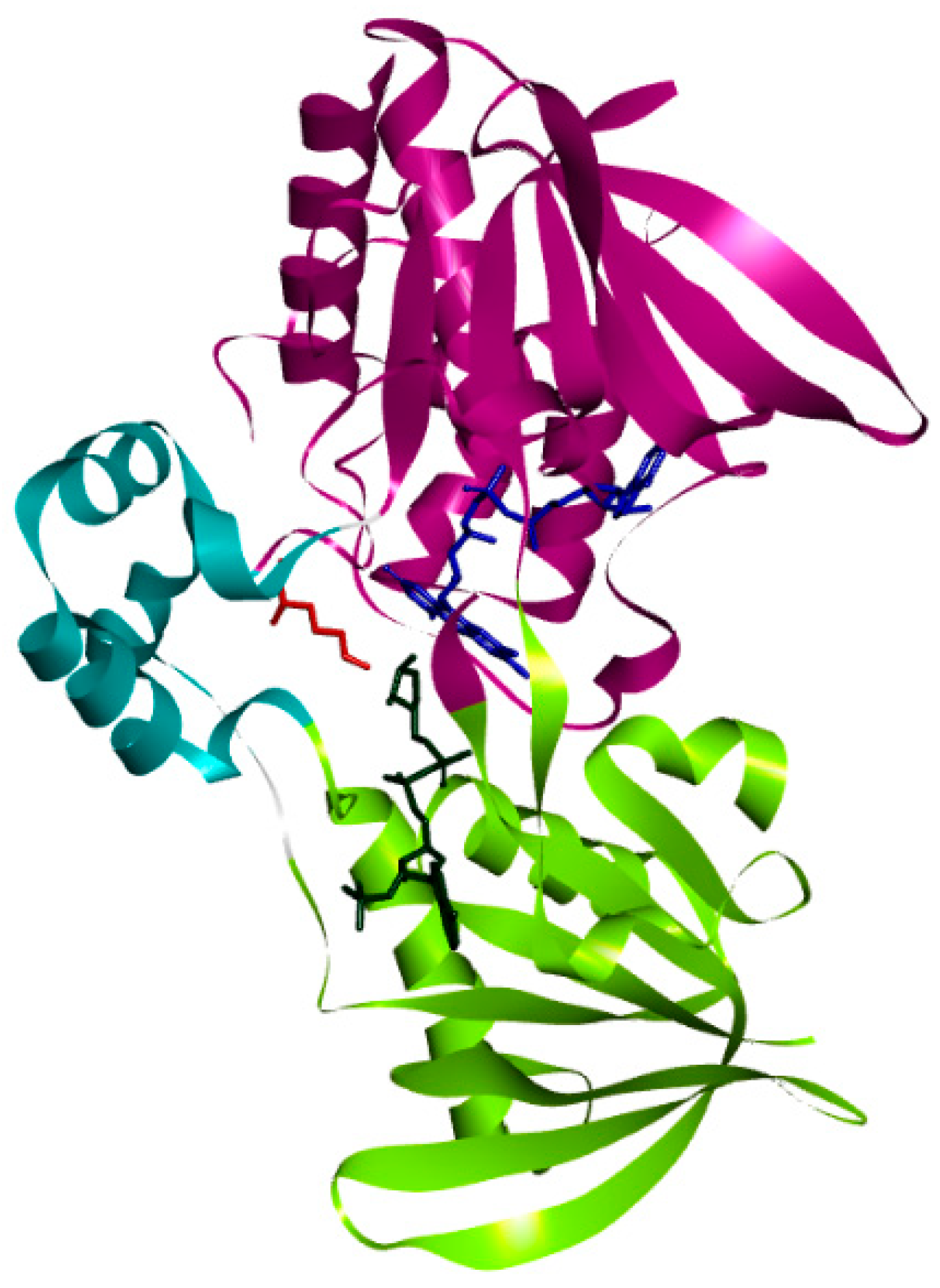
3.1. Structure of PvdA
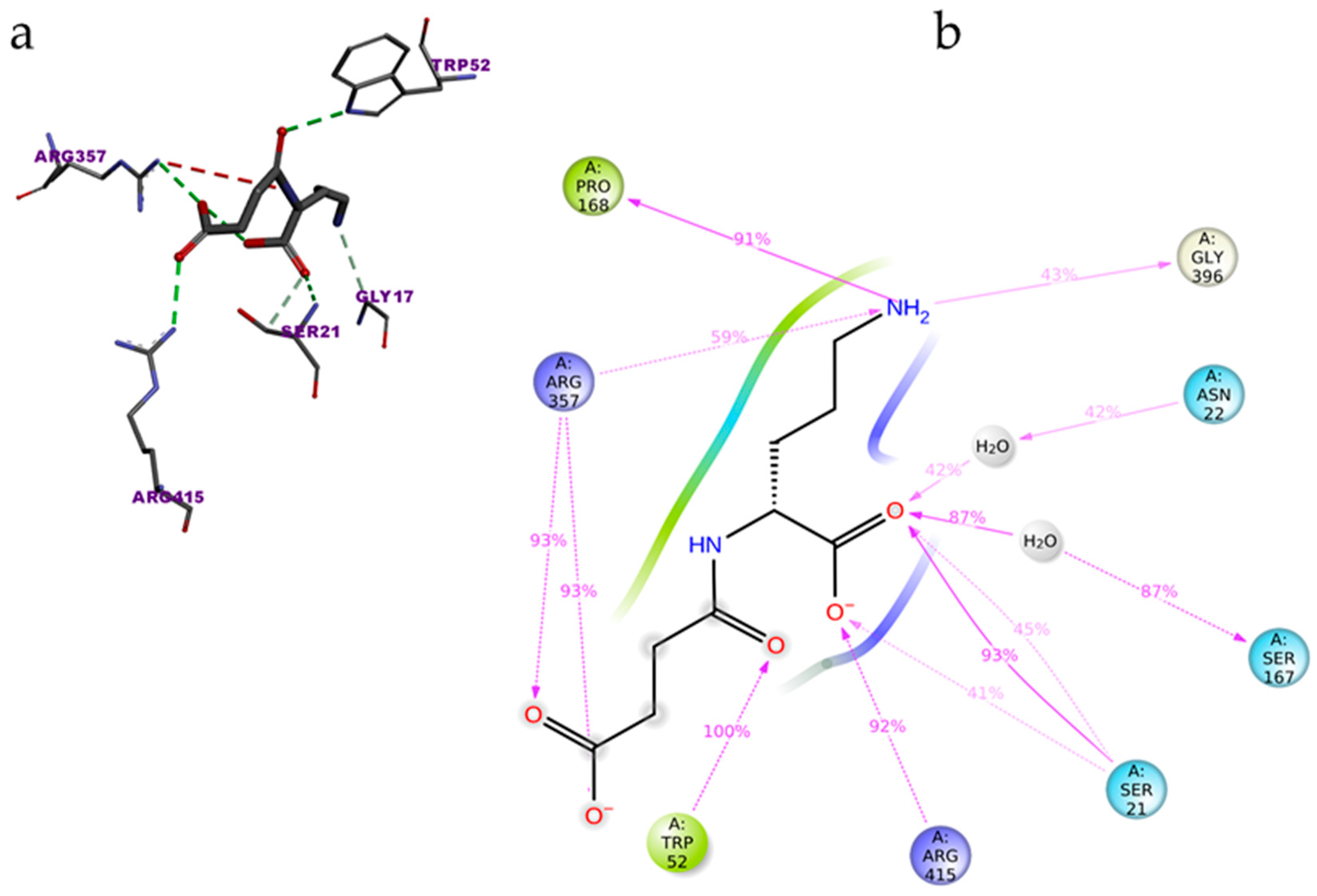
3.2. Top Binders of PvdA
3.3. ADME Properties and Drug-Likeness
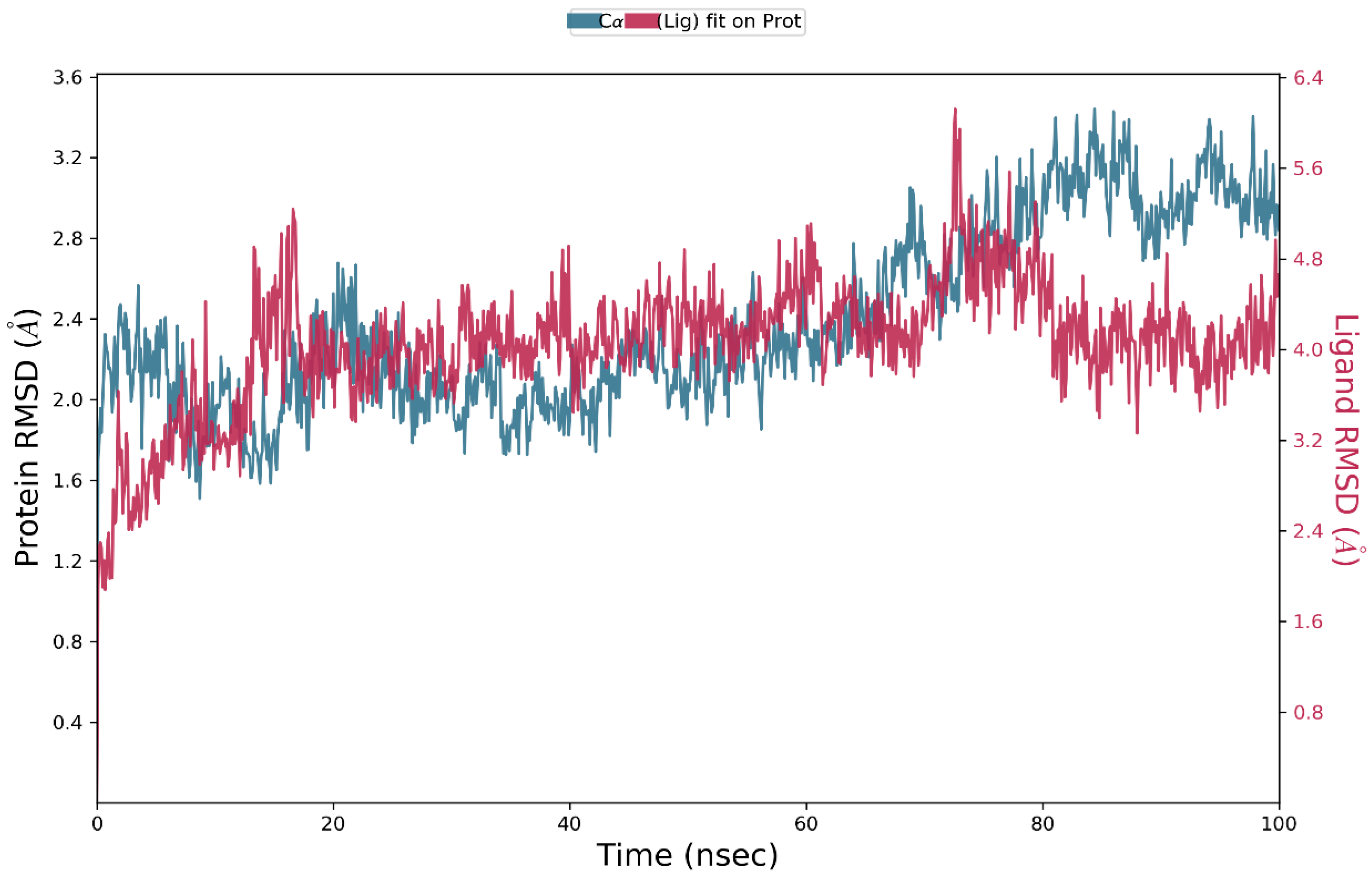
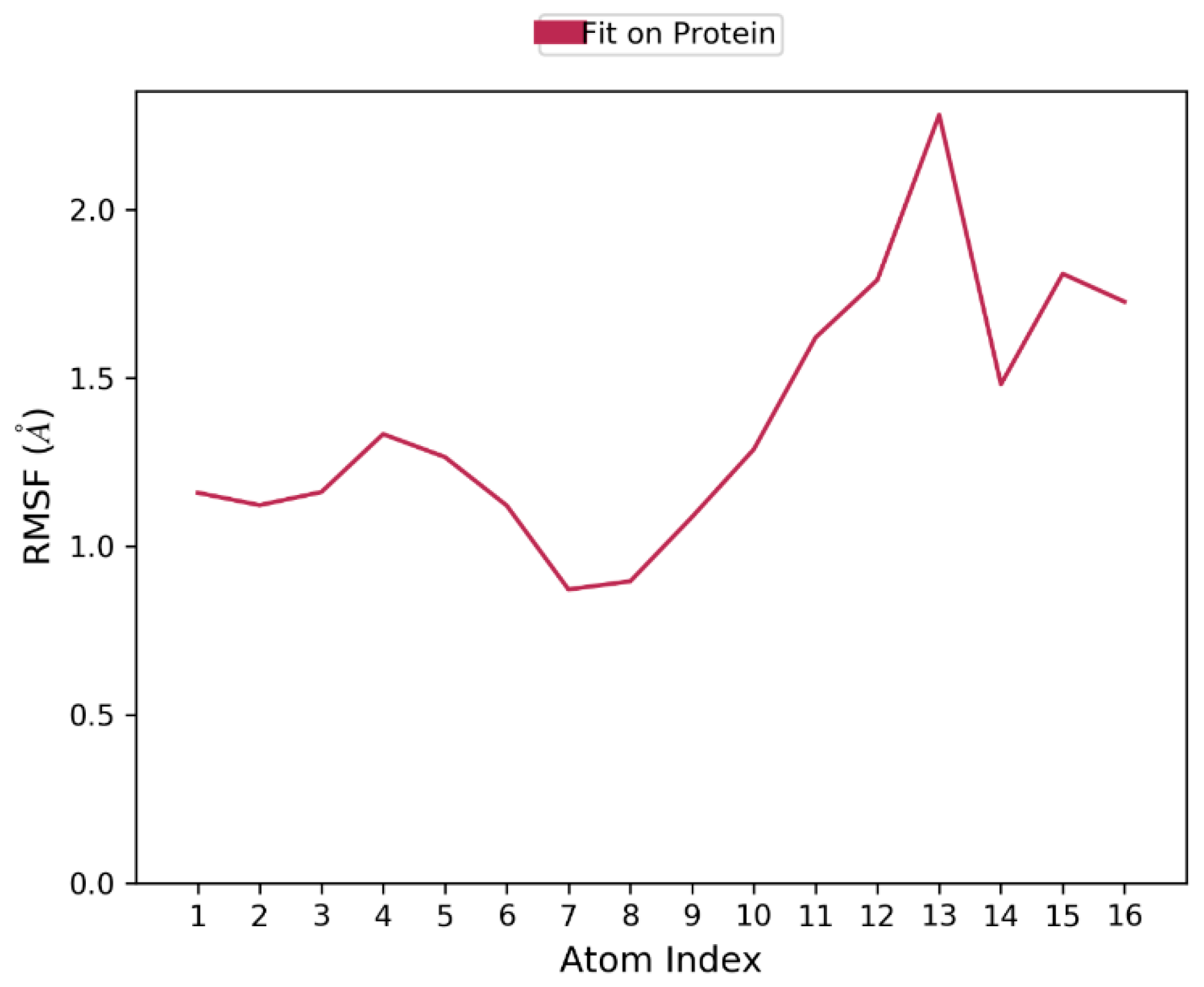
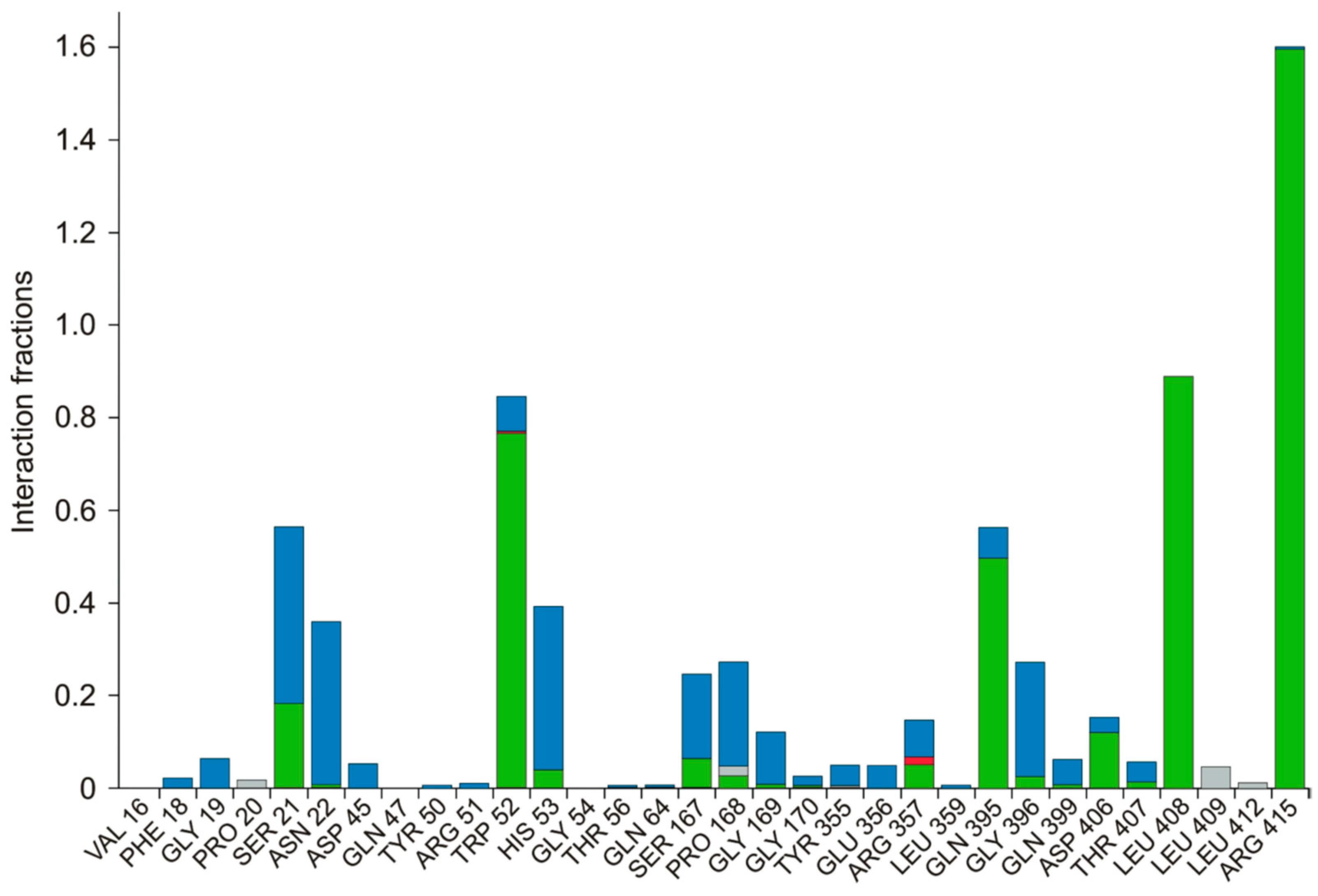
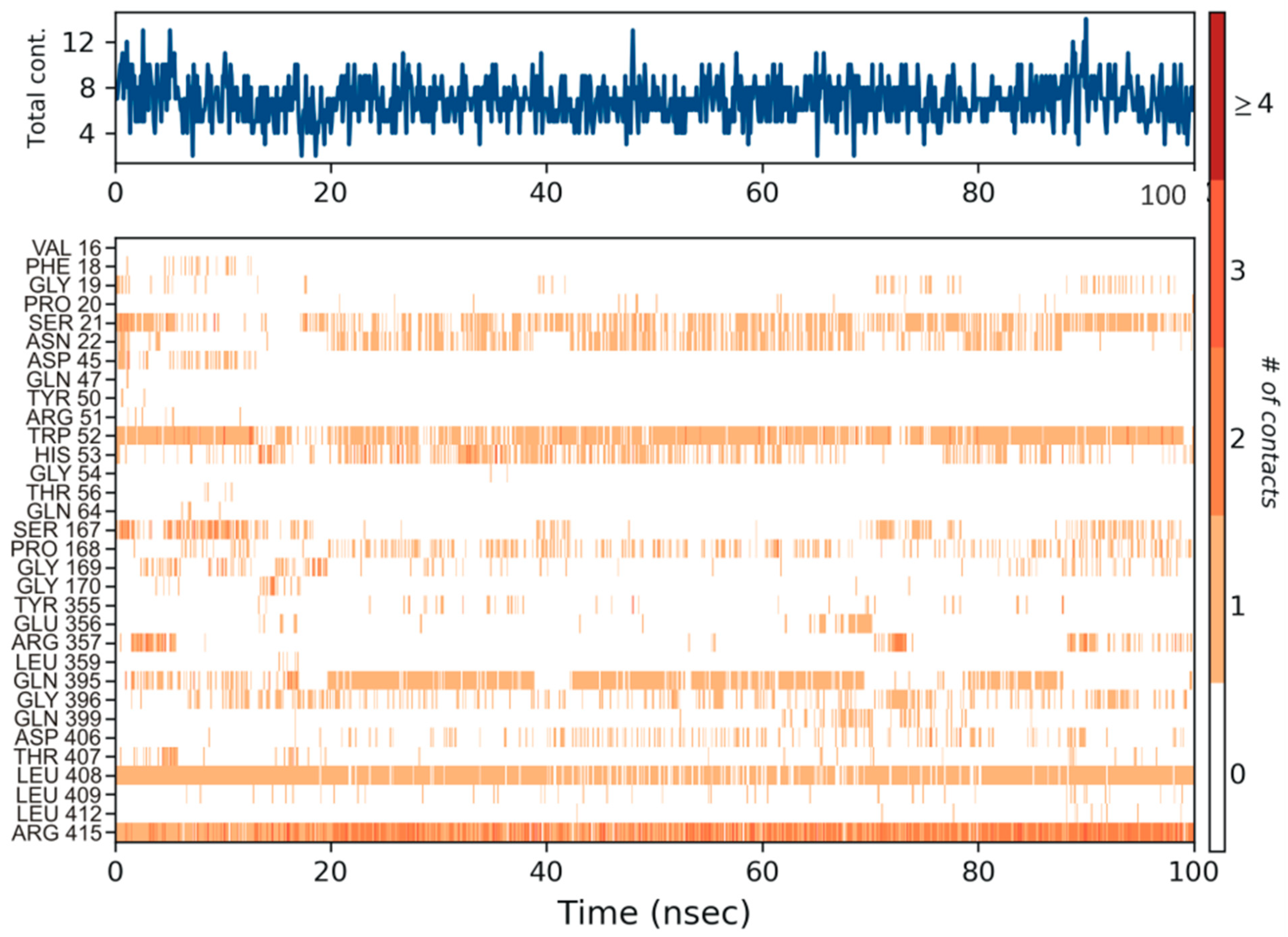
3.4. Molecular Dynamics Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baron, C. Antivirulence drugs to target bacterial secretion systems. Curr. Opin. Microbiol. 2010, 13, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Calvert, M.B.; Jumde, V.R.; Titz, A. Pathoblockers or antivirulence drugs as a new option for the treatment of bacterial infections. Beilstein J. Org. Chem. 2018, 14, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, F.; Baldelli, V.; Halliday, N.; Pantalone, P.; Polticelli, F.; Fiscarelli, E.; Williams, P.; Visca, P.; Leoni, L.; Rampioni, G. Identification of FDA-approved drugs as antivirulence agents targeting the pqs quorum-sensing system of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e01296-18. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.L.; Simeonov, A. Targeting iron assimilation to develop new antibacterials. Expert Opin. Drug Discov. 2012, 7, 831–847. [Google Scholar] [CrossRef]
- Smith, D.J.; Lamont, I.L.; Anderson, G.J.; Reid, D.W. Targeting iron uptake to control Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 2013, 42, 1723–1736. [Google Scholar] [CrossRef]
- Litton, E.; Lim, J. Iron metabolism: An emerging therapeutic target in critical illness. Annu. Update Intensive Care Emerg. Med. 2019, 2019, 573–584. [Google Scholar]
