
Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies


Abstract
:1. Introduction
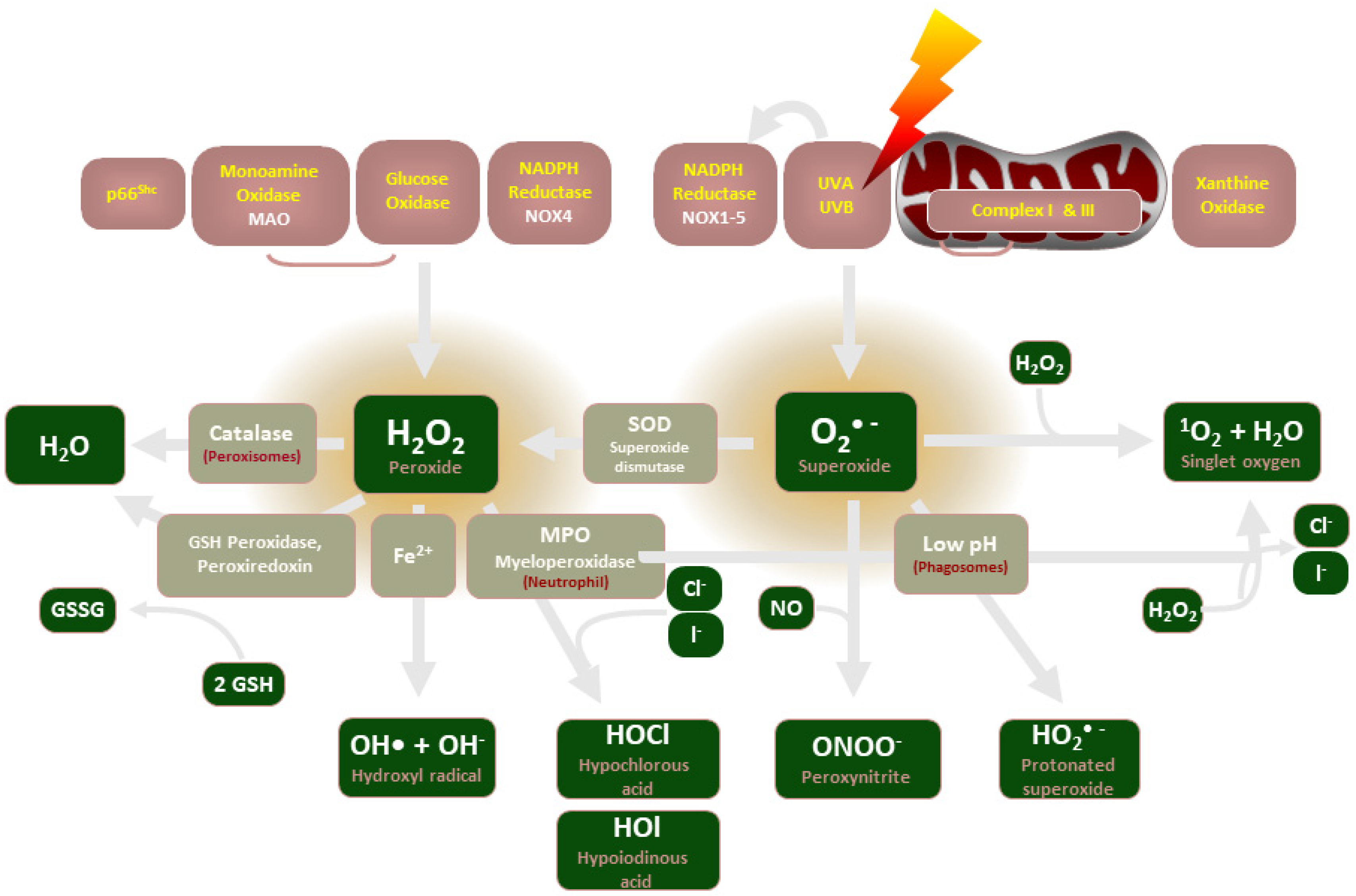
2. Role of NADPH Oxidases
2.1. NADPH Oxidase Description
2.2. ROS and NADPH Oxidase in Recent Experimental Studies
2.3. NADPH Oxidases and ROS in Disease
2.4. NADPH Oxidase-Produced H2O2 Mediates Vascular Tone in Healthy Subjects during Exercise
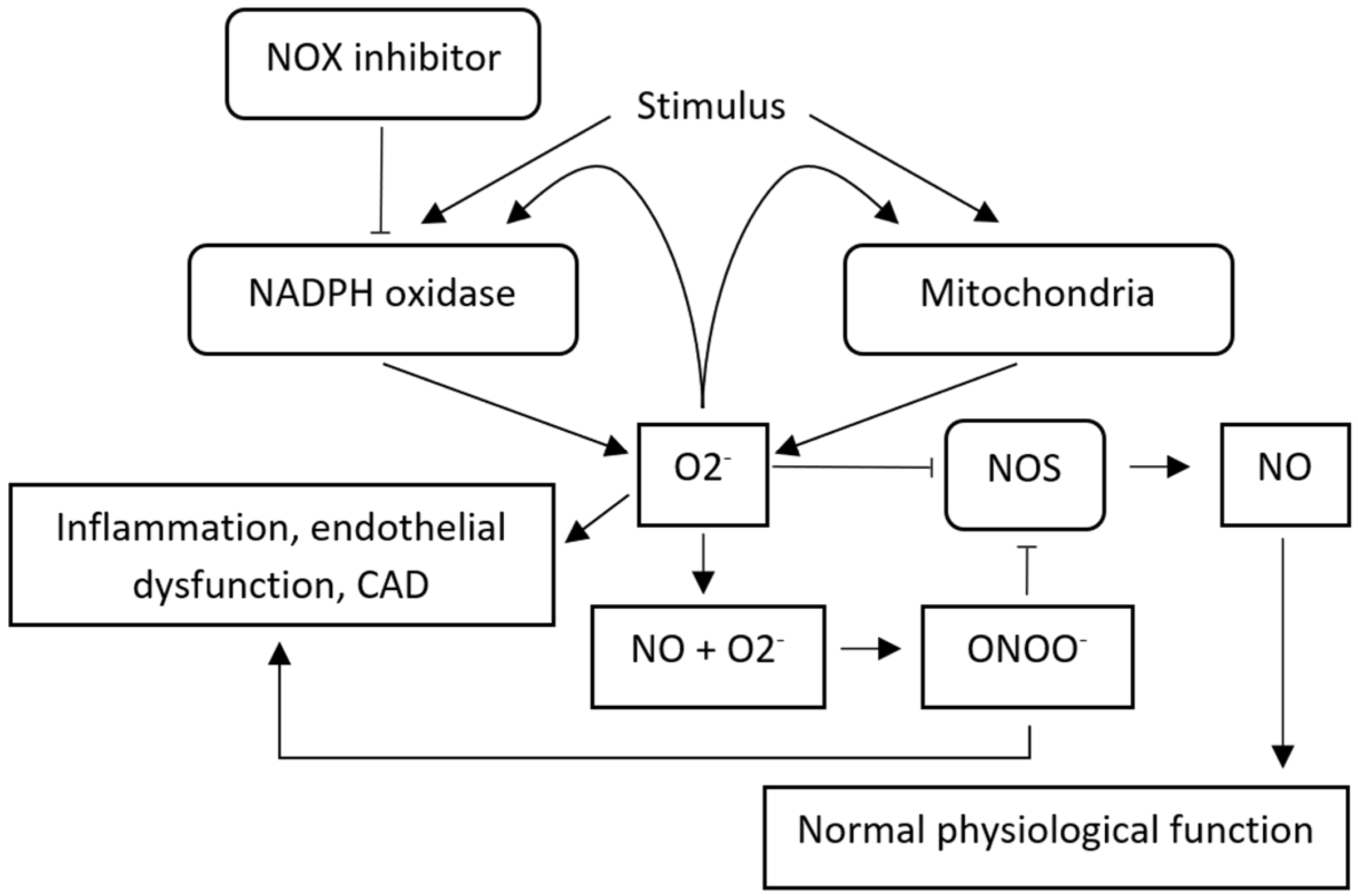
2.5. RIRR, Endothelial Dysfunction and Angiogenesis
3. Overview of Clinical Trials
3.1. Clinical Trials Targeting NADPH Oxidases to Improve Patient Outcomes
3.2. A Trial of Setanaxib in Patients with Primary Biliary Cholangitis (PBC) and Liver Stiffness
3.3. GKT137831 in IPF Patients with Idiopathic Pulmonary Fibrosis (GKT137831)
3.4. Microvascular Dysfunction in Obesity
3.5. Endothelial Dysfunction and Oxidative Stress in Children with Sleep Disordered Breathing
3.6. Oral GKT137831 in Patients with Type 2 Diabetes and Albuminuria
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, J.A.; Larion, S.; Mintz, J.D.; de Chantemèle, E.J.B.; Fulton, D.J.; Stepp, D.W. Genetic deletion of NADPH oxidase 1 rescues microvascular function in mice with metabolic disease. Circ. Res. 2017, 121, 502–511. [Google Scholar] [CrossRef]
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Loschen, G.; Azzi, A.; Richter, C.; Flohé, L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett. 1974, 42, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [Green Version]
- Blough, N.V.; Zafiriou, O.C. Reaction of superoxide with nitric oxide to form peroxonitrite in alkaline aqueous solution. Inorg. Chem. 1985, 24, 3502–3504. [Google Scholar] [CrossRef]
- Bartesaghi, S.; Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 2017, 14, 618–625. [Google Scholar] [CrossRef]
- White, C.R.; Brock, T.A.; Chang, L.Y.; Crapo, J.; Briscoe, P.; Ku, D.; Bradley, W.A.; Gianturco, S.H.; Gore, J.; Freeman, B.A. Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 1044–1048. [Google Scholar] [CrossRef] [Green Version]
- Van Der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 2018, 118, 1338–1408. [Google Scholar] [CrossRef]
- Jenkins, D.J.; Kitts, D.; Giovannucci, E.L.; Sahye-Pudaruth, S.; Paquette, M.; Blanco Mejia, S.; Patel, D.; Kavanagh, M.; Tsirakis, T.; Kendall, C.W.; et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2020, 112, 1642–1652. [Google Scholar] [CrossRef]
