
Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation


Abstract
:1. Introduction
2. Materials and Methods
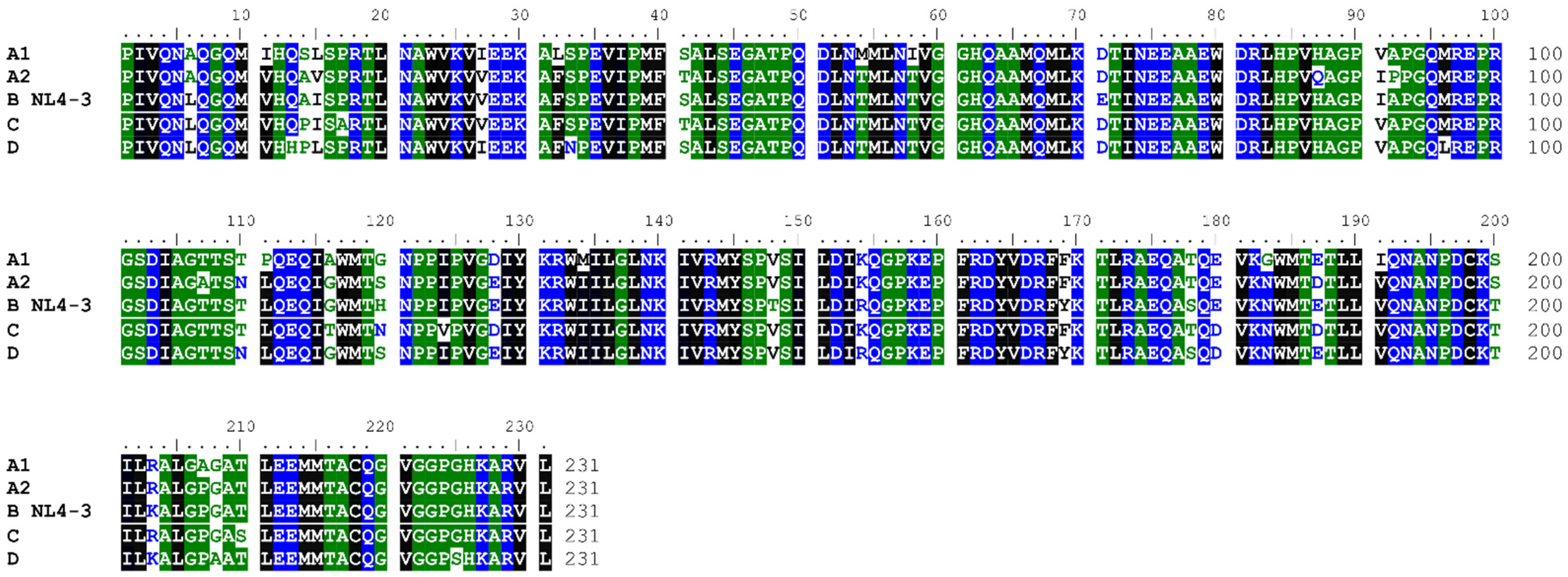
2.1. Alignment of CA Sequences from Diverse Clades
2.2. Construction of Monomeric CA Expression Vectors from Diverse Clades
2.3. Overproduction and Purification of Monomeric CA Proteins
2.4. In Vitro Assembly Assay
2.5. Modeling and Generation of the HIV-1 CA Proteins from Diverse Clades
2.6. Overproduction and Purification of Hexameric CA Protein
2.7. Peptides
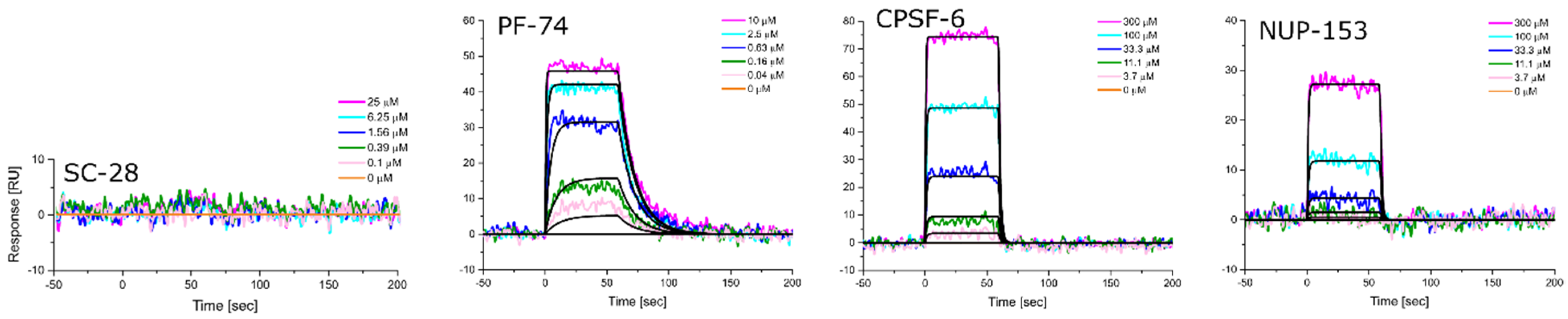
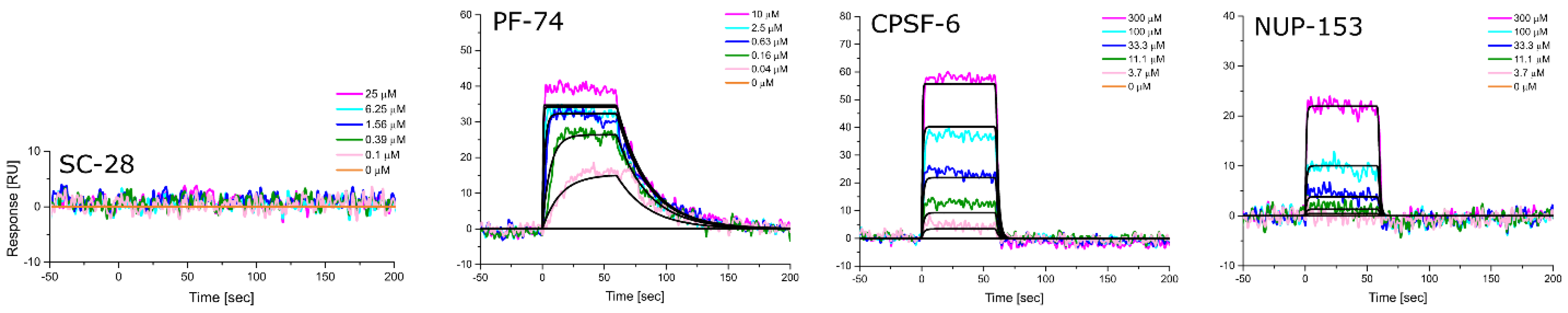
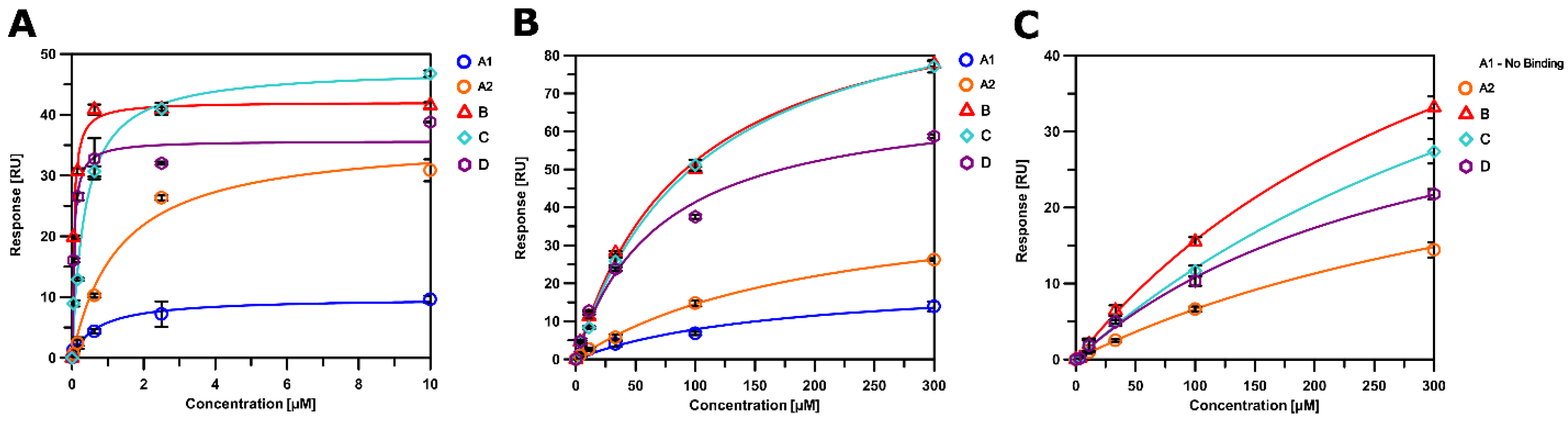
2.8. SPR Characterization
2.9. Docking Calculations for PF-74
2.10. Docking Calculations for NUP-153
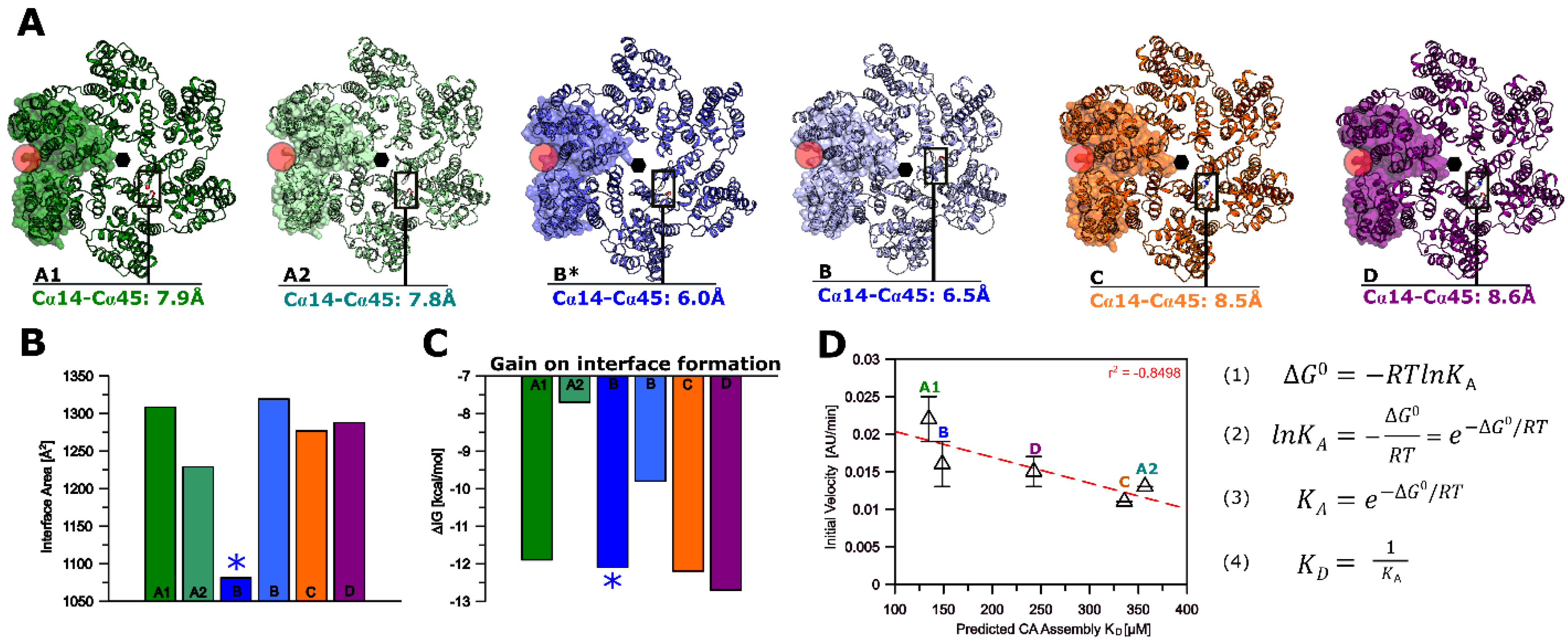
2.11. Interface and Interprotomer Pocket Analysis of the HIV-1 CA Hexamer
3. Results
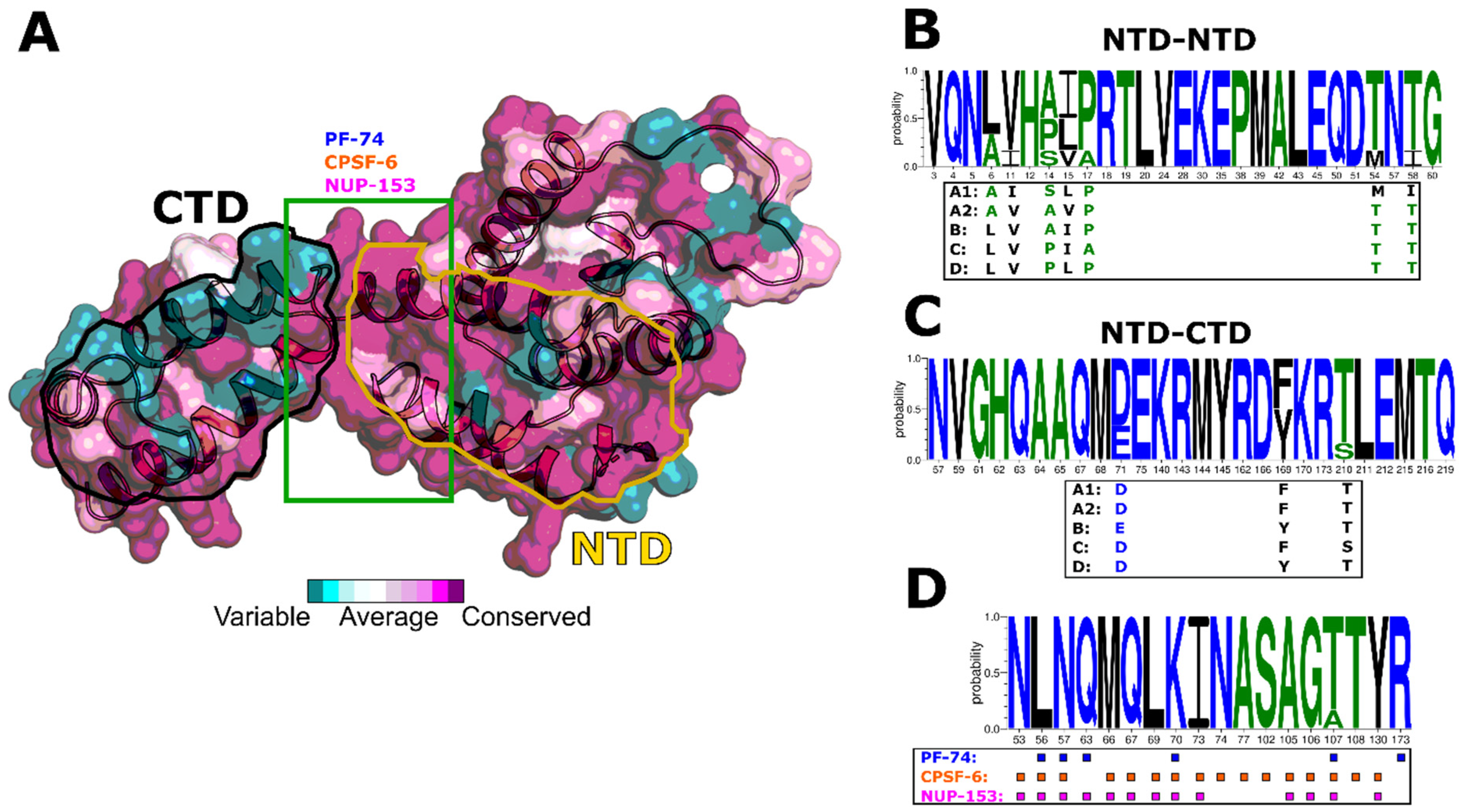
3.1. HIV-1 CA Protein Conservation
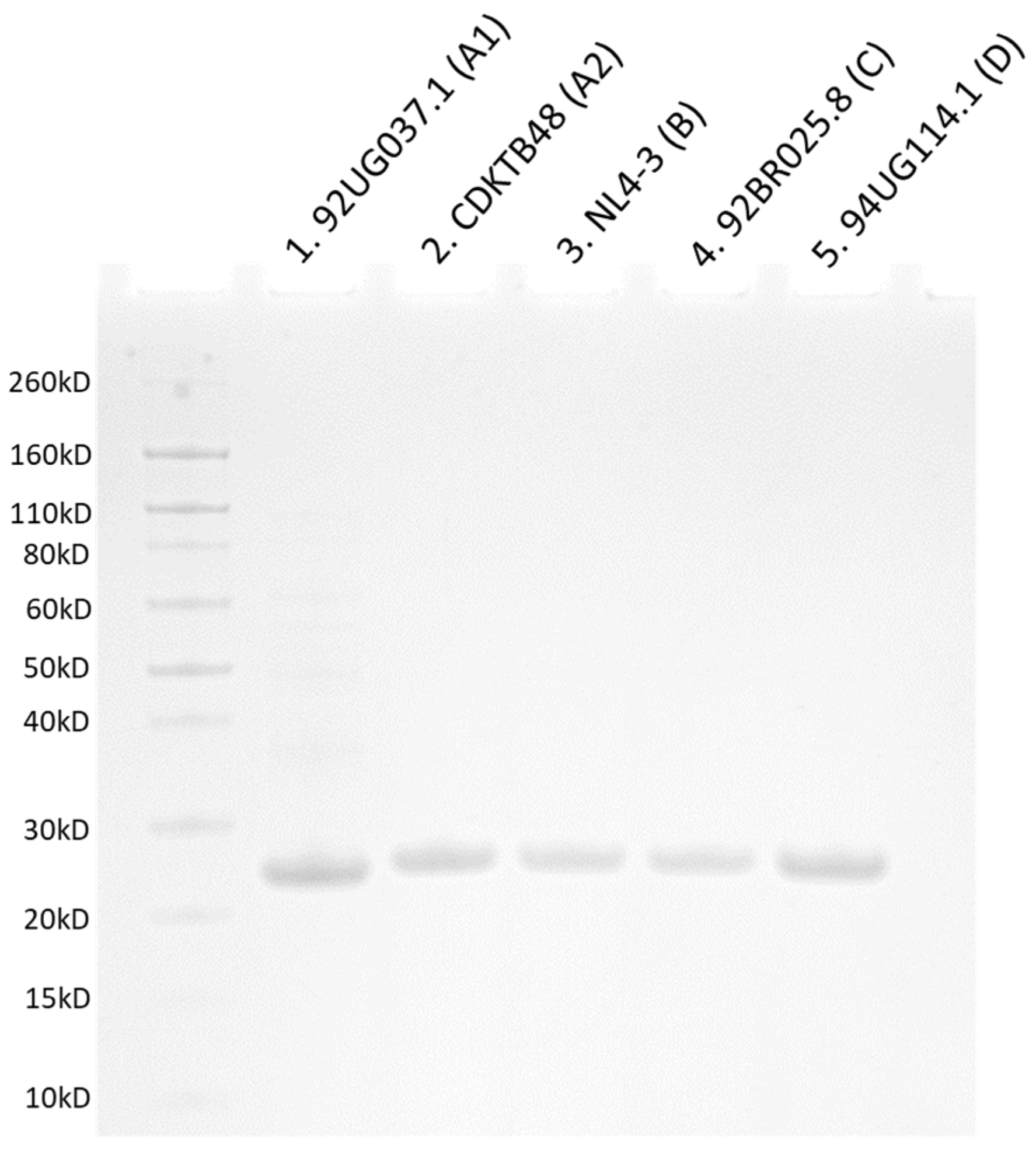
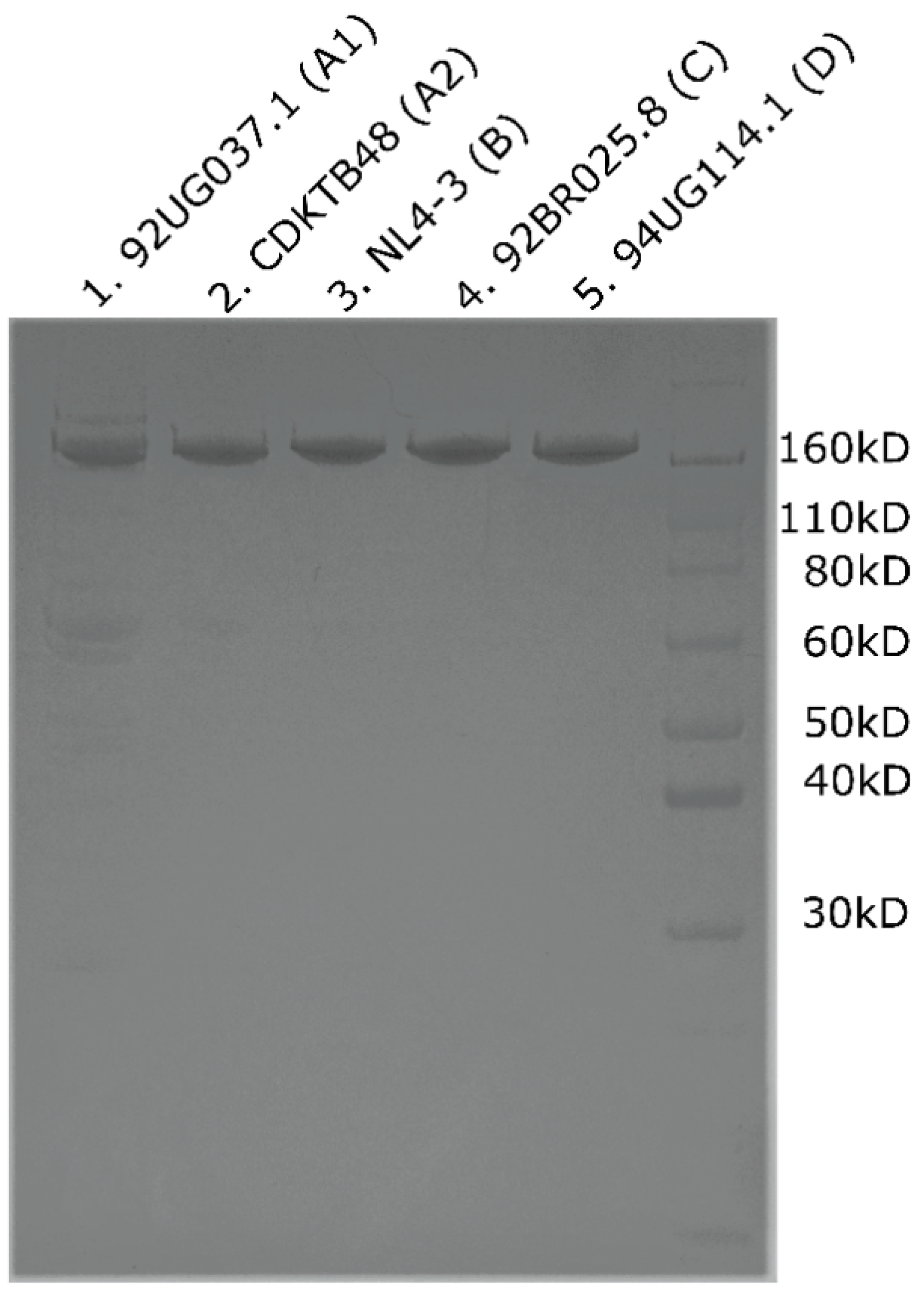
3.2. Overproduction and Purification of CA Monomeric Proteins from Diverse Clades
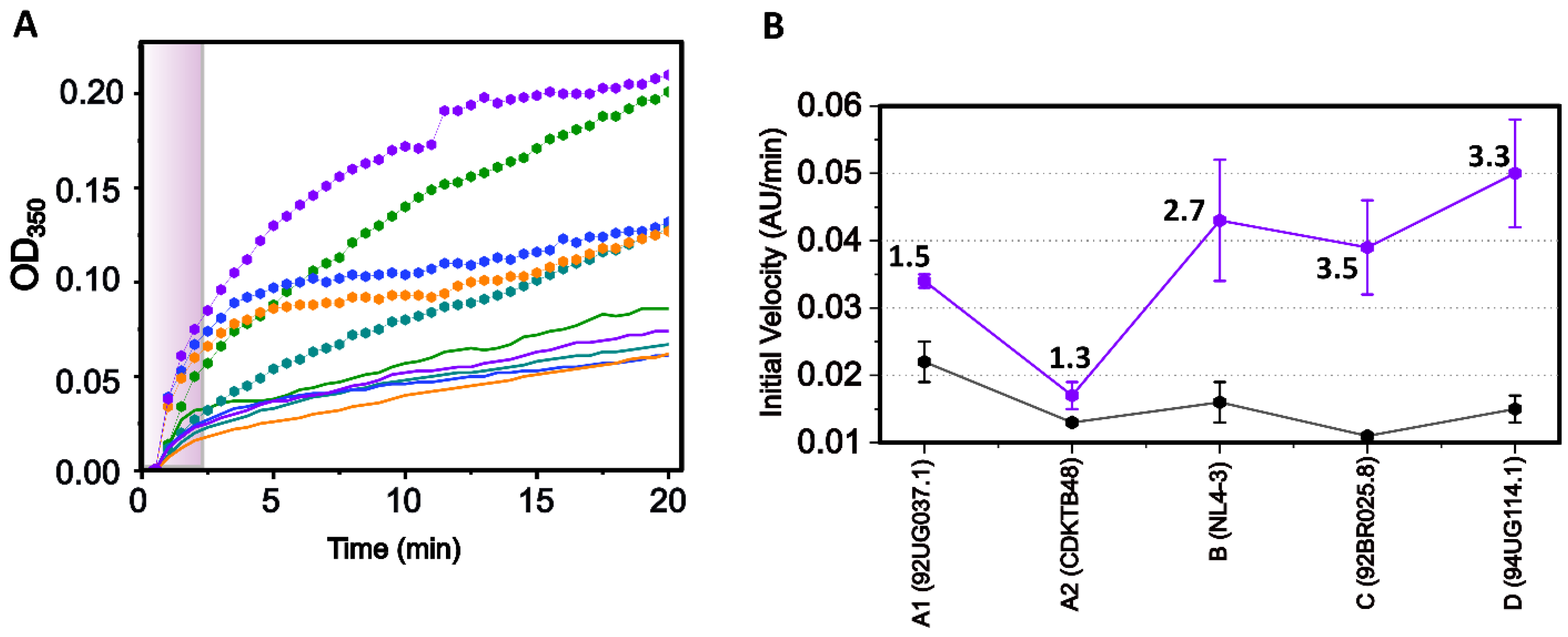
3.3. In Vitro Assembly of CA Monomeric Proteins from Diverse Clades
3.4. Overproduction and Purification of CA Hexameric Proteins from Diverse Clades
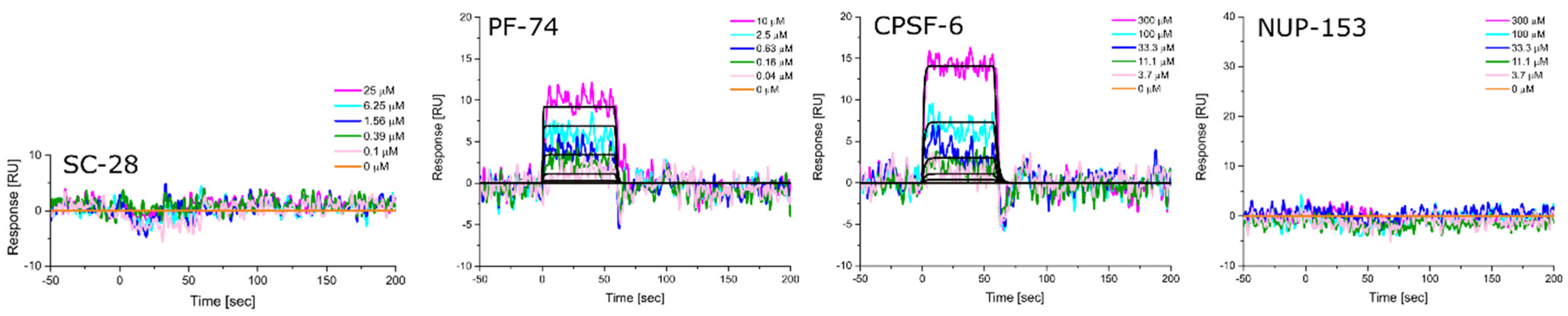
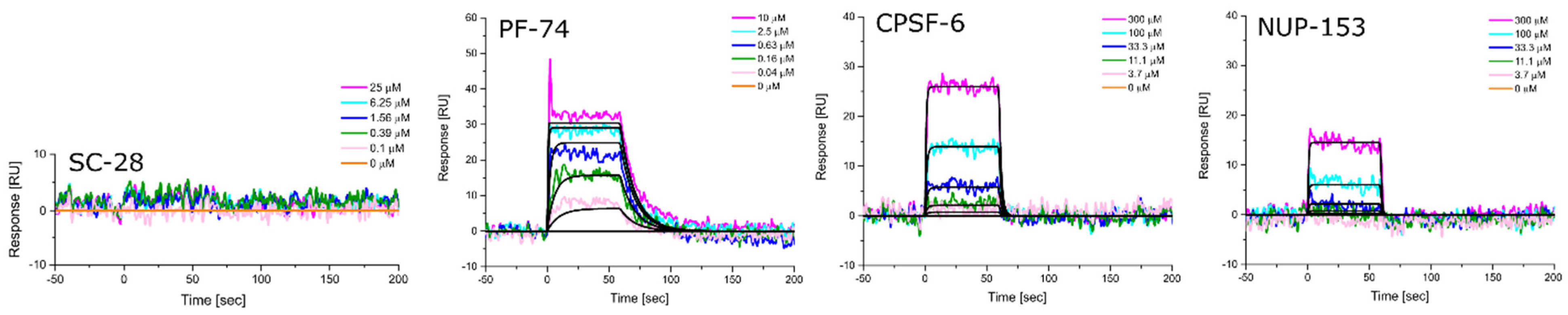
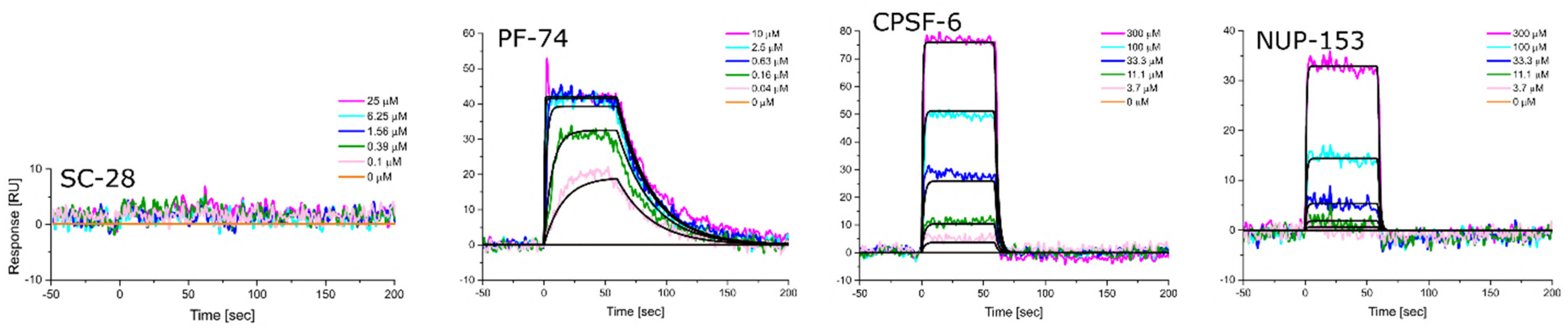
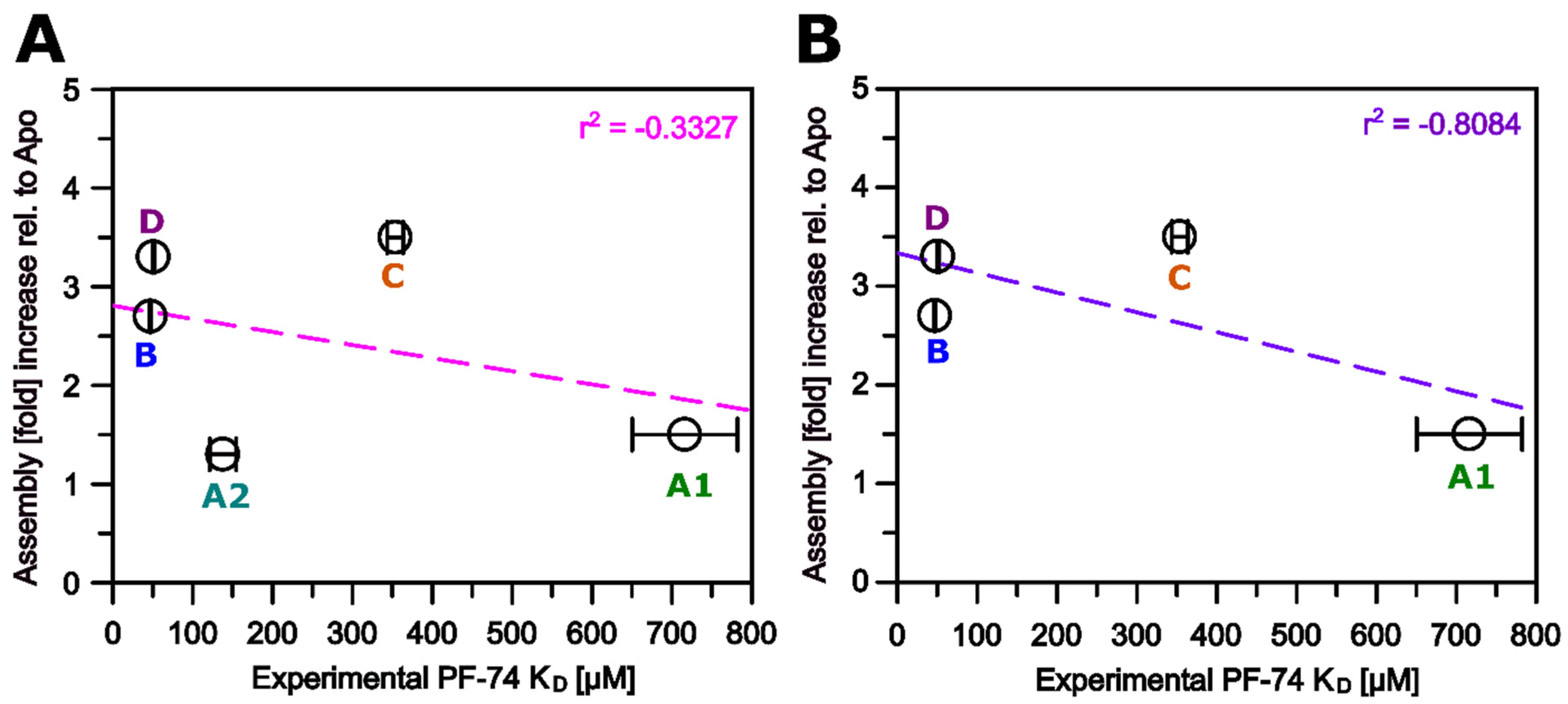
3.5. Altered Substrate Recognition across Hexameric CA Proteins from Diverse Clades
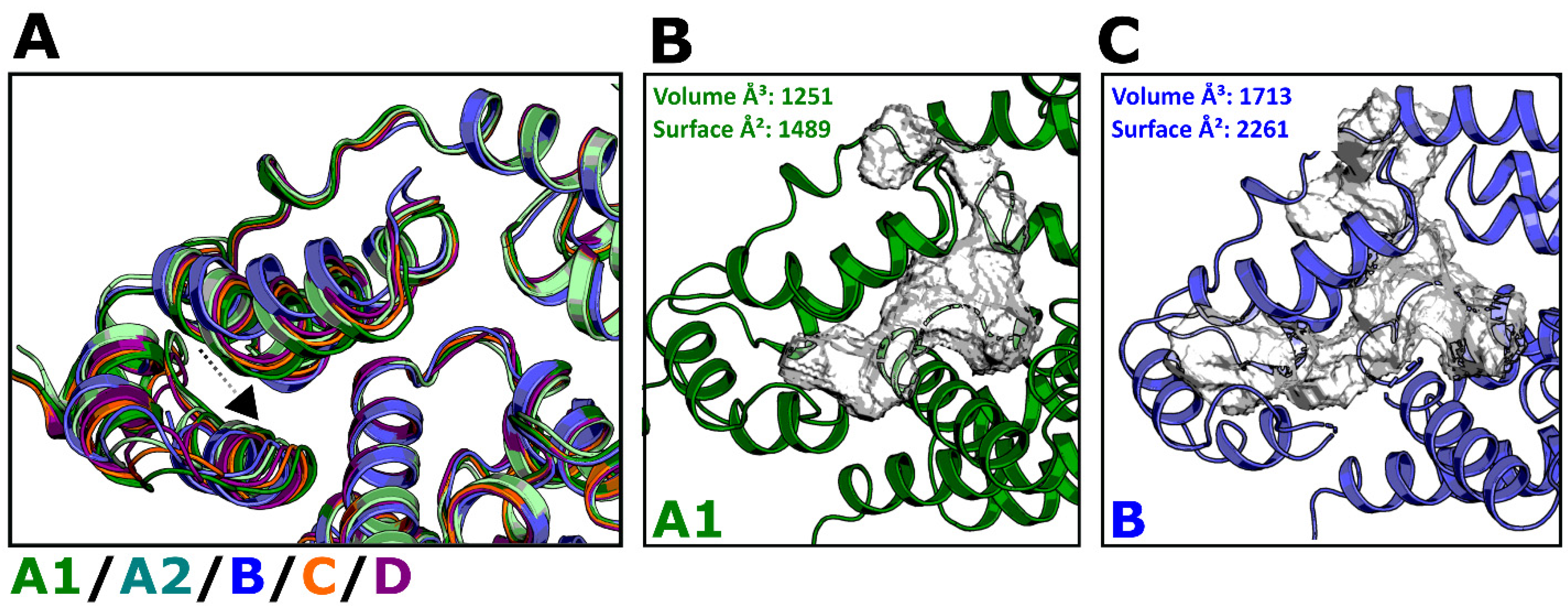
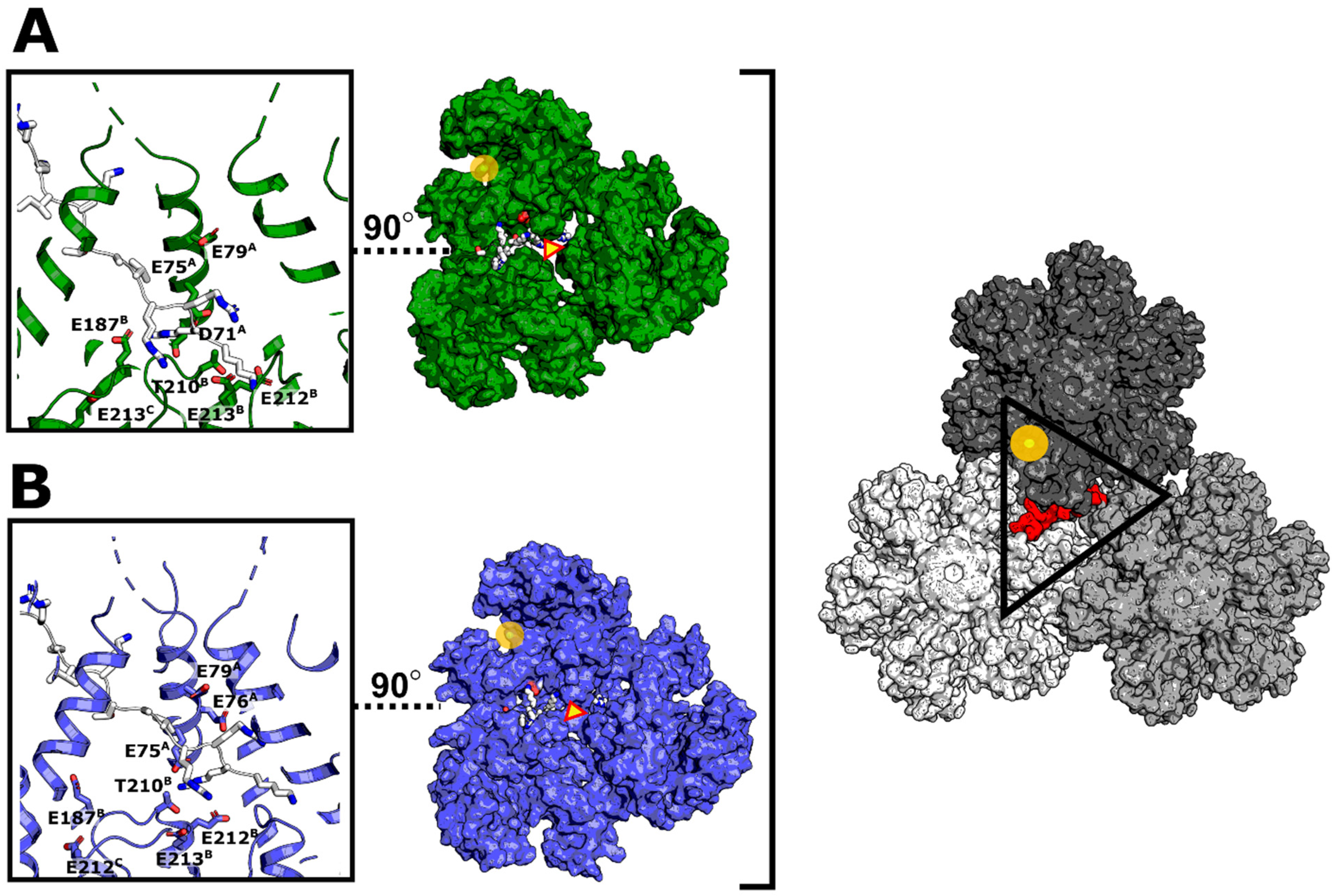
3.6. Modeling of Non-Subtype-B CA Proteins—Implications for Assembly
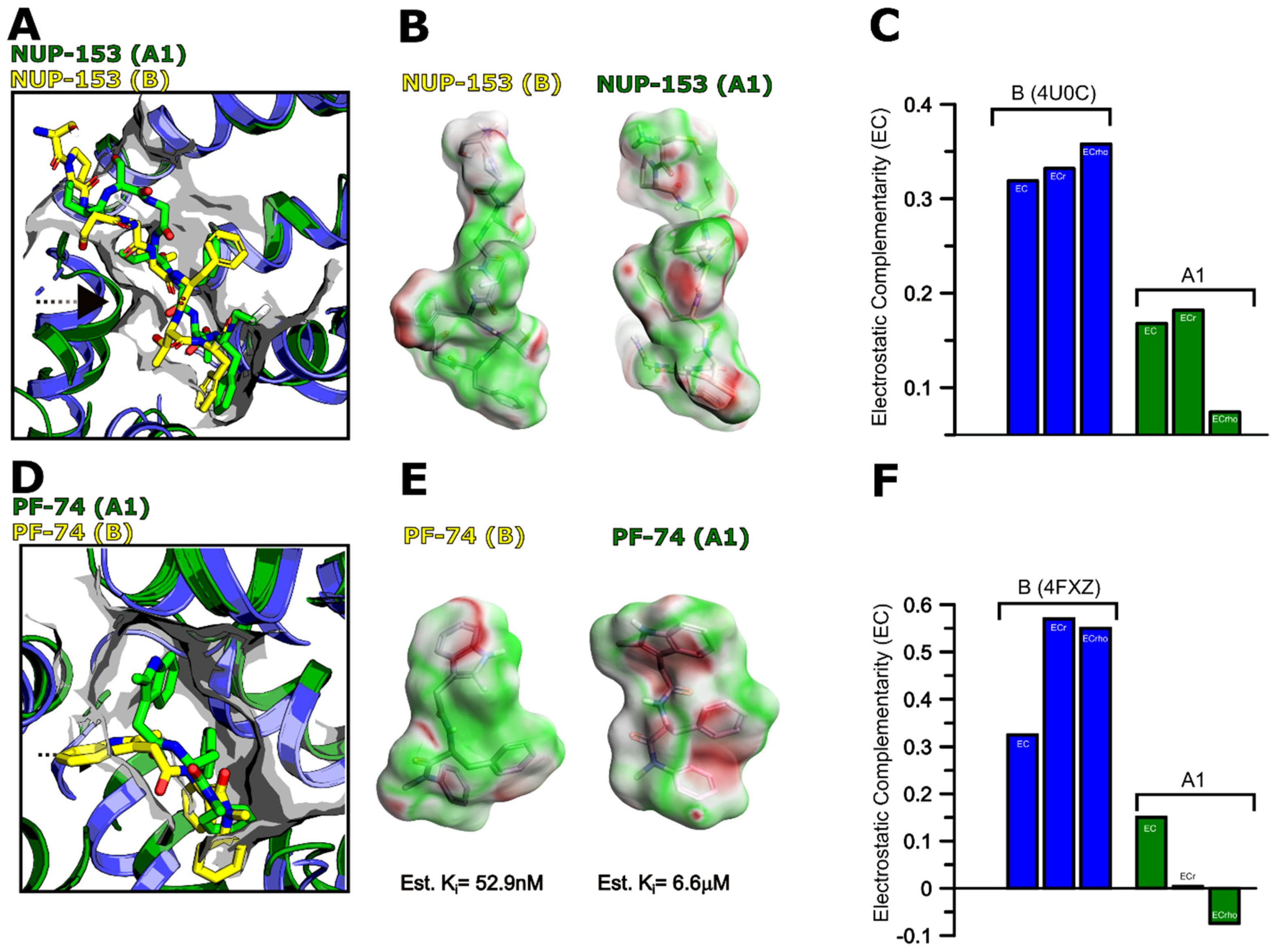
3.7. Modeling of Non-Subtype-B CA Proteins—Implications for PF-74 and NUP-153 Binding
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gamble, T.R.; Vajdos, F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; I Sundquist, W.; Hill, C.P. Crystal Structure of Human Cyclophilin A Bound to the Amino-Terminal Domain of HIV-1 Capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Ganser-Pornillos, B.K.; Cheng, A.; Yeager, M. Structure of Full-Length HIV-1 CA: A Model for the Mature Capsid Lattice. Cell 2007, 131, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; I Sundquist, W. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitti, R.K.; Lee, B.M.; Walker, J.; Summers, M.F.; Yoo, S.; Sundquist, W.I. Structure of the Amino-Terminal Core Domain of the HIV-1 Capsid Protein. Science 1996, 273, 231–235. [Google Scholar] [CrossRef]
