
Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges

 , , , and
, , , and
Abstract
:1. Introduction
2. Rationale of Nanopharmaceutical Development for Cancer Therapy
3. Targeting Tumor Microenvironment in Cancer Therapy
3.1. Targeting Cancer Stem Cells in TME
3.2. Targeting Tumor Stroma in TME
3.2.1. Angiogenic Targets
3.2.2. Drug Chemoresistance Targets
3.3. Disseminated Tumor Cell Targets
4. Modes of Targeting: From Size to Multicomplexity DDSs
5. Approved Nanopharmaceuticals for Clinical Uses in Cancer Therapy
5.1. Lipid-Based Approved Nanopharmaceuticals
5.2. Protein-Based Approved Nanopharmaceuticals
5.3. Metallic-Based Approved Nanopharmaceuticals
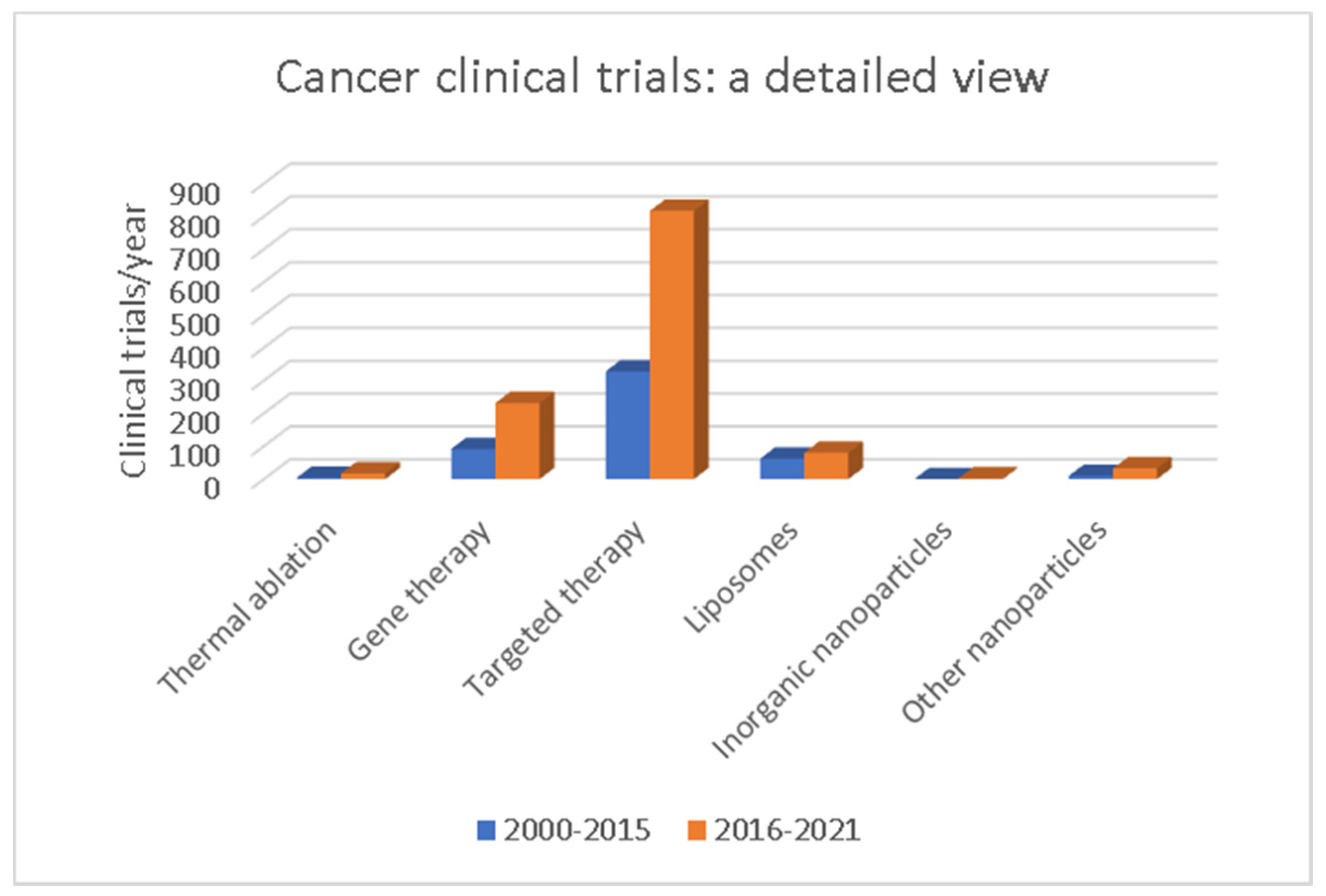
6. Nanopharmaceuticals in Clinical Trials for Cancer Therapy
7. Future Perspectives of Nanopharmaceuticals Targeting TME
8. Challenges in the Clinical Translation of Nanopharmaceuticals
8.1. Costs, Production, and Toxicology Limitations
8.2. Clinical Translation of Nanopharmaceuticals
9. FDA and EMA Regulatory Guidelines
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach. In The Development and Causes of Cancer, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9963/ (accessed on 1 January 2020).
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [Green Version]
- Misra, R.; Acharya, S.; Sahoo, S.K. Cancer nanotechnology: Application of nanotechnology in cancer therapy. Drug Discov. Today 2010, 19, 842–850. [Google Scholar] [CrossRef]
- Martinelli, C.; Pucci, C.; Ciofani, G. Nanostructured carriers as innovative tools for cancer diagnosis and therapy. APL Bioeng. 2019, 3, 011502. [Google Scholar] [CrossRef] [Green Version]
- Yingchoncharoen, P.; Kalinowski, D.S.; Richardson, D.R. Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come. Pharmacol. Rev. 2016, 68, 701–787. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef]
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer nanomedicine: From targeted delivery to combination therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Zhou, Y.; Xie, Y.; Song, P.; Ma, X. Signaling Pathways Associated with Cancer Stem Cells Play a Significant Role in Immunotherapy Resistance. J. Oncol. Res. 2019, 1, 1–10. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T. Cancer Stem Cells (CSCs) as a Rational Therapeutic Cancer Target, and Screening for CSC-targeting Drugs. Yakugaku Zasshi 2017, 137, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target. Ther. 2020, 5, 1–35. [Google Scholar] [CrossRef] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, D.G.; Rycaj, K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin. Cancer Biol. 2018, 52, 94–106. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug resistance driven by cancer stem cells and their niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenço, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018, 20, 1193–1202. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the TME in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef]
- Hayat, H.; Hayat, H.; Francis, B.; Gudi, D.M.; Bishop, J.O.; Wang, P. Concise Review: The Role of Stem Cells in Cancer Progression and Therapy. Onco Targets Ther. 2021, 14, 2761–2772. [Google Scholar] [CrossRef]
- Medema, J.P. Targeting the Colorectal Cancer Stem Cell. N. Engl. J. Med. 2017, 377, 888–890. [Google Scholar] [CrossRef]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Zhang, C.L.; Huang, T.; Wu, B.L.; He, W.X.; Liu, D. Stem cells in cancer therapy: Opportunities and challenges. Oncotarget 2017, 8, 75756–75766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K.; Senthebane, D.A.; Ganz, C.; Thomford, N.E.; Wonkam, A.; Dandara, C. Advances in Therapeutic Targeting of Cancer Stem Cells within the Tumor Microenvironment: An Updated Review. Cells 2020, 9, 1896. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, N.; Verma, V.; Kumar, M.; Kumar, A.; Singh, B. Nanomedicine in cancer stem cell therapy: From fringe to forefront. Cell Tissue Res. 2018, 374, 427–438. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.J.; Lei, K.F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer active targeting by nanoparticles: A comprehensive review of literature. J. Cancer Res. Clin. Oncol. 2015, 141, 769–784. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.A.M.A.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Domanska, U.M.; Boer, J.C.; Timmer-Bosscha, H.; van Vugt, M.A.; Hoving, H.D.; Kliphuis, N.M.; Rosati, S.; van der Poel, H.G.; de Jong, I.J.; de Vries, E.G.; et al. CXCR4 inhibition enhances radiosensitivity, while inducing cancer cell mobilization in a prostate cancer mouse model. Clin. Exp. Metastasis 2014, 31, 829–839. [Google Scholar] [CrossRef]