- Breidenstein, E.B.; de la Fuente-Núñez, C.; Hancock, R.E. Pseudomonas aeruginosa: All roads lead to resistance. Trends Microbiol. 2011, 19, 419–426. [Google Scholar] [CrossRef]
- Cox, C.D.; Adams, P. Siderophore activity of pyoverdin for Pseudomonas aeruginosa. Infect. Immun. 1985, 48, 130–138. [Google Scholar] [CrossRef]
- Cox, C.D.; Rinehart, K.L.; Moore, M.L.; Cook, J.C. Pyochelin: Novel structure of an iron-chelating growth promoter for Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1981, 78, 4256–4260. [Google Scholar] [CrossRef]
- Meyer, J.M.; Neely, A.; Stintzi, A.; Georges, C.; Holder, I.A. Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect. Immun. 1996, 64, 518–523. [Google Scholar] [CrossRef]
- Takase, H.; Nitanai, H.; Hoshino, K.; Otani, T. Impact of Siderophore Production on Pseudomonas aeruginosa Infections in Immunosuppressed Mice. Infect. Immun. 2000, 68, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.V.; Kirienko, D.R.; Larkins-Ford, J.; Wählby, C.; Ruvkun, G.; Ausubel, F.M. Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe 2013, 13, 406–416. [Google Scholar] [CrossRef]
- Minandri, F.; Imperi, F.; Frangipani, E.; Bonchi, C.; Visaggio, D.; Facchini, M.; Pasquali, P.; Bragonzi, A.; Visca, P. Role of iron uptake systems in Pseudomonas aeruginosa virulence and airway infection. Infect. Immun. 2016, 84, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, N.V.; Ausubel, F.M.; Ruvkun, G. Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2015, 112, 1821–1826. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Kirienko, D.R.; Webster, P.; Fisher, A.L.; Kirienko, N.V. Pyoverdine, a siderophore from Pseudomonas aeruginosa, translocates into C. elegans, removes iron, and activates a distinct host response. Virulence 2018, 9, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, D.R.; Kang, D.; Kirienko, N.V. Novel pyoverdine inhibitors mitigate Pseudomonas aeruginosa pathogenesis. Front. Microbiol. 2019, 9, 3317. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Olucha, J.; Meneely, K.M.; Chilton, A.S.; Lamb, A.L. Two structures of an N-hydroxylating flavoprotein monooxygenase: Ornithine hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 31789–31798. [Google Scholar] [CrossRef] [PubMed]
- Al-Masoudi, N.A.; Elias, R.S.; Saeed, B. Molecular docking studies of some antiviral and antimalarial drugs via bindings to 3cl-protease and polymerase enzymes of the novel coronavirus (Sars-cov-2). Biointerface Res. Appl. Chem. 2020, 10, 6444–6459. [Google Scholar]
- Yedidi, R.S.; Liu, Z.; Kovari, I.A.; Woster, P.M.; Kovari, L.C. P1 and P1′ para-fluoro phenyl groups show enhanced binding and favorable predicted pharmacological properties: Structure-based virtual screening of extended lopinavir analogs against multi-drug resistant HIV-1 protease. J. Mol. Graph. Model. 2014, 47, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Meneely, K.M.; Lamb, A.L. Biochemical characterization of a flavin adenine dinucleotide-dependent monooxygenase, ornithine hydroxylase from Pseudomonas aeruginosa, suggests a novel reaction mechanism. Biochemistry 2007, 46, 11930–11937. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Chinnamanyakar, R.; Ramanathan, E.M. Anti-cancer and antimicrobial activity, in-silico ADME and docking studies of biphenyl pyrazoline derivatives. Biointerface Res. Appl. Chem. 2021, 11, 8266–8282. [Google Scholar]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Tuckerman, M.; Berne, B.J.; Martyna, G.