- Mirmiran, P.; Hosseini-Esfahani, F.; Esfandiar, Z.; Hosseinpour-Niazi, S.; Azizi, F. Associations between dietary antioxidant intakes and cardiovascular disease. Sci. Rep. 2022, 12, 1504. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.; Cooper, M.E.; et al. NADPH Oxidase, NOX1, Mediates Vascular Injury in Ischemic Retinopathy. Antioxid. Redox Signal. 2014, 20, 2726–2740. [Google Scholar] [CrossRef]
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887. [Google Scholar] [CrossRef] [Green Version]
- De Figueiredo, A.S.P.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Szanto, I.; Rubbia-Brandt, L.; Kiss, P.; Steger, K.; Banfi, B.; Kovari, E.; Herrmann, F.; Hadengue, A.; Krause, K.-H. Expression ofNOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 2005, 207, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef]
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef] [Green Version]
- Helfinger, V.; Palfi, K.; Weigert, A.; Schröder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFκB-Dependent Manner. Oxidative Med. Cell. Longev. 2019, 2019, 3264858. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.D.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ. Res. 2021, 128, 1320–1322. [Google Scholar] [CrossRef]
- Fusco, R.; Siracusa, R.; Gugliandolo, E.; Peritore, A.F.; D’Amico, R.; Cordaro, M.; Crupi, R.; Impellizzeri, D.; Gomiero, C.; Cuzzocrea, S.; et al. Micro Composite Palmitoylethanolamide/Rutin Reduces Vascular Injury through Modulation of the Nrf2/HO−1 and NF-kB Pathways. Curr. Med. Chem. 2021, 28, 6287–6302. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef] [Green Version]
- Fry, E.A.; Inoue, K. c-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65. [Google Scholar] [CrossRef]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Martínez, M.P.; López-Oliva, M.E.; Rodríguez, C.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Medina, J.S.; et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018, 19, 92–104. [Google Scholar] [CrossRef]
- Larsen, B.T.; Bubolz, A.H.; Mendoza, S.A.; Pritchard, K.A.; Gutterman, D.D. Bradykinin-Induced Dilation of Human Coronary Arterioles Requires NADPH Oxidase–Derived Reactive Oxygen Species. Arter. Thromb. Vasc. Biol. 2009, 29, 739–745. [Google Scholar] [CrossRef]
- Xie, Y.; Nishijima, Y.; Zinkevich, N.S.; Korishettar, A.; Fang, J.; Mathison, A.J.; Zimmermann, M.T.; Wilcox, D.A.; Gutterman, D.D.; Shen, Y.; et al. NADPH oxidase 4 contributes to TRPV4-mediated endothelium-dependent vasodilation in human arterioles by regulating protein phosphorylation of TRPV4 channels. Basic Res. Cardiol. 2022, 117, 24. [Google Scholar] [CrossRef]