- Kelly, B.N.; Howard, B.R.; Wang, H.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Implications for viral capsid assembly from crystal structures of HIV-1 Gag(1-278) and CA(N)(133-278). Biochemistry 2006, 45, 11257–11266. [Google Scholar] [CrossRef]
- Kelly, B.N.; Kyere, S.; Kinde, I.; Tang, C.; Howard, B.R.; Robinson, H.; Sundquist, W.I.; Summers, M.F.; Hill, C.P. Structure of the Antiviral Assembly Inhibitor CAP-1 Complex with the HIV-1 CA Protein. J. Mol. Biol. 2007, 373, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Worthylake, D.K.; Wang, H.; Yoo, S.; Sundquist, W.I.; Hill, C.P. Structures of the HIV-1 capsid protein dimerization domain at 2.6 A resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 85–92. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-Ray Structures of the Hexameric Building Block of the HIV Capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [Green Version]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Gres, A.T.; Kirby, K.A.; KewalRamani, V.N.; Tanner, J.J.; Pornillos, O.; Sarafianos, S.G. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 2015, 349, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Lascano, J.; Uchil, P.D.; Mothes, W.; Luban, J. TRIM5 Retroviral Restriction Activity Correlates with the Ability to Induce Innate Immune Signaling. J. Virol. 2015, 90, 308–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalskyy, D.B.; Ivanov, D.N. Recognition of the HIV Capsid by the TRIM5α Restriction Factor Is Mediated by a Subset of Pre-Existing Conformations of the TRIM5α SPRY Domain. Biochemistry 2014, 53, 1466–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grutter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tipper, C.; Sodroski, J. Role of TRIM5alpha RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade, and restriction of simian immunodeficiency virus. J. Virol. 2011, 85, 8116–8132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganser-Pornillos, B.K.; Chandrasekaran, V.; Pornillos, O.; Sodroski, J.G.; Sundquist, W.I.; Yeager, M. Hexagonal assembly of a restricting TRIM5α protein. Proc. Natl. Acad. Sci. USA 2011, 108, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Black, L.R.; Aiken, C. TRIM5α Disrupts the Structure of Assembled HIV-1 Capsid Complexes In Vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- Langelier, C.R.; Sandrin, V.; Eckert, D.M.; Christensen, D.E.; Chandrasekaran, V.; Alam, S.L.; Aiken, C.; Olsen, J.C.; Kar, A.K.; Sodroski, J.G.; et al. Biochemical Characterization of a Recombinant TRIM5α Protein That Restricts Human Immunodeficiency Virus Type 1 Replication. J. Virol. 