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Cheng, A.; Merz, K.M. Application of the nosé−hoover chain algorithm to the study of protein dynamics. J. Phys. Chem. 1996, 100, 1927–1937. [Google Scholar] [CrossRef]
- Kalibaeva, G.; Ferrario, M.; Ciccotti, G. Constant pressure-constant temperature molecular dynamics: A correct constrained NPT ensemble using the molecular virial. Mol. Phys. 2003, 101, 765–778. [Google Scholar] [CrossRef]
- Kumar, B.K.; Faheem, N.; Sekhar, K.V.G.C.; Ojha, R.; Prajapati, V.K.; Pai, A.; Murugesan, S. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J. Biomol. Struct. Dyn. 2022, 40, 1363–1386. [Google Scholar] [CrossRef]
- Dreyer, G.B.; Metcalf, B.W.; ATomaszek, T.; Carr, T.J.; Chandler, A.C.; Hyland, L.; AFakhoury, S.; Magaard, V.W.; Moore, M.L.; EStrickler, J. Inhibition of human immunodeficiency virus 1 protease in vitro: Rational design of substrate analogue inhibitors. Proc. Natl. Acad. Sci. USA 1989, 86, 9752–9756. [Google Scholar] [CrossRef]
- Tomasselli, A.G.; Olsen, M.K.; Hui, J.O.; Staples, D.J.; Sawyer, T.K.; Heinrikson, R.L.; Tomich, C.S.C. Substrate analog inhibition and active site titration of purified recombinant HIV-1 protease. Biochemistry 1990, 29, 264–269. [Google Scholar] [CrossRef]
- Coggins, B.E.; Li, X.; McClerren, A.L.; Hindsgaul, O.; Raetz, C.R.H.; Zhou, P. Structure of the LpxC deacetylase with a bound substrate-analog inhibitor. Nat. Struct. Mol. Biol. 2003, 10, 645–651. [Google Scholar] [CrossRef]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Jann, A.; Stalon, V.; Wauven, C.V.; Leisinger, T.; Haas, D. N-Succinylated intermediates in an arginine catabolic pathway of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1986, 83, 4937–4941. [Google Scholar] [CrossRef] [PubMed]