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Damico, R.; Zulueta, J.J.; Hassoun, P.M. Pulmonary Endothelial Cell NOX. Am. J. Respir. Cell Mol. Biol. 2012, 47, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827. [Google Scholar] [CrossRef]
- Nelkine, L.; Vrolijk, M.F.; Drent, M.; Bast, A. Role of antioxidants in the treatment of gastroesophageal reflux disease-associated idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2020, 26, 363–371. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1832, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhang, X.; Zhang, F.-C.; Zong, J.-B.; Zhang, W.; Zhao, Y. Aberrant TGF-β1 signaling contributes to the development of primary biliary cirrhosis in murine model. World J. Gastroenterol. 2013, 19, 5828–5836. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Gokulan, R.C.; Horvat, A.; Yermalitskaya, L.; Korolkova, O.; Washington, K.M.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep. 2017, 7, 9956. [Google Scholar] [CrossRef] [Green Version]
- Gole, H.; Tharp, D.L.; Bowles, D.K. Upregulation of Intermediate-Conductance Ca2+-Activated K+ Channels (KCNN4) in Porcine Coronary Smooth Muscle Requires NADPH Oxidase 5 (NOX5). PLoS ONE 2014, 2, e105337. [Google Scholar] [CrossRef]
- Minnis, P.; Henry, K.; Keane, M.P. Reflux in idiopathic pulmonary fibrosis: Table 1. QJM Int. J. Med. 2015, 109, 7–10. [Google Scholar] [CrossRef] [Green Version]
- Sukumar, P.; Viswambharan, H.; Imrie, H.; Cubbon, R.M.; Yuldasheva, N.; Gage, M.; Galloway, S.; Skromna, A.; Kandavelu, P.; Santos, C.X.; et al. Nox2 NADPH Oxidase Has a Critical Role in Insulin Resistance–Related Endothelial Cell Dysfunction. Diabetes 2013, 62, 2130–2134. [Google Scholar] [CrossRef] [Green Version]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Banfi, B.; Sobey, C.G.; Faraci, F.M. Role of nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J. Appl. Physiol. 2012, 113, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 969. [Google Scholar] [CrossRef]
- Durand, M.J.; Dharmashankar, K.; Bian, J.T.; Das, E.; Vidovich, M.; Gutterman, D.D.; Phillips, S.A. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension 2015, 65, 140. [Google Scholar] [CrossRef] [Green Version]
- Zinkevich, N.S.; Fancher, I.S.; Gutterman, D.D.; Phillips, S.A. Roles of NADPH oxidase and mitochondria in flow-induced vasodilation of human adipose arterioles: ROS-induced ROS release in coronary artery disease. Microcirculation 2017, 24, e12380. [Google Scholar] [CrossRef]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine (U.S.). A Trial of Setanaxib in Patients with Primary Biliary Cholangitis (PBC) and Liver Stiffness. Identifier NCT05014672. 2022. Available online: https://clinicaltrials.gov/ct2/show/record/NCT05014672 (accessed on 22 May 2022).