- Domanska, U.M.; Timmer-Bosscha, H.; Nagengast, W.B.; Oude Munnink, T.H.; Kruizinga, R.C.; Ananias, H.J.; Kliphuis, N.M.; Huls, G.; De Vries, E.G.; de Jong, I.J.; et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia 2012, 14, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Tayoun, T.; Faugeroux, V.; Oulhen, M.; Aberlenc, A.; Pawlikowska, P.; Farace, F. CTC-Derived Models: A Window into the Seeding Capacity of Circulating Tumor Cells (CTCs). Cells 2019, 8, 1145. [Google Scholar] [CrossRef] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massagué, J. Targeting metastatic cancer. Nat. Med. 2021, 27, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Hu, Y.; Zhou, Q. The CXCL12-CXCR4 Signaling Axis Plays a Key Role in Cancer Metastasis and is a Potential Target for Developing Novel Therapeutics against Metastatic Cancer. Curr. Med. Chem. 2020, 27, 5543–5561. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, M.V.; Unzueta, U.; Aviñó, A.; Gallardo, A.; Álamo, P.; Sala, R.; Sánchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruehauf, S. Current clinical indications for plerixafor. Transfus. Med. Hemother. 2013, 40, 246–250. [Google Scholar] [CrossRef] [Green Version]
- Thomas, O.S.; Weber, W. Overcoming Physiological Barriers to Nanoparticle Delivery-Are We There Yet? Front. Bioeng. Biotechnol. 2019, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Upreti, M.; Jyoti, A.; Sethi, P. Tumor microenvironment and nanotherapeutics. Transl. Cancer Res. 2013, 2, 309. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with Penetration of Nanoparticles across Cell and Tissue Barriers: A Review of Current Status and Future Prospects. Nano Today 2014, 9, 223. [Google Scholar] [CrossRef]
- Xu, S.; Olenyuk, B.Z.; Okamoto, C.T.; Hamm-Alvarez, S.F. Targeting receptor-mediated endocytotic pathways with nanoparticles: Rationale and advances. Adv. Drug Deliv. Rev. 2013, 65, 121–138. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Kauffman, S. How to escape the cancer attractor: Rationale and limitations of multi-target drugs. Semin. Cancer Biol. 2013, 23, 270–278. [Google Scholar] [CrossRef]
- Torchilin, V.P. Drug targeting. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S81–S91. [Google Scholar] [CrossRef]
- Pethe, A.M.; Yadav, K.S. Polymers, responsiveness and cancer therapy. Artif. Cells Nanomed. Biotechnol. 2019, 47, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.G.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Passive and active drug targeting: Drug delivery to tumors as an example. Handb. Exp. Pharmacol. 2010, 197, 3–53. [Google Scholar] [CrossRef]
- Heshmati, N.; Dabbaghianamiri, M.; Tunnell, J.W.; Betancourt, T. Design of Smart Nanomedicines for Effective Cancer Treatment. Int. J. Pharm. 2022, 621, 121791. [Google Scholar] [CrossRef] [PubMed]
- Vácha, R.; Martinez-Veracoechea, F.J.; Frenkel, D. Receptor-mediated endocytosis of nanoparticles of various shapes. Nano Lett. 2011, 11, 5391–5395. [Google Scholar] [CrossRef]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody fragments as nanoparticle targeting ligands: A step in the right direction. Chem. Sci. 2016, 8, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, P.R.; Hutabarat, R.M.; Thompson, K.M. Discovery and development of therapeutic aptamers. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 237–257. [Google Scholar] [CrossRef]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, A.; Deshpande, P.; Torchilin, V. Stimuli-sensitive nanopreparations for combination cancer therapy. J. Control. Release 2014, 190, 352–370. [Google Scholar] [CrossRef]
- Rahim, M.A.; Jan, N.; Khan, S.; Shah, H.; Madni, A.; Khan, A.; Jabar, A.; Khan, S.; Elhissi, A.; Hussain, Z. Recent Advancements in Stimuli Responsive Drug Delivery Platforms for Active and Passive Cancer Targeting. Cancers 2021, 13, 670. [Google Scholar] [CrossRef]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 7, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Fomina, N.; McFearin, C.; Sermsakdi, M.; Edigin, O.; Almutairi, A. UV and near-IR triggered release from polymeric nanoparticles. J. Am. Chem. Soc. 2010, 132, 9540–9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, T.P.; Moreira, J.A.; Monterio, F.J.; Laranjeira, M.S. Nanomaterials in cancer: Reviewing the combination of hyperthermia and triggered chemotherapy. J. Control. Release 2022, S0168–S3659, 240–241. [Google Scholar] [CrossRef]
- Farjadian, F.; Ghasemi, A.; Gohari, O.; Roointan, A.; Karim, M.; Hamblin, M.R. Nanopharmaceuticals and nanomedicines currently on the market: Challenges and opportunities. Nanomedicine 2019, 14, 93–126. [Google Scholar] [CrossRef]
- Cattel, L.; Ceruti, M.; Dosio, F. From conventional to stealth liposomes: A new frontier in cancer chemotherapy. J. Chemother. 2004, 16, 94–97. [Google Scholar] [CrossRef]
- Immordino, M.L.; Brusa, P.; Arpicco, S.; Stella, B.; Dosio, F.; Cattel, L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J. Control. Release 2003, 91, 417–429. [Google Scholar] [CrossRef]
- Jensen, G.M.; Hodgson, D.F. Opportunities and challenges in commercial pharmaceutical liposome applications. Adv. Drug Deliv. Rev. 2020, 154, 2–12. [Google Scholar] [CrossRef]
- Tereshkina, Y.A.; Torkhovskaya, T.I.; Tikhonova, E.G.; Kostryukova, L.V.; Sanzhakov, M.A.; Korotkevich, E.I.; Khudoklinova, Y.Y.; Orlova, N.A.; Kolesanova, E.F. Nanoliposomes as drug delivery systems: Safety concerns. J. Drug Target. 2022, 30, 313–325. [Google Scholar] [CrossRef] [PubMed]
- DOXIL approved by FDA. AIDS Patient Care 1995, 9, 306. Available online: https://pubmed.ncbi.nlm.nih.gov/11361446/ (accessed on 4 April 2022).