- Szucs, T. Cilazapril. A review. Drugs 1991, 41, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Horibata, Y.; Morimoto, Y.; Tateno, S.; Kawasoe, Y.; Niwa, K. Syndrome of inappropriate secretion of antidiuretic hormone associated with angiotensin-converting enzyme inhibitor administration. Pediatr. Cardiol. 2013, 34, 1261–1263. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Name of the Drug | Structure | Binding Energy (kcal mol−1) |
|---|---|---|---|
| 1. | (2s)-2,8-Diaminooctanoic Acid |  | −5.2 |
| 2. | 2,6-Diaminopimelic Acid |  | −5.1 |
| 3. | 2-Methylleucine |  | −4.9 |
| 4. | Allo-Isoleucine |  | −4.8 |
| 5. | 4-Carboxy-4-Aminobutanal |  | −4.6 |
| 6. | d-Leucine |  | −4.6 |
| 7. | 2-Aminopimelic Acid |  | −4.6 |
| 8. | d-Lysine |  | −4.6 |
| 9. | 2-Amino-6-Oxo-Hexanoic Acid |  | −4.5 |
| 10. | d-Glutamine |  | −4.5 |
| 11. | 6-hydroxy-l-norleucine |  | −4.4 |
| 12. | Norvaline |  | −4.4 |
| 13. | 5-Hydroxy Norvaline |  | −4.3 |
| 14. | Alpha-Aminobutyric Acid |  | −4.1 |
| 15. | Delta-Amino Valeric Acid |  | −3.9 |
| S. No. | Name of the Drug | Structure | Binding Energy (kcal mol−1) |
|---|---|---|---|
| 1. | N-2-Succinyl ornithine |  | −12.8 |
| 2. | Trypanothione |  | −11.7 |
| 3. | Talotrexin |  | −11.0 |
| 4. | Davunetide |  | −11.0 |
| 5. | CTT-1057 |  | −9.4 |
| 6. | Cilazapril |  | −9.1 |
| 7. | S-P-Nitrobenzyloxycarbonylglutathione |  | −8.3 |
| 8. | Glutathione disulfide |  | −8.1 |