- National Library of Medicine (U.S.). GKT137831 in IPF Patients with Idiopathic Pulmonary Fibrosis. Identifier NCT03865927. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03865927 (accessed on 22 May 2022).
- National Library of Medicine (U.S.). Microvascular Dysfunction in Obesity. Identifier NCT04087655. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT04087655 (accessed on 22 May 2022).
- La Favor, J.D.; Dubis, G.S.; Yan, H.; White, J.D.; Nelson, M.A.; Anderson, E.J.; Hickner, R.C. Microvascular Endothelial Dysfunction in Sedentary, Obese Humans Is Mediated by NADPH Oxidase. Arter. Thromb. Vasc. Biol. 2016, 36, 2412–2420. [Google Scholar] [CrossRef] [Green Version]
- Loffredo, L.; Zicari, A.M.; Occasi, F.; Perri, L.; Carnevale, R.; Angelico, F.; Del Ben, M.; Martino, F.; Nocella, C.; Savastano, V.; et al. Endothelial dysfunction and oxidative stress in children with sleep disordered breathing: Role of NADPH oxidase. Atherosclerosis 2015, 240, 222–227. [Google Scholar] [CrossRef]
- Fonseca, V.A. Defining and Characterizing the Progression of Type 2 Diabetes. Diabetes Care 2009, 32 (Suppl. 2), S151–S156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cosmo, S.; Viazzi, F.; Pacilli, A.; Giorda, C.; Ceriello, A.; Gentile, S.; Russo, G.; Rossi, M.C.; Nicolucci, A.; Guida, P.; et al. Predictors of chronic kidney disease in type 2 diabetes. Medicine 2016, 95, e4007. [Google Scholar] [CrossRef]
- Athavale, A.; Roberts, D.M. Management of proteinuria: Blockade of the renin-angiotensin-aldosterone system. Aust. Prescr. 2020, 43, 121–125. [Google Scholar] [CrossRef]
- Fox, C.S.; Matsushita, K.; Woodward, M.; Bilo, H.J.; Chalmers, J.; Heerspink, H.J.L.; Lee, B.J.; Perkins, R.M.; Rossing, P.; Sairenchi, T.; et al. Associations of kidney disease measures with mortality and end-stage renal disease in individuals with and without diabetes: A meta-analysis. Lancet 2012, 380, 1662–1673, Erratum in Lancet 2013, 381, 374. [Google Scholar] [CrossRef] [Green Version]
- National Library of Medicine (U.S.). Safety and Efficacy of Oral GKT137831 in Patient with Type 2 Diabetes and Albuminuria. Identifier NCT02010242. 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02010242 (accessed on 22 May 2022).