2008, 82, 11682–11694. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Pathak, V.K.; Peng, W.; Hu, W.S. Capsid proteins from human immunodeficiency virus type 1 and simian immunodeficiency virus SIVmac can coassemble into mature cores of infectious viruses. J. Virol. 2008, 82, 8253–8261. [Google Scholar] [CrossRef] [Green Version]
- Sokolskaja, E.; Luban, J. Cyclophilin, TRIM5, and innate immunity to HIV-1. Curr. Opin. Microbiol. 2006, 9, 404–408. [Google Scholar] [CrossRef]
- Javanbakht, H.; Yuan, W.; Yeung, D.F.; Song, B.; Diaz-Griffero, F.; Li, Y.; Li, X.; Stremlau, M.; Sodroski, J. Characterization of TRIM5α trimerization and its contribution to human immunodeficiency virus capsid binding. Virology 2006, 353, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Berthoux, L.; Sebastian, S.; Sokolskaja, E.; Luban, J. Cyclophilin A is required for TRIM5α-mediated resistance to HIV-1 in Old World monkey cells. Proc. Natl. Acad. Sci. USA 2005, 102, 14849–14853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.; Smith, M.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Jacques, D.; McEwan, W.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.; Aiken, C.; James, L.C. Host Cofactors and Pharmacologic Ligands Share an Essential Interface in HIV-1 Capsid That Is Lost upon Disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.; Selwood, D.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.; James, L.C. CPSF6 Defines a Conserved Capsid Interface that Modulates HIV-1 Replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Matreyek, K.; Yücel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 Phenylalanine-Glycine Motifs Engage a Common Binding Pocket within the HIV-1 Capsid Protein to Mediate Lentiviral Infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef] [Green Version]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelek, M.; Di Nunzio, F.; Henriques, R.; Charneau, P.; Arhel, N.; Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA 2012, 109, 8564–8569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaller, T.; Ocwieja, K.