- Barenholz, Y. Doxil®-the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Makwana, V.; Karanjia, J.; Haselhorst, T.; Anoopkumar-Dukie, S.; Rudrawar, S. Liposomal doxorubicin as targeted delivery platform: Current trends in surface functionalization. Int. J. Pharm. 2021, 593, 120117. [Google Scholar] [CrossRef]
- Perez, A.T.; Domenech, G.H.; Frankel, C.; Vogel, C.L. Pegylated liposomal doxorubicin (Doxil) for metastatic breast cancer: The Cancer Research Network, Inc., experience. Cancer Investig. 2002, 20, 22–29. [Google Scholar] [CrossRef]
- Rom, J.; Bechstein, S.; Domschke, C.; Golatta, M.; Mayer, C.; Heil, J.; Thum, J.; Smetanay, K.; Windemuth-Kieselbach, C.; Wallwiener, M.; et al. Efficacy and toxicity profile of pegylated liposomal doxorubicin (Caelyx) in patients with advanced breast cancer. Anti-Cancer Drugs 2014, 25, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Aubel-Sadron, G.; Londos-Gagliardi, D. Daunorubicin and doxorubicin, anthracycline antibiotics, a physicochemical and biological review. Biochimie 1984, 66, 333–352. [Google Scholar] [CrossRef]
- Fassas, A.; Anagnostopoulos, A. The use of liposomal daunorubicin (DaunoXome) in acute myeloid leukemia. Leuk. Lymphoma 2005, 46, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Kager, L.; Pötschger, U.; Bielack, S. Review of mifamurtide in the treatment of patients with osteosarcoma. Ther. Clin. Risk Manag. 2010, 6, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzogani, K.; Straube, M.; Hoppe, U.; Kiely, P.; O’Dea, G.; Enzmann, H.; Salmon, P.; Salmonson, T.; Pignatti, F. The European Medicines Agency approval of 5-aminolaevulinic acid (Ameluz) for the treatment of actinic keratosis of mild to moderate intensity on the face and scalp: Summary of the scientific assessment of the Committee for Medicinal Products for Human Use. J. Dermatolog. Treat. 2014, 25, 371–374. [Google Scholar] [CrossRef]
- Legha, S.S. Vincristine neurotoxicity. Pathophysiology and management. Med. Toxicol. 1986, 1, 421–427. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Liposomal Vincristine (Marqibo) for Rare Leukemia. Available online: https://www.cancernetwork.com/view/fda-approves-liposomal-vincristine-marqibo-rare-leukemia (accessed on 4 April 2022).
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Frampton, J.E. Liposomal Irinotecan: A Review in Metastatic Pancreatic Adenocarcinoma. Drugs 2020, 80, 1007–1018. [Google Scholar] [CrossRef]
- Hong, S.; Choi, D.W.; Kim, H.N.; Park, C.G.; Lee, W.; Park, H.H. Protein-Based Nanoparticles as Drug Delivery Systems. Pharmaceutics 2020, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.T.; Ma, J.; Kankala, R.K.; Yu, Q.; Wang, B.; Chen, A.Z. Recent Advances in Fabrication of Well-Organized Protein-Based Nanostructures. ACS Appl. Bio Mater. 2021, 4, 4039–4048. [Google Scholar] [CrossRef]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA drug approval summary: Pegaspargase (oncaspar) for the first-line treatment of children with acute lymphoblastic leukemia (ALL). Oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef]
- Heo, Y.A.; Syed, Y.Y.; Keam, S.J. Pegaspargase: A Review in Acute Lymphoblastic Leukaemia. Drugs 2019, 79, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, M.J.; Eisenberg, D. Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein. Sci. 1994, 3, 1464–1475. [Google Scholar] [CrossRef]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, Q.; Zhang, H.; Bronson, R.T.; Madsen, J.C.; Sachs, D.H.; Huang, C.A.; Wang, Z. Ontak-like human IL-2 fusion toxin. J. Immunol. Methods 2017, 448, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, K. Current treatment strategies and emerging therapies for cutaneous lymphoma. J. Dermatol. 2022, 49, 223–231. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine [NLM]. Available online: https://clinicaltrials.gov/ (accessed on 3 January 2022).
- Sartor, O. Eligard: Leuprolide acetate in a novel sustained-release delivery system. Urology 2003, 61, 25–31. [Google Scholar] [CrossRef]
- Van Tellingen, O.; Huizing, M.T.; Nannan Panday, V.R.; Schellens, J.H.M.; Nooijen, W.J.; Beijnen, J.H. Cremophor EL causes (pseudo-) non-linear pharmacokinetics of paclitaxel in patients. Br. J. Cancer 1999, 81, 330. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.B. Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1994, 5, S3–S6. [Google Scholar]
- Yardley, D.A. nab-Paclitaxel mechanisms of action and delivery. J. Control. Release 2013, 170, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Kundranda, M.N.; Niu, J. Albumin-bound paclitaxel in solid tumors: Clinical development and future directions. Drug Des. Devel. Ther. 2015, 9, 3767–3777. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, R.; Li, C.; Song, Y.; Liu, G.; Huang, Q.; Yu, L.; Zhu, D.; Lu, C.; Lu, A.; et al. Nab-paclitaxel promotes the cancer-immunity cycle as a potential immunomodulator. Am. J. Cancer Res. 2021, 11, 3445. [Google Scholar]
- Li, C.H.; Karantza, V.; Aktan, G.; Lala, M. Current treatment landscape for patients with locally recurrent inoperable or metastatic triple-negative breast cancer: A systematic literature review. Breast Cancer Res. 2019, 21, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab emtansine (T-DM1): An antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Lozza, I.; Torres-Suárez, A.I. Actively Targeted Nanomedicines in Breast Cancer: From Pre-Clinical Investigation to Clinic. Cancers 2022, 14, 1198. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, I.D. Monoclonal antibodies to the myeloid stem cells: Therapeutic implications of CMA-676, a humanized anti-CD33 antibody calicheamicin conjugate. Leukemia 2000, 14, 474–475. [Google Scholar] [CrossRef] [Green Version]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef]