| 9. | S-Hydroxymethyl Glutathione |  | −7.6 |
| 10. | Glutathione |  | −7.2 |
| 11. | Argininosuccinate |  | −6.9 |
| 12. | Glutathione Sulfinate |  | −6.6 |
| 13. | N-(Phosphonoacetyl)-l-Ornithine |  | −6.3 |
| 14. | N-Alpha-l-Acetyl-Arginine |  | −6.0 |
| 15. | Gamma-Glutamylcysteine |  | −6.0 |
| 16. | N-omega-nitro-l-arginine methyl ester |  | −6.0 |
| 17. | S-methyl-glutathione |  | −5.9 |
| 18. | N-Acetyl-l-Citrulline |  | −5.8 |
| 19. | Nitroarginine |  | −5.8 |
| 20. | N3, N4-Dimethylarginine |  | −5.6 |
| 21. | l-Eflornithine |  | −5.5 |
| 22. | 5-N-Allyl-Arginine |  | −5.5 |
| 23. | Glutamine t-butyl ester |  | −5.5 |
| 24. | N, N-dimethylarginine |  | −5.4 |
| 25. | Aceglutamide |  | −5.4 |
| 26. | Tilarginine |  | −5.4 |
| 27. | l-Citrulline |  | −5.3 |
| 28. | Glutamine hydroxamate |  | −5.2 |
| 29. | N5-Methylglutamine |  | −4.9 |
| 30. | l-Thiocitrulline |  | −4.9 |
| Name of the Protein | l-Ornithine-N5-Monooxygenase (or) Ornithine Hydroxylase |
|---|---|
| Number of amino acids | 463 |
| PDB Id | 3S5W |
| Resolution | 1.9 Å |
| Number of chains | 2 (Homodimer) |
| Number of Domains | Three FAD Binding Domain NADPH Binding Domain Ornithine Binding Domain |
| Amino acids interacting with natural ligand (N5- hydroxyl ornithine) | Lys69, Asn254, Phe257, Asn284, Ser410 |
| Lipinski (Lipinski et al., 1997) | Ghose Ghose, (Viswanadhan, Wendoloski, 1999) | Veber (Veber et al., 2002) | Egan (Egan, Merz, Baldwin, 2000) | Muegge (Muegge, Heald, Brittelli, 2001) |
|---|---|---|---|---|
| MW ≤ 500 MLOGP ≤ 4.15 N or O ≤ 10 NH or OH ≤ 5 | 160 ≤ MW ≤ 480 −0.4 ≤ WLOGP ≤ 5.6 40 ≤ MR ≤ 130 20 ≤ atoms ≤ 70 | Rotatable bonds ≤ 10 TPS ≤ 140 | WLOGP ≤ 5.88 TPSA ≤ 131.6 | 200 ≤ MW ≤ 600 −2 ≤ XLOGP ≤ 5 TPSA ≤ 150 Num. rings ≤ 7 Num. carbon > 4 Num. heteroatoms > 1 Num. rotatable bonds ≤ 15 H-bond acc. ≤ 10 H-bond don. ≤ 5 |