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 Capsid-Cyclophilin Interactions Determine Nuclear Import Pathway, Integration Targeting and Replication Efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Engelman, A. The Requirement for Nucleoporin NUP153 during Human Immunodeficiency Virus Type 1 Infection Is Determined by the Viral Capsid. J. Virol. 2011, 85, 7818–7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichel, K.; Price, A.J.; Schaller, T.; Towers, G.J.; Freund, S.M.; James, L.C. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 2013, 10, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, B.; Buffone, C.; Opp, S.; Di Nunzio, F.; Vieira, D.A.D.S.A.; Brandariz-Nuñez, A.; Diaz-Griffero, F. Restriction of HIV-1 Requires the N-Terminal Region of MxB as a Capsid-Binding Motif but Not as a Nuclear Localization Signal. J. Virol. 2015, 89, 8599–8610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Pan, Q.; Liang, Z.; Qiao, W.; Cen, S.; Liang, C. The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 2015, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Buffone, C.; Schulte, B.; Opp, S.; Diaz-Griffero, F. Contribution of MxB Oligomerization to HIV-1 Capsid Binding and Restriction. J. Virol. 2015, 89, 3285–3294. [Google Scholar] [CrossRef] [Green Version]
- Fribourgh, J.L.; Nguyen, H.; Matreyek, K.; Alvarez, F.; Summers, B.J.; Dewdney, T.G.; Aiken, C.; Zhang, P.; Engelman, A.; Xiong, Y. Structural Insight into HIV-1 Restriction by MxB. Cell Host Microbe 2014, 16, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Fricke, T.; E White, T.; Schulte, B.; De Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nuñez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Hou, G.; Zhang, H.; Suiter, C.L.; Ahn, J.; Byeon, I.-J.L.; Perilla, J.R.; Langmead, C.J.; Hung, I.; Gor’Kov, P.L.; et al. Dynamic allostery governs cyclophilin A–HIV capsid interplay. Proc. Natl. Acad. Sci. USA 2015, 112, 14617–14622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setiawan, L.C.; Kootstra, N.A. Adaptation of HIV-1 to rhTrim5α-mediated restriction in vitro. Virology 2015, 486, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, Z.; Lee, K.; Ndjomou, J.; Xu, H.; Oztop, I.; Matous, J.; Takemura, T.; Unutmaz, D.; Engelman, A.; Hughes, S.H.; et al. Human Immunodeficiency Virus Type 1 Capsid Mutation N74D Alters Cyclophilin A Dependence and Impairs Macrophage Infection. J. Virol. 2012, 86, 4708–4714. [Google Scholar] [CrossRef] [Green Version]
- Mascarenhas, A.P.; Musier-Forsyth, K. The capsid protein of human immunodeficiency virus: Interactions of HIV-1 capsid with host protein factors. FEBS J. 2009, 276, 6118–6127. [Google Scholar] [CrossRef] [PubMed]