- Lewis Phillips, G.D.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- DiJoseph, J.F.; Goad, M.E.; Dougher, M.M.; Boghaert, E.R.; Kunz, A.; Hamann, P.R.; Damle, N.K. Potent and specific antitumor efficacy of CMC-544, a CD22-targeted immunoconjugate of calicheamicin, against systemically disseminated B-cell lymphoma. Clin. Cancer Res. 2004, 10, 8620–8629. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, G.; Beers, R.; Margulies, I.; Kreitman, R.J.; Pastan, I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res. 2002, 8, 995–1002. [Google Scholar]
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Arrojo, R.; Liu, D.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. Bioconjug. Chem. 2015, 26, 919–931. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Zhao, M.; Li, H.; Fan, L.; Ma, Y.; Gong, H.; Lai, W.; Fang, Q.; Hu, Z. Quantitative proteomic analysis to the first commercialized liposomal paclitaxel nano-platform Lipusu revealed the molecular mechanism of the enhanced anti-tumor effect. Artif. Cells Nanomed. Biotechnol. 2018, 46, S147–S155. [Google Scholar] [CrossRef] [Green Version]
- Ahn, H.K.; Jung, M.; Sym, S.J.; Shin, D.B.; Kang, S.M.; Kyung, S.Y.; Park, J.W.; Jeong, S.H.; Cho, E.K. A phase II trial of Cremorphor EL-free paclitaxel (Genexol-PM) and gemcitabine in patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2014, 74, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranade, A.A.; Joshi, D.A.; Phadke, G.K.; Patil, P.P.; Kasbekar, R.B.; Apte, T.G.; Dasare, R.R.; Mengde, S.D.; Parikh, P.M.; Bhattacharyya, G.S.; et al. Clinical and economic implications of the use of nanoparticle paclitaxel (Nanoxel) in India. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, v6–v12. [Google Scholar] [CrossRef]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Mahmoudi, K.; Bouras, A.; Bozec, D.; Ivkov, R.; Hadjipanayis, C. Magnetic hyperthermia therapy for the treatment of glioblastoma: A review of the therapy’s history, efficacy and application in humans. Int. J. Hyperth. 2018, 34, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera Gil, P.; Hühn, D.; del Mercato, L.L.; Sasse, D.; Parak, W.J. Nanopharmacy: Inorganic nanoscale devices as vectors and active compounds. Pharmacol. Res. 2010, 62, 115–125. [Google Scholar] [CrossRef]
- Havel, H.A. Where Are the Nanodrugs? An Industry Perspective on Development of Drug Products Containing Nanomaterials. AAPS J. 2016, 18, 1351–1353. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742. [Google Scholar]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Salvioni, L.; Rizzuto, M.A.; Bertolini, J.A.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty Years of Cancer Nanomedicine: Success, Frustration, and Hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef] [Green Version]
- Bukhari, S.N.A. Emerging Nanotherapeutic Approaches to Overcome Drug Resistance in Cancers with Update on Clinical Trials. Pharmaceutics 2022, 14, 866. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, O.L.; Sjaastad, K.; Radomski, M.W.; Volkov, Y.; Prina-Mello, A. Magnetic Nanoparticles in Cancer Theranostics. Theranostics 2015, 5, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Jokerst, J.V.; Gambhir, S.S. Molecular imaging with theranostic nanoparticles. Acc. Chem. Res. 2011, 44, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, D.; Quake, S.R.; McCabe, E.R.B.; Chng, W.J.; Chow, E.K.; Ding, X.; Gelb, B.D.; Ginsburg, G.S.; Hassenstab, J.; Ho, C.M.; et al. Enabling Technologies for Personalized and Precision Medicine. Trends Biotechnol. 2020, 38, 497–518. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.; Liu, Y.; Gao, Z.; Huang, W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharm. Sin. B 2021, 11, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Ertas, Y.N.; Abedi Dorcheh, K.; Akbari, A.; Jabbari, E. Nanoparticles for Targeted Drug Delivery to Cancer Stem Cells: A Review of Recent Advances. Nanomaterials 2021, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Caserta, C.A.; Romeo, E.; Rumio, C. Antibody-Drug Conjugates (ADC) Against Cancer Stem-Like Cells (CSC)-Is There Still Room for Optimism? Front. Oncol. 2019, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Chen, Y.; Zhang, Z.; Tang, B.; Zhou, Z.; Chen, H. Nanoparticle-Based RNAi Therapeutics Targeting Cancer Stem Cells: Update and Prospective. Pharmaceutics 2021, 13, 2116. [Google Scholar] [CrossRef]
- Suo, X.; Zhang, J.; Zhang, Y.; Liang, X.J.; Zhang, J.; Liu, D. A nano-based thermotherapy for cancer stem cell-targeted therapy. J. Mater. Chem. B 2020, 8, 3985–4001. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sonanin, D.; Maurer, A.; Daldrup-Link, H.E. The yin and yang of imaging tumor associated macrophages with PET and MRI. Theranostics 2019, 9, 7730–7748. [Google Scholar] [CrossRef]
- Resnik, D.B.; Tinkle, S.S. Ethics in nanomedicine. Nanomedicine 2007, 2, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Aillon, K.L.; Xie, Y.; El-Gendy, N.; Berkland, C.J.; Forrest, M.L. Effects of nanomaterial physicochemical properties on in vivo toxicity. Adv. Drug Deliv. Rev. 2009, 61, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef]
- Fornaguera, C.; García-Celma, M.J. Personalized Nanomedicine: A Revolution at the Nanoscale. J. Pers. Med. 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Kiminami, H.; Yamashita, A.; Abe, Y.; Yoshino, K.; Suzuki, S. Assessment of the effects of sterilization methods on protein drug stability by elucidating decomposition mechanism and material analysis. Int. J. Pharm. 2015, 484, 51–56. [Google Scholar] [CrossRef]
- Li, Y.; Boraschi, D. Endotoxin contamination: A key element in the interpretation of nanosafety studies. Nanomedicine 2016, 11, 269–287. [Google Scholar] [CrossRef] [Green Version]