| Name of the Compound | Molecular Weight (g × mol−1) | H-Bond Acceptors | H-Bond Donors | Rotatable Bonds | LogPo/w | GI Absorption | BBB Permeation | Bioavailability Score |
|---|---|---|---|---|---|---|---|---|
| N2- Succinyl-l-ornithine | 232.23 | 6 | 4 | 9 | −1.18 | High | No | 0.56 |
| Trypanothione | 723.86 | 13 | 11 | 33 | −4.20 | Low | No | 0.17 |
| Talotrexin | 573.56 | 10 | 7 | 14 | 0.18 | Low | No | 0.11 |
| Davunetide | 824.92 | 13 | 10 | 29 | −3.07 | Low | No | 0.17 |
| CTT-1057 | 706.61 | 16 | 9 | 28 | −0.12 | Low | No | 0.11 |
| Cilazapril | 417.50 | 7 | 2 | 9 | 1.37 | High | No | 0.55 |
| Name of the Compound | Lipinski | Ghose | Veber | Egan | Muegge |
|---|---|---|---|---|---|
| N-2-Succinyl ornithine | Yes; 0 violation | No; 1 violation: WLOGP < −0.4 | Yes; 0 violation | Yes; 0 violation | No; 1 violation: XLOGP3 < −2 |
| Trypanothione | No; 3 violations: MW > 500, N or O > 10, NH or OH > 5 | No; 4 violations: MW > 480, WLOGP < −0.4, MR > 130, #atoms > 70 | No; 2 violations: Rotors > 10, TPSA > 140 | No; 1 violation: TPSA > 131.6 | No; 6 violations: MW > 600, XLOGP3 < −2, TPSA > 150, Rotors > 15, H-acc > 10, H-don > 5 |
| Talotrexin | No; 3 violations: MW > 500, N or O > 10, NH or OH > 5 | No; 2 violations: MW > 480, MR > 130 | No; 2 violations: Rotors > 10, TPSA > 140 | No; 1 violation: TPSA > 131.6 | No; 2 violations: TPSA > 150, H-don > 5 |