- Vajdos, F.F.; Yoo, S.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci. 2008, 6, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Myszka, D.G.; Yeh, C.-Y.; McMurray, M.; Hill, C.P.; I Sundquist, W. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef] [Green Version]
- Colgan, J.; E Yuan, H.; Franke, E.K.; Luban, J. Binding of the human immunodeficiency virus type 1 Gag polyprotein to cyclophilin A is mediated by the central region of capsid and requires Gag dimerization. J. Virol. 1996, 70, 4299–4310. [Google Scholar] [CrossRef] [Green Version]
- Thali, M.; A Bukovsky, A.; Kondo, E.; Rosenwlrth, B.; Walsh, C.T.; Sodroski, J.; Göttlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef]
- Perrier, M.; Bertine, M.; Le Hingrat, Q.; Joly, V.; Visseaux, B.; Collin, G.; Landman, R.; Yazdanpanah, Y.; Descamps, D.; Charpentier, C. Prevalence of gag mutations associated with in vitro resistance to capsid inhibitor GS-CA1 in HIV-1 antiretroviral-naive patients. J. Antimicrob. Chemother. 2017, 72, 2954–2955. [Google Scholar] [CrossRef] [PubMed]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV Capsid is a Tractable Target for Small Molecule Therapeutic Intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Ferhadian, D.; Sowd, G.A.; Serrao, E.; Shi, J.; Halambage, U.D.; Teng, S.; Soto, J.; Siddiqui, M.A.; Engelman, A.N.; et al. Roles of Capsid-Interacting Host Factors in Multimodal Inhibition of HIV-1 by PF74. J. Virol. 2016, 90, 5808–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef] [Green Version]
- Dick, A.; Cocklin, S. Subtype Differences in the Interaction of HIV-1 Matrix with Calmodulin: Implications for Biological Functions. Biomolecules 2021, 11, 1294. [Google Scholar] [CrossRef]
- Gao, F.; Robertson, D.L.; Morrison, S.G.; Hui, H.; Craig, S.; Decker, J.; Fultz, P.N.; Girard, M.; Shaw, G.M.; Hahn, B.H.; et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. J. Virol. 1996, 70, 7013–7029. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Vidal, N.; Li, Y.; Trask, S.A.; Chen, Y.; Kostrikis, L.G.; Ho, D.D.; Kim, J.; Oh, M.-D.; Choe, K.; et al. Evidence of Two Distinct Subsubtypes within the HIV-1 Subtype A Radiation. AIDS Res. Hum. Retrovir. 2001, 17, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Morrison, S.G.; Robertson, D.L.; Thornton, C.L.; Craig, S.; Karlsson, G.; Sodroski, J.; Morgado, M.; Galvao-Castro, B.; von Briesen, H.; et al. Molecular cloning and analysis of functional envelope genes from human immunodeficiency virus type 1 sequence subtypes A through G. The WHO and NIAID Networks for HIV Isolation and Characterization. J. Virol. 