- Smulders, S.; Kaiser, J.P.; Zuin, S.; Van Landuyt, K.L.; Golanski, L.; Vanoirbeek, J.; Wick, P.; Hoet, P.H. Contamination of nanoparticles by endotoxin: Evaluation of different test methods. Part. Fibre Toxicol. 2012, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Vasir, J.K.; Labhasetwar, V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv. Drug Deliv. Rev. 2007, 59, 718–728. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–743. [Google Scholar]
- Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012, 14, 282–295. [Google Scholar] [CrossRef] [Green Version]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- FDA U.S. Food and Drug Administration. Available online: https://www.fda.gov/science-research/nanotechnology-programs-fda/nanotechnology-guidance-documents (accessed on 9 May 2022).
- EMA European Medicine Agency. Available online: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines (accessed on 9 May 2022).
- Innovation Task Force. Available online: https://www.tsa.gov/itf (accessed on 9 May 2022).
- Northfelt, D.W.; Dueck, A.C.; Flynn, T.P.; Zander, P.J.; Stella, P.J.; Melnik, M.; Pavey, E.S.; Perez, E.A. Phase II trial combining nab-paclitaxel (NP), gemcitabine (G), and bevacizumab (B) in patients (pts) with metastatic breast cancer (MBC): NCCTG N0735. ASCO Annu. Meet. I 2011, 29 (Suppl. 15), 1126. [Google Scholar] [CrossRef]
- Roy, V.; Laplant, B.R.; Gross, G.G.; Bane, C.L.; Palmieri, F.M. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane®) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Ann. Oncol. 2009, 20, 449. [Google Scholar] [CrossRef]
- Yardley, D.; Burris, H.; Peacock, N.; Raefsky, E.; Melnik, M.; Inhorn, R.; Shipley, D.; Hainsworth, J. A pilot study of adjuvant nanoparticle albumin-bound (nab) paclitaxel and cyclophosphamide, with trastuzumab in HER2-positive patients, in the treatment of early-stage breast cancer. Breast Cancer Res. Treat. 2010, 123, 471–475. [Google Scholar] [CrossRef]
- Nahleh, Z.A.; Barlow, W.E.; Hayes, D.F.; Schott, A.F.; Gralow, J.R.; Sikov, W.M.; Perez, E.A.; Chennuru, S.; Mirshahidi, H.R.; Corso, S.W.; et al. SWOG S0800 (NCI CDR0000636131): Addition of bevacizumab to neoadjuvant nab-paclitaxel with dose-dense doxorubicin and cyclophosphamide improves pathologic complete response (pCR) rates in inflammatory or locally advanced breast cancer. Breast Cancer Res. Treat. 2016, 158, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Prager, D.; Hermann, R.; Fehrenbacher, L.; Johnson, B.E.; Sandler, A.; Kris, M.G.; Tran, H.T.; Klein, P.; Li, X.; et al. TRIBUTE: A phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 5892–5899. [Google Scholar] [CrossRef]
- Bertino, E.M.; Williams, T.M.; Nana-Sinkam, S.P.; Shilo, K.; Chatterjee, M.; Mo, X.; Rahmani, M.; Phillips, G.S.; Villalona-Calero, M.A.; Otterson, G.A. Stromal Caveolin-1 Is Associated With Response and Survival in a Phase II Trial of nab-Paclitaxel With Carboplatin for Advanced NSCLC Patients. Clin. Lung Cancer 2015, 16, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrózek, E.; Layman, R.; Ramaswamy, B.; Lustberg, M.; Vecchione, A.; Knopp, M.V.; Shapiro, C.L. Phase II trial of neoadjuvant weekly nanoparticle albumin-bound paclitaxel, carboplatin, and biweekly bevacizumab therapy in women with clinical stage II or III HER2-negative breast cancer. Clin. Breast Cancer 2014, 14, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsh, V. nab-paclitaxel for the management of patients with advanced non-small-cell lung cancer. Expert Rev. Anticancer Ther. 2014, 14, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Hosein, P.J.; de Lima Lopes, G., Jr.; Pastorini, V.H.; Gomez, C.; Macintyre, J.; Zayas, G.; Reis, I.; Montero, A.J.; Merchan, J.R.; Rocha Lima, C.M. A phase II trial of nab-Paclitaxel as second-line therapy in patients with advanced pancreatic cancer. Am. J. Clin. Oncol. 2013, 36, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Connolly, R.M.; Leal, J.P.; Goetz, M.P.; Zhang, Z.; Zhou, X.C.; Jacobs, L.K.; Mhlanga, J.; Joo, H.O.; Carpenter, J.; Storniolo, A.M.; et al. TBCRC 008: Early change in 18F-FDG uptake on PET predicts response to preoperative systemic therapy in human epidermal growth factor receptor 2-negative primary operable breast cancer. J. Nucl. Med. 2015, 56, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, E.J.; Lee-Ma, A.; Troxell, L.S.; Chan, J. Low-dose filgrastim in patients with breast cancer treated with docetaxel, doxorubicin, and cyclophosphamide. Am. J. Health Syst. Pharm. 2008, 65, 1552–1555. [Google Scholar] [CrossRef]
- Hamilton, E.; Kimmick, G.; Hopkins, J.; Marcom, P.K.; Rocha, G.; Welch, R.; Broadwater, G.; Blackwell, K. Nab-paclitaxel/bevacizumab/carboplatin chemotherapy in first-line triple negative metastatic breast cancer. Clin. Breast Cancer 2013, 13, 416–420. [Google Scholar] [CrossRef]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T.; et al. A first in human phase 1 study of CG0070, a GM-CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J. Urol. 2012, 88, 2391–2397. [Google Scholar] [CrossRef]
- Symonds, L.; Linden, H.; Gadi, V.; Korde, L.; Rodler, E.; Gralow, J.; Redman, M.; Baker, K.; Wu, Q.V.; Jenkins, I.; et al. Combined Targeted Therapies for First-line Treatment of Metastatic Triple Negative Breast Cancer-A Phase II Trial of Weekly Nab-Paclitaxel and Bevacizumab Followed by Maintenance Targeted Therapy With Bevacizumab and Erlotinib. Clin. Breast Cancer 2019, 19, e283–e296. [Google Scholar] [CrossRef]
- Teneriello, M.G.; Tseng, P.C.; Crozier, M.; Encarnacion, C.; Hancock, K.; Messing, M.J.; Boehm, K.A.; Williams, A.; Asmar, L. Phase II evaluation of nanoparticle albumin-bound paclitaxel in platinum-sensitive patients with recurrent ovarian, peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2009, 27, 1426–1431. [Google Scholar] [CrossRef]
- Quintanilha, J.C.F.; Wang, J.; Sibley, A.B.; Jiang, C.; Etheridge, A.S.; Shen, F.; Jiang, G.; Mulkey, F.; Patel, J.N.; Hertz, D.L.; et al. Bevacizumab-induced hypertension and proteinuria: A genome-wide study of more than 1000 patients. Br. J. Cancer 2021, 126, 265–274. [Google Scholar] [CrossRef]