- Gray, S.P.; Shah, A.M.; Smyrnias, I. NADPH oxidase 4 and its role in the cardiovascular system. Vasc. Biol. 2019, 1, H59–H66. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, G.; Szyndralewiez, C.; Molango, S.; Carnesecchi, S.; Heitz, F.; Wiesel, P.; Wood, J.M. Therapeutic potential of NADPH oxidase 1/4 inhibitors. J. Cereb. Blood Flow Metab. 2016, 174, 1647–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielonka, J.; Zielonka, M.; VerPlank, L.; Cheng, G.; Hardy, M.; Ouari, O.; Ayhan, M.M.; Podsiadły, R.; Sikora, A.; Lambeth, J.D.; et al. Mitigation of NADPH Oxidase 2 Activity as a Strategy to Inhibit Peroxynitrite Formation. J. Biol. Chem. 2016, 291, 7029–7044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Khan, S.R. NADPH oxidase: A therapeutic target for hyperoxaluria-induced oxidative stress—An update. Future Med. Chem. 2019, 11, 2975–2978. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Intervention | Setanaxib—A NOX1 and NOX4 Inhibitor | GKT137831—A NOX1 and NOX4 Inhibitor | Aerobic Exercise | Adenotonsillectomy (AT) | GKT137831—A NOX1 and NOX4 Inhibitor |
|---|---|---|---|---|---|
| Regimen | 1200–1600 mg daily for 52 weeks | 400 mg twice daily orally for 24 weeks | Intervals of 70–90% max HR for 30 min 3 times a week for 8 weeks | - | 100 mg twice daily for first 6 weeks of treatment, and 200 mg twice daily for next 6 weeks |
| Target | Reduce liver inflammation and bile duct injury in patients with primary biliary cholangitis (PBC) | Reduce pulmonary injury in patients with idiopathic pulmonary fibrosis (IPF) | Reduce NOX isoform expression and mitochondrial ROS thereby improving endothelial function | Reverse adverse cardiovascular effects of obstructive sleep apnea and NADPH oxidase-associated endothelial function | Evaluate efficacy of oral GKT137831 in Type-II diabetic patients with residual albuminuria |
| Status | Recruiting | Ongoing | Ongoing | Completed | Completed |
| (Estimated) start date | 1-December-2021 | 7-September-2020 | 20-November-2019 | 12-February-2012 | 2013 |
| (Estimated) completion date | 16-September-2024 | 31-July-2023 | 31-July-2022 | 1-May-2014 | 1-March-2015 |
| No. of patients | 318 | 60 | 25 | 15 | 200 |
| Results | - | - | - | See Section 3.5 | See Section 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sylvester, A.L.; Zhang, D.X.; Ran, S.; Zinkevich, N.S. Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies. Biomolecules 2022, 12, 823. https://doi.org/10.3390/biom12060823
Sylvester AL, Zhang DX, Ran S, Zinkevich NS. Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies. Biomolecules. 2022; 12(6):823. https://doi.org/10.3390/biom12060823
Chicago/Turabian StyleSylvester, Anthony L., David X. Zhang, Sophia Ran, and Natalya S. Zinkevich. 2022. "Inhibiting NADPH Oxidases to Target Vascular and Other Pathologies: An Update on Recent Experimental and Clinical Studies" Biomolecules 12, no. 6: 823. https://doi.org/10.3390/biom12060823