1996, 70, 1651–1667. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Robertson, D.L.; Carruthers, C.D.; Morrison, S.G.; Jian, B.; Chen, Y.; Barré-Sinoussi, F.; Girard, M.; Srinivasan, A.; Abimiku, A.G.; et al. A Comprehensive Panel of Near-Full-Length Clones and Reference Sequences for Non-Subtype B Isolates of Human Immunodeficiency Virus Type 1. J. Virol. 1998, 72, 5680–5698. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [Green Version]
- Kortagere, S.; Madani, N.; Mankowski, M.K.; Schön, A.; Zentner, I.; Swaminathan, G.; Princiotto, A.; Anthony, K.; Oza, A.; Sierra, L.-J.; et al. Inhibiting Early-Stage Events in HIV-1 Replication by Small-Molecule Targeting of the HIV-1 Capsid. J. Virol. 2012, 86, 8472–8481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, I.; Hohenberg, H.; Wilk, T.; Wiegers, K.; Grättinger, M.; Müller, B.; Fuller, S.; Kräusslich, H.-G. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 2000, 19, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, I.; Hohenberg, H.; Krausslich, H.-G. In Vitro Assembly Properties of Purified Bacterially Expressed Capsid Proteins of Human Immunodeficiency Virus. Eur. J. Biochem. 1997, 249, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, L.S.; E Agresta, B.; A Carter, C. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 1992, 66, 4874–4883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Schwedler, U.K.; Stemmler, T.L.; Klishko, V.Y.; Li, S.; Albertine, K.H.; Davis, D.R.; Sundquist, W.I. Proteolytic refolding of the HIV-1 capsid protein amino-terminus facilitates viral core assembly. EMBO J. 1998, 17, 1555–1568. [Google Scholar] [CrossRef] [Green Version]
- Lanman, J.; Sexton, J.; Sakalian, M.; Prevelige, P.E., Jr. Kinetic Analysis of the Role of Intersubunit Interactions in Human Immunodeficiency Virus Type 1 Capsid Protein Assembly In Vitro. J. Virol. 2002, 76, 6900–6908. [Google Scholar] [CrossRef] [Green Version]