- Conlin, A.K.; Seidman, A.D.; Bach, A.; Lake, D.; Dickler, M.; D’Andrea, G.; Traina, T.; Danso, M.; Brufsky, A.M.; Saleh, M.; et al. Phase II trial of weekly nanoparticle albumin-bound paclitaxel with carboplatin and trastuzumab as first-line therapy for women with HER2-overexpressing metastatic breast cancer. Clin. Breast Cancer 2010, 10, 281–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.; Davies, A.M.; Sangha, R.S.; Lau, D.; Lara, P., Jr.; Chew, H.K.; Beckett, L.; Mack, P.C.; Riess, J.W.; Gandara, D.R.; et al. Phase I/II trial of pemetrexed plus nab-paclitaxel in advanced solid tumor patients with emphasis on non-small cell lung cancer. Investig. New Drugs 2013, 31, 1587–1591. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.L.; Savin, M.A.; Edelman, G.; Pippen, J.E.; Robert, N.J.; Geister, B.V.; Kirby, R.L.; Clawson, A.; O’Shaughnessy, J.A. Phase II study of weekly albumin-bound paclitaxel for patients with metastatic breast cancer heavily pretreated with taxanes. Clin. Breast Cancer 2007, 7, 850–856. [Google Scholar] [CrossRef]
- Lammers, P.E.; Lu, B.; Horn, L.; Shyr, Y.; Keedy, V. nab-Paclitaxel in Combination With Weekly Carboplatin With Concurrent Radiotherapy in Stage III Non-Small Cell Lung Cancer. Oncologist 2015, 20, 491–492. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.L.; Ray, A.; Van Brocklin, M.; Burnett, D.M.; Bowen, R.C.; Dyess, D.L.; Butler, T.W.; Dumlao, T.; Khong, H.T. A phase I trial of azacitidine and nanoparticle albumin bound paclitaxel in patients with advanced or metastatic solid tumors. Oncotarget 2016, 8, 52413–52419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaklamani, V.G.; Siziopikou, K.; Scholtens, D.; Lacouture, M.; Gordon, J.; Uthe, R.; Meservey, C.; Hansen, N.; Khan, S.A.; Jeruss, J.S.; et al. Pilot neoadjuvant trial in HER2 positive breast cancer with combination of nab-paclitaxel and lapatinib. Breast Cancer Res. Treat. 2012, 132, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.G.; Hughes, R.; Sumer, B.D.; Myers, L.L.; Truelson, J.M.; Khan, S.A.; Ma, T.W.; Xie, Y.; Yordy, J.S.; Cooley, S.; et al. A Phase I/II Study of Nab-Paclitaxel, Cisplatin, and Cetuximab With Concurrent Radiation Therapy for Locally Advanced Squamous Cell Cancer of the Head and Neck. Cancer Investig. 2017, 35, 23–31. [Google Scholar] [CrossRef]
- Yardley, D.A.; Hart, L.; Bosserman, L.; Salleh, M.N.; Waterhouse, D.M.; Hagan, M.K.; Richards, P.; DeSilvio, M.L.; Mahoney, J.M.; Nagarwala, Y. Phase II study evaluating lapatinib in combination with nab-paclitaxel in HER2-overexpressing metastatic breast cancer patients who have received no more than one prior chemotherapeutic regimen. Breast Cancer Res. Treat. 2013, 137, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Mukai, H.; Saeki, T.; Ro, J.; Lin, Y.C.; Nagai, S.E.; Lee, K.S.; Watanabe, J.; Ohtani, S.; Kim, S.B.; et al. A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients. Br. J. Cancer 2019, 120, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Sohal, D.P.S.; Duong, M.; Ahmad, S.A.; Gandhi, N.S.; Beg, M.S.; Wang-Gillam, A.; Wade, J.L.; Chiorean, E.G.; Guthrie, K.A.; Lowy, A.M.; et al. Efficacy of Perioperative Chemotherapy for Resectable Pancreatic Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 421–427. [Google Scholar] [CrossRef]
- Kanwal, M.; Smahel, M.; Olsen, M.; Smahelova, J.; Tachezy, R. Aspartate β-hydroxylase as a target for cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 163. [Google Scholar] [CrossRef]
- Adkins, D.; Ley, J.; Oppelt, P.; Gay, H.A.; Daly, M.; Paniello, R.C.; Jackson, R.; Pipkorn, P.; Rich, J.; Zevallos, J.; et al. Impact on Health-Related Quality of Life of Induction Chemotherapy Compared With Concurrent Cisplatin and Radiation Therapy in Patients With Head and Neck Cancer. Clin. Oncol. R. Coll. Radiol. 2019, 31, e123–e131. [Google Scholar] [CrossRef]
- Salazar, L.G.; Lu, H.; Reichow, J.L.; Childs, J.S.; Coveler, A.L.; Higgins, D.M.; Waisman, J.; Allison, K.H.; Dang, Y.; Disis, M.L.; et al. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol. 2017, 3, 969–973. [Google Scholar] [CrossRef]
- Yardley, D.A.; Coleman, R.; Conte, P.; Cortes, J.; Brufsky, A.; Shtivelband, M.; Young, R.; Bengala, C.; Ali, H.; Eakel, J.; et al. nab-Paclitaxel plus carboplatin or gemcitabine versus gemcitabine plus carboplatin as first-line treatment of patients with triple-negative metastatic breast cancer: Results from the tnAcity trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1763–1770. [Google Scholar] [CrossRef]
- Jani, A.; Horowitz, D.P. Radiation Therapy Deviations in Trial of Locally Advanced Pancreatic Cancer. JAMA 2016, 316, 1409. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, L.; Hu, J.; Wang, D.; Hu, C.; Zhou, J.; Wu, L.; Cao, L.; Liu, J.; Zhang, H.; et al. Pembrolizumab Plus Chemotherapy for Chinese Patients With Metastatic Squamous NSCLC in KEYNOTE-407. JTO Clin. Res. Rep. 2021, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Vicente, D.; Tafreshi, A.; Robinson, A.; Soto Parra, H.; Mazières, J.; Hermes, B.; Cicin, I.; Medgyasszay, B.; Rodríguez-Cid, J.; et al. A Randomized, Placebo-Controlled Trial of Pembrolizumab Plus Chemotherapy in Patients With Metastatic Squamous NSCLC: Protocol-Specified Final Analysis of KEYNOTE-407. J. Thorac. Oncol. 2020, 15, 1657–1669. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Foster, C.C.; Blair, E.A.; Karrison, T.G.; Agrawal, N.; Melotek, J.M.; Portugal, L.; Brisson, R.J.; Dekker, A.; Kochanny, S.; et al. OPTIMA: A phase II dose and volume de-escalation trial for human papillomavirus-positive oropharyngeal cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennari, A.; Sun, Z.; Hasler-Strub, U.; Colleoni, M.; Kennedy, M.J.; Von Moos, R.; Cortés, J.; Vidal, M.J.; Hennessy, B.; Walshe, J.; et al. A randomized phase II study evaluating different maintenance schedules of nab-paclitaxel in the first-line treatment of metastatic breast cancer: Final results of the IBCSG 42-12/BIG 2-12 SNAP trial. Ann. Oncol. 