| Davunetide | No; 3 violations: MW > 500, N or O > 10, NH or OH > 5 | No; 4 violations: MW > 480, WLOGP < −0.4, MR > 130, #atoms > 70 | No; 2 violations: Rotors > 10, TPSA > 140 | No; 1 violation: TPSA > 131.6 | No; 6 violations: MW > 600, XLOGP3 < −2, TPSA > 150, Rotors > 15, H-acc > 10, H-don > 5 |
| CTT-1057 | No; 3 violations: MW > 500, N or O > 10, NH or OH > 5 | No; 4 violations: MW > 480, WLOGP < −0.4, MR > 130, #atoms > 70 | No; 2 violations: Rotors > 10, TPSA > 140 | No; 1 violation: TPSA > 131.6 | No; 6 violations : MW > 600, XLOGP3 < −2, TPSA > 150, Rotors > 15, H-acc > 10, H-don > 5 |
| Cilazapril | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation | Yes; 0 violation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosy, J.C.; Babkiewicz, E.; Maszczyk, P.; Mrówka, P.; Kumar, B.K.; Murugesan, S.; Kunjiappan, S.; Sundar, K. l-Ornithine-N5-monooxygenase (PvdA) Substrate Analogue Inhibitors for Pseudomonas aeruginosa Infections Treatment: Drug Repurposing Computational Studies. Biomolecules 2022, 12, 887. https://doi.org/10.3390/biom12070887
Rosy JC, Babkiewicz E, Maszczyk P, Mrówka P, Kumar BK, Murugesan S, Kunjiappan S, Sundar K. l-Ornithine-N5-monooxygenase (PvdA) Substrate Analogue Inhibitors for Pseudomonas aeruginosa Infections Treatment: Drug Repurposing Computational Studies. Biomolecules. 2022; 12(7):887. https://doi.org/10.3390/biom12070887
Chicago/Turabian StyleRosy, Joseph Christina, Ewa Babkiewicz, Piotr Maszczyk, Piotr Mrówka, Banoth Karan Kumar, Sankaranarayanan Murugesan, Selvaraj Kunjiappan, and Krishnan Sundar. 2022. "l-Ornithine-N5-monooxygenase (PvdA) Substrate Analogue Inhibitors for Pseudomonas aeruginosa Infections Treatment: Drug Repurposing Computational Studies" Biomolecules 12, no. 7: 887. https://doi.org/10.3390/biom12070887