- Kortagere, S.; Xu, J.P.; Mankowski, M.K.; Ptak, R.G.; Cocklin, S. Structure–Activity Relationships of a Novel Capsid Targeted Inhibitor of HIV-1 Replication. J. Chem. Inf. Model. 2014, 54, 3080–3090. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Pornillos, O.; Ganser-Pornillos, B.K.; Banumathi, S.; Hua, Y.; Yeager, M. Disulfide Bond Stabilization of the Hexameric Capsomer of Human Immunodeficiency Virus. J. Mol. Biol. 2010, 401, 985–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sun, L.; Meuser, M.E.; Zalloum, W.A.; Xu, S.; Huang, T.; Cherukupalli, S.; Jiang, X.; Ding, X.; Tao, Y.; et al. Design, synthesis, and mechanism study of dimerized phenylalanine derivatives as novel HIV-1 capsid inhibitors. Eur. J. Med. Chem. 2021, 226, 113848. [Google Scholar] [CrossRef] [PubMed]
- Bravman, T.; Bronner, V.; Lavie, K.; Notcovich, A.; Papalia, G.A.; Myszka, D.G. Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor. Anal. Biochem. 2006, 358, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform. 2009, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Aportela, E.; Lopez-Blanco, J.R.; Chacon, P. FRODOCK 2.0: Fast protein—protein docking server. Bioinformatics 2016, 32, 2386–2388. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the Topology of Active Sites: On the Prediction of Pockets and Subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [Google Scholar] [CrossRef]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wu, C.; Freniere, C.; Tripler, T.N.; Xiong, Y. Nuclear Import of HIV-1. Viruses 2021, 13, 2242. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; He, M.; Tang, S.; Hewlett, I.; Tan, Z.; Li, J.; Jin, Y.; Yang, M. Synthesis and antiviral activities of novel acylhydrazone derivatives targeting HIV-1 capsid protein. Bioorg. Med. Chem. Lett. 2009, 19, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Loeliger, E.; Kinde, I.; Kyere, S.; Mayo, K.; Barklis, E.; Sun, Y.; Huang, M.; Summers, M.F. Antiviral Inhibition of the HIV-1 Capsid Protein. J. Mol. Biol. 2003, 327, 1013–1020. [Google Scholar] [CrossRef]
- Xu, J.P.; Francis, A.C.; E Meuser, M.; Mankowski, M.; Ptak, R.G.; A Rashad, A.; Melikyan, G.B.; Cocklin, S. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J. Drug Des. Res. 2018, 5. [Google Scholar]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.P.; Branson, J.D.; Lawrence, R.; Cocklin, S. Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer. Bioorg. Med. Chem. Lett. 2016, 26, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhou, J.; Halambage, U.D.; Shah, V.B.; Burse, M.J.; Wu, H.; Blair, W.S.; Butler, S.L.; Aiken, C. Compensatory Substitutions in the HIV-1 Capsid Reduce the Fitness Cost Associated with Resistance to a Capsid-Targeting Small-Molecule Inhibitor. J. Virol. 