2018, 29, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Alberts, D.S.; Blessing, J.A.; Landrum, L.M.; Warshal, D.P.; Martin, L.P.; Rose, S.L.; Bonebrake, A.J.; Ramondetta, L.M. Phase II trial of nab-paclitaxel in the treatment of recurrent or persistent advanced cervix cancer: A gynecologic oncology group study. Gynecol. Oncol. 2012, 127, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.S.; Bodman-Smith, M.D.; Dalgleish, A.G.; Fischer, M.D. Phase I/II study of topical imiquimod and intralesional interleukin-2 in the treatment of accessible metastases in malignant melanoma. Br. J. Dermatol. 2007, 156, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Sanoff, H.K.; Moon, D.H.; Moore, D.T.; Boles, J.; Bui, C.; Blackstock, W.; O’Neil, B.H.; Subramaniam, S.; McRee, A.J.; Carlson, C.; et al. Phase I/II trial of nano-camptothecin CRLX101 with capecitabine and radiotherapy as neoadjuvant treatment for locally advanced rectal cancer. Nanomedicine 2019, 18, 189–195. [Google Scholar] [CrossRef]
- Werner, T.L.; Ray, A.; Lamb, J.G.; VanBrocklin, M.; Hueftle, K.; Cohen, A.L.; Beck, A.C.; Buys, S.S.; Dyess, D.L.; Butler, T.W.; et al. A Phase I Study of Neoadjuvant Chemotherapy With Nab-Paclitaxel, Doxorubicin, and Cyclophosphamide in Patients With Stage II to III Breast Cancer. Clin. Breast Cancer 2017, 17, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Kottschade, L.A.; Suman, V.J.; Perez, D.G.; McWilliams, R.R.; Kaur, J.S.; Amatruda, T.T.; Geoffroy, F.J.; Gross, H.M.; Cohen, P.A.; Jaslowski, A.J.; et al. A randomized phase 2 study of temozolomide and bevacizumab or nab-paclitaxel, carboplatin, and bevacizumab in patients with unresectable stage IV melanoma: A North Central Cancer Treatment Group study, N0775. Cancer 2013, 119, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Guminski, A.D.; Lee, A.; Lumba, S.; Maher, R. Nab-paclitaxel salvage chemotherapy for metastatic ocular melanoma. J. Clin. Oncol. 2012, 30, e19042. [Google Scholar] [CrossRef]
- Kottschade, L.A.; Suman, V.J.; Amatruda, T.; McWilliams, R.R.; Mattar, B.I.; Nikcevich, D.A.; Behrens, R.; Fitch, T.R.; Jaslowski, A.J.; Markovic, S.N. A phase II trial of nab-paclitaxel (ABI-007) and carboplatin in patients with unresectable stage IV melanoma: A North Central Cancer Treatment Group Study, N057E(1). Cancer 2011, 117, 1704–1710. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Feliu, J.; Jorge Fernández, M.; Macarulla, T.; Massuti, B.; Albero, A.; González González, J.F.; Quintero-Aldana, G.; Delgado-Mingorance, J.I.; Fernández Montes, A.; García Piernavieja, C.; et al. Phase II clinical trial of nab-paclitaxel plus gemcitabine in elderly patients with previously untreated locally advanced or metastatic pancreatic adenocarcinoma: The BIBABRAX study. Cancer Chemother. Pharmacol. 2021, 87, 543–553. [Google Scholar] [CrossRef]
- Spitler, L.E.; Boasberg, P.; O’Day, S.; Hamid, O.; Cruickshank, S.; Mesko, S.; Weber, R.W. Phase II study of nab-paclitaxel and bevacizumab as first-line therapy for patients with unresectable stage III and IV melanoma. Am. J. Clin. Oncol. 2015, 38, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Luo, L.; Zeng, L.; Xing, J.; Xia, Y.; Sun, S.; Zhang, L.; Yu, Z.; Yao, J.; Yu, Z.; et al. Porous Gold Nanoshells on Functional NH2 -MOFs: Facile Synthesis and Designable Platforms for Cancer Multiple Therapy. Small 2018, 14, e1801851. [Google Scholar] [CrossRef] [PubMed]
- Clive, S.; Gardiner, J.; Leonard, R.C. Miltefosine as a topical treatment for cutaneous metastases in breast carcinoma. Cancer Chemother. Pharmacol. 1999, 44, S29–S30. [Google Scholar] [CrossRef] [PubMed]
- Jazrawi, A.; Pantiora, E.; Abdsaleh, S.; Bacovia, D.V.; Eriksson, S.; Leonhardt, H.; Wärnberg, F.; Karakatsanis, A. Magnetic-Guided Axillary UltraSound (MagUS) Sentinel Lymph Node Biopsy and Mapping in Patients with Early Breast Cancer. A Phase 2, Single-Arm Prospective Clinical Trial. Cancers 2021, 13, 4285. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
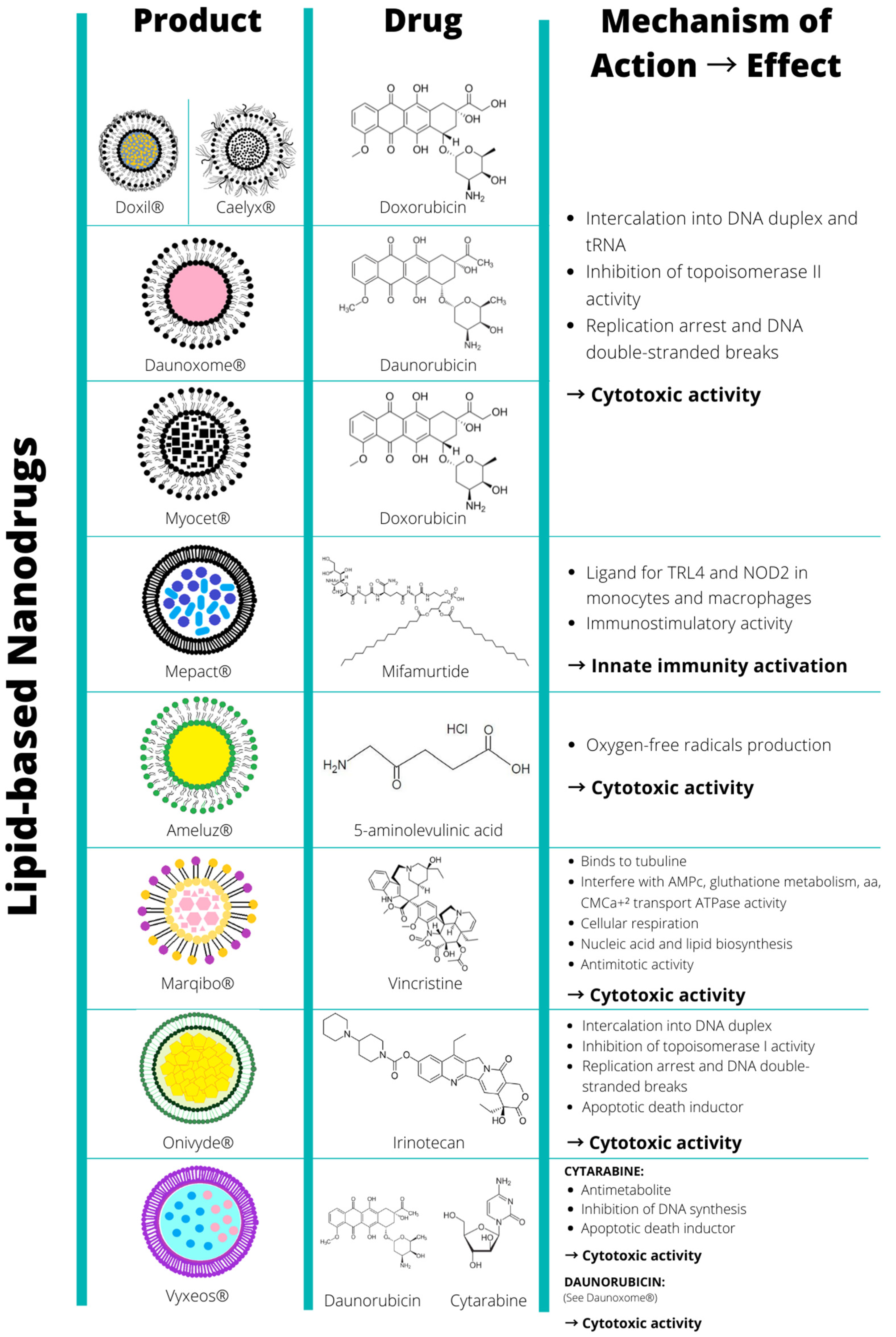
| Nanostructure | ProductTM | Nanotechnology Platform | Drug | Nanoformulation Advantages | TME Targeting | Indication | Company | Approval (Year) |
|---|---|---|---|---|---|---|---|---|
| Lipid based nanoparticles | Doxil | PEGylatyed STEALTH® liposomes composed of MPEG-DSPE, HSPC, CHO. | Doxorubicin | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ cardiotoxicity | Cancer and stroma cells | Kaposi’s sarcoma, ovarian Ca, multiple myeloma | Ortho Biotech | FDA (1995) |
| Caelyx | PEGylated liposomal doxorubicin composed of MPEG-DSPE, HSPC, CHO | Doxorubicin | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ cardiotoxicity | Cancer and stroma cells | Metastatic breast, Ca., ovarian Ca., Kaposi’s sarcoma, and multiple myeloma | Schering-Plough | EMA (1996) | |
| DaunoXome | Citrate salt of daunorubicin encapsulated in non-pegylated liposomes composed of DSPC and CHO (2:1 MR) | Daunorubicin | ↓ protein binding ↑ blood circulation time ↑ tumor uptake (EPR) ↓ cardiotoxicity | Cancer and stroma cells | Kaposi’s sarcoma | Galen | FDA (1996) | |
| Myocet | Liposomal doxorubicin (non-PEGylated) composed of PC, CHO, citric acid, and NaOH | Doxorubicin | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ cardiotoxicity | Cancer and stroma cells | Metastatic breast Ca. | Teva UK | EMA (2000) | |
| Mepact | Liposomal mifamurtide (fully synthetic analogue of a component of Mycobacterium sp. cell wall) composed of POGP, DGPS, MS | Mifamurtide | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ toxicity | Macrophages | Osteosarcoma | Millenium | EMA (2009) | |
| Ameluz | Gel containing 5-aminolevulinic acid, E211, SoyPC, and PG | 5-aminolevulinic acid | sustained release ↓ toxicity | Cancer and stroma cells | Superficial and/or nodular basal cell carcinoma | Biofrontera Bioscience GmbH | EMA (2011) | |