2015, 89, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Rankovic, S.; Ramalho, R.; Aiken, C.; Rousso, I. PF74 Reinforces the HIV-1 Capsid To Impair Reverse Transcription-Induced Uncoating. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Tuyishime, M.; Danish, M.; Princiotto, A.; Mankowski, M.K.; Lawrence, R.; Lombart, H.-G.; Esikov, K.; Berniac, J.; Liang, K.; Ji, J.; et al. Discovery and optimization of novel small-molecule HIV-1 entry inhibitors using field-based virtual screening and bioisosteric replacement. Bioorg. Med. Chem. Lett. 2014, 24, 5439–5445. [Google Scholar] [CrossRef] [Green Version]
- Tuyishime, M.; Lawrence, R.; Cocklin, S. Core chemotype diversification in the HIV-1 entry inhibitor class using field-based bioisosteric replacement. Bioorg. Med. Chem. Lett. 2015, 26, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuser, M.E.; Rashad, A.A.; Ozorowski, G.; Dick, A.; Ward, A.B.; Cocklin, S. Field-Based Affinity Optimization of a Novel Azabicyclohexane Scaffold HIV-1 Entry Inhibitor. Molecules 2019, 24, 1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 Clade | Initial Velocity Apo (AU/min) | r2 | Initial Velocity PF-74 (AU/min) | r2 |
|---|---|---|---|---|
| A1 (92UG037.1) | 0.022 ± 0.003 | 0.966 | 0.034 ± 0.001 | 0.996 |
| A2 (CDKTB48) | 0.013 ± 0.000 | 0.990 | 0.017 ± 0.002 | 0.983 |
| B (NL4-3) | 0.016 ± 0.003 | 0.945 | 0.043 ± 0.009 | 0.925 |
| C (92BR025.8) | 0.011 ± 0.000 | 0.929 | 0.039 ± 0.007 | 0.930 |
| D (94UG114.1) | 0.015 ± 0.002 | 0.954 | 0.050 ± 0.008 | 0.955 |
| HIV-1 Clade | KD [nM] Equilibrium | ±SD | kon [M−1s−1] | ±SD | koff [s−1] | ±SD | KD [nM] Kinetic/Global | ±SD |
|---|---|---|---|---|---|---|---|---|
| A1 | 716.3 | 66.1 | 6.15 × 105 | 1.80 × 105 | 6.47 × 10−1 | 2.82 × 10−1 | 946.5 | 313.5 |
| A2 | 138.0 | 16.5 | 6.69 × 105 | 1.78 × 105 | 9.16 × 10−2 | 1.42 × 10−2 | 139.3 | 14.6 |
| B (NL4.3) | 47.0 | 0.6 | 7.56 × 105 | 4.55 × 104 | 3.63 × 10−2 | 6.92 × 10−3 | 47.9 | 1.2 |
| C | 353.7 | 10.0 | 2.28 × 105 | 2.20 × 104 | 7.54 × 10−2 | 6.92 × 10−3 | 330.0 | 14.7 |
| D | 51.0 | 1.3 | 7.73 × 105 | 4.01 × 104 | 3.95 × 10−2 | 5.25 × 10−3 | 51.0 | 4.1 |
| HIV-1 Clade | KD [μM] Equilibrium | ±SD | kon [M−1s−1] | ±SD | koff [s−1] | ±SD | KD [μM] Kinetic/Global | ±SD |
|---|---|---|---|---|---|---|---|---|
| A1 | 181.0 | 55.9 | 1.85 × 103 | 9.29 × 101 | 4.77 × 10−1 | 3.27 × 10−2 | 257.3 | 7.8 |
| A2 | 197.0 | 35.4 | 3.68 × 103 | 1.33 × 103 | 7.46 × 10−1 | 1.73 × 10−1 | 209.7 | 28.2 |
| B (NL4.3) | 93.8 | 3.3 | 5.96 × 103 | 1.28 × 103 | 5.89 × 10−1 | 1.28 × 10−1 | 98.6 | 4.