| Marqibo | Liposomal vincristine composed of SM and CHO | Vincristine | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ toxicity | Cancer and stroma cells | Acute lymphoid leukaemia | Spectrum | FDA (2012) | |
| Onivyde | Nanoliposomes composed of DSPC, CHO, MPEG-2000-DSPE | Irinotecan | ↑ blood circulation time ↑ tumor uptake (EPR) ↓ toxicity | Cancer and stroma cells | Pancreatic Ca., Colorectal Ca. | Merrimack | FDA (2015) | |
| Vyxeos | Nanoliposomes composed of DSPC, DEPG, and CHO | Daunorubicin Cytarabine | ↑ blood circulation time, ↑ accumulation in bone marrow | Cancer and stroma cells | Acute myeloid leukaemia | Jazz Pharmaceuticals | EMA (2018) | |
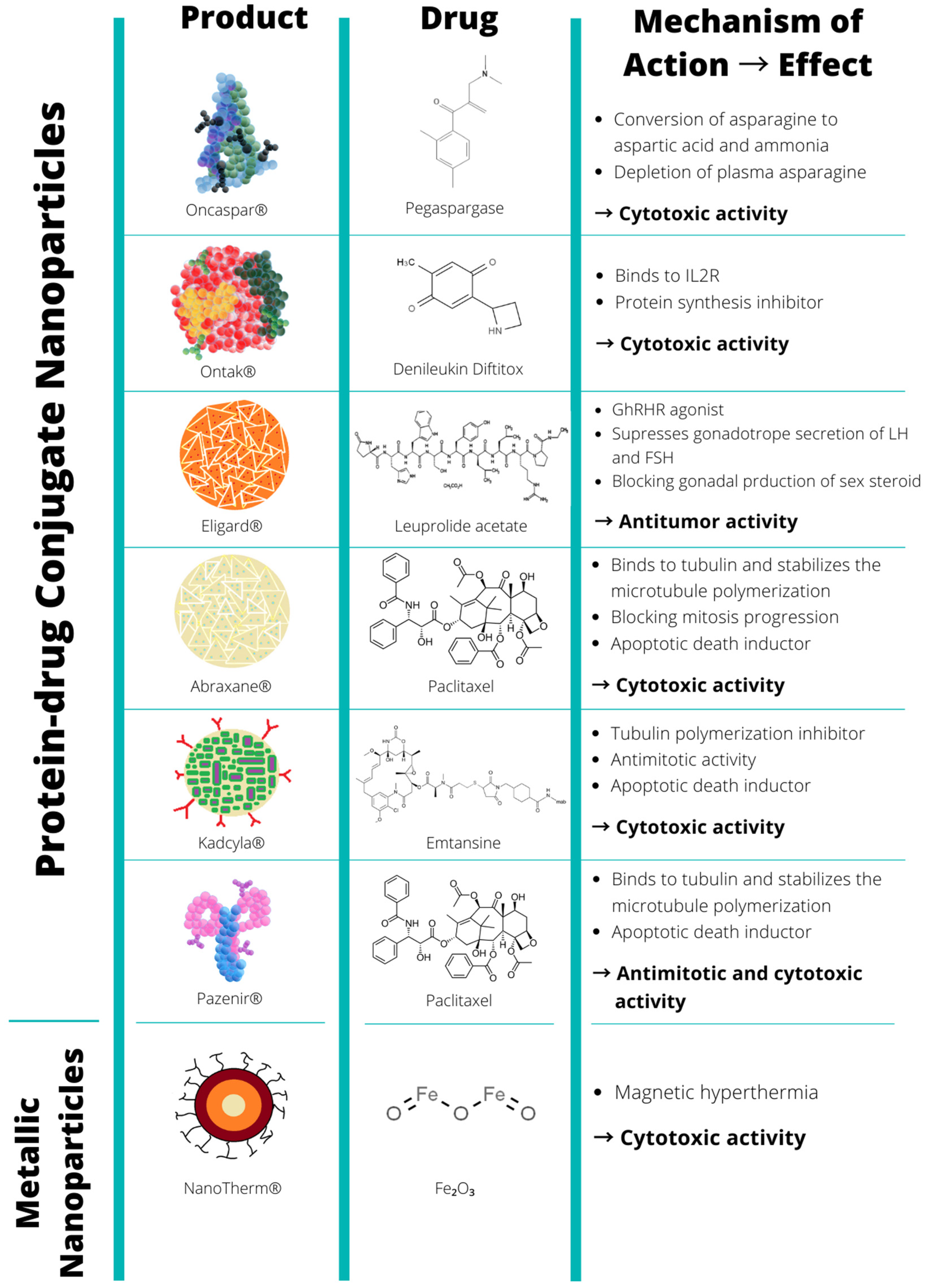
| Protein-drug conjugates | Oncaspar | Covalent conjugate of L-asparaginase with mPEG, MSP, Na2HPO4, Heptahydrate, and NaCl | Pegaspargase | ↑ blood circulation time ↑ tumor uptake (EPR) | Cancer cells | Acute lymphoblastic leukaemia | Les Laboratoires Servier | FDA (1994) |
| Ontak | Recombinant cytotoxic protein composed of diphtheria toxin fragments A and B (Met1-Thr387)-His and human IL-2 (Ala1-Thr133) | Denileukin Diftitox | ↑ blood circulation time ↑ tumor uptake (EPR) ↑ selectivity ↓ severe toxicity | Activated T-cells | Cutaneous T-cell lymphoma | Les laboratoires Servier | FDA (1999) | |
| Eligard | Polymeric matrix of leuprorelin acetate composed of PLGA (85:15) NMP and LA | Leuprorelin acetate | ↑ blood circulation time ↑ tumor uptake (EPR) | Cancer cells | Prostate cancer | Recordati Industria Chimica e Farmaceutica | FDA (2002) | |
| Abraxane | Colloidal suspension without solvent of paclitaxel bound to albumin (active substance) in the form of a spherical nanoparticle | Paclitaxel | ↑ Solubility ↑ blood circulation time ↑ tumor uptake (EPR) ↓ severe toxicity | Cancer and stroma cells | Breast Ca. Non-small lung Ca., Pancreatic Ca. | American Biosciencem, Inc. | FDA (2005) | |
| Kadcyla | Trastuzumab, covalently linked to DM1 via the stable thioether linker MCC | DM1 (or Emtansine) | ↑ blood circulation time ↑ tumor uptake (EPR) ↑ selectivity ↓ toxicity | Cancer cells | HER2+ breast Ca. | Roche Genentech | EMA (2013) FDA (2013) | |
| Pazenir | Paclitaxel formulated as albumin bound nanoparticles. Powder for dispersion for infusion | Paclitaxel | ↑ Solubility ↑ blood circulation time ↑ tumor uptake (EPR) ↓ severe toxicity | Cancer cells | Metastatic breast Ca., metastatic adenocarcinoma of the pancreas, non-small cell lung Ca. | Ratiopharm GmbH | EMA (2019) | |
| Metallic nanoparticles | NanoTherm | Nanoparticles of superparamagnetic iron oxide coated with amino silane | Fe2O3 | ↑ blood circulation time ↑ tumor uptake (EPR) -heat production under stimulation with EMF -teranostic properties | Residual cancer and stroma cells | Glioblastoma, prostate, and pancreatic Ca. | Magforce | EMA (2013) |
| Drug | ProductTM | Molecular Target | Cell Targeting | Company | Approval (Year) | Indication |
|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | Mylotarg | CD33 | Myeloid stem cells, myeloblasts, monoblasts, monocytes/macrophages, granulocyte precursors, and mast cells | Pfizer/Wyeth | 2017; 2000 | Relapsed acute myelogenous leukemia (AML) |
| Brentuximab vedotin | Adcetris | CD30 | Lymphoid cells | Seattle Genetics, Millennium/ Takeda | 2011 | Relapsed HL and relapsed sALCL |
| Inotuzumab ozogamicin | Besponsa | CD22 | B-cells | Pfizer/Wyeth | 2017 | Relapsed or refractory CD22-positive B-cell precursor acute lymphoblastic leukemia |
| Moxetumomab pasudotox | Lumoxiti | CD22 | Leukemia cells | Astrazeneca | 2018 | Relapsed or refractory hairy cell leukemia (HCL) |
| Polatuzumab vedotin-piiq | Polivy | CD79 | B-cells | Genentech, Roche | 2019 | Relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) |
| Enfortumab vedotin | Padcev | Nectin-4 | Cancer cells | Astellas/ Seattle Genetics | 2019 | Locally advanced or metastatic urothelial cancer who have received a PD-1 or PD-L1 inhibitor, and a Paclitaxel-containing therapy |
| Trastuzumab deruxtecan | Enhertu | HER2 | Cancer cells | AstraZeneca/ Daiichi Sankyo | 2019 | Unresectable or metastatic HER2-positive breast cancer patients who have received two or more prior anti-HER2-based regimens |
| Sacituzumab govitecan | Trodelvy | Trop-2 | Cancer cells | Immunomedics | 2020 | Metastatic triple-negative breast cancer (mTNBC) patients who have received at least two prior therapies (for patients with relapsed or refractory metastatic disease) |
| Belantamab mafodotin-blmf | Blenrep | BCMA | B-cells | GlaxoSmithKline | 2020 | Relapsed or refractory multiple myeloma |
| Loncastuximab tesirine-lpyl | Zynlonta | CD19 | B cells and follicular dendritic cells | ADC Therapeutics | 2021 | Large B-cell lymphoma |
| Tisotumab vedotin-tftv | Tivdak | Tissue factor | Cancer and stroma cells of TME | Seagen Inc | 2021 | Recurrent or metastatic cervical cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, F.; Caruana, P.; De la Fuente, N.; Español, P.; Gámez, M.; Balart, J.; Llurba, E.; Rovira, R.; Ruiz, R.; Martín-Lorente, C.; et al. Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges. Biomolecules 2022, 12, 784. https://doi.org/10.3390/biom12060784
Rodríguez F, Caruana P, De la Fuente N, Español P, Gámez M, Balart J, Llurba E, Rovira R, Ruiz R, Martín-Lorente C, et al. Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges. Biomolecules. 2022; 12(6):784. https://doi.org/10.3390/biom12060784
Chicago/Turabian StyleRodríguez, Francisco, Pablo Caruana, Noa De la Fuente, Pía Español, María Gámez, Josep Balart, Elisa Llurba, Ramón Rovira, Raúl Ruiz, Cristina Martín-Lorente, and et al. 2022. "Nano-Based Approved Pharmaceuticals for Cancer Treatment: Present and Future Challenges" Biomolecules 12, no. 6: 784. https://doi.org/10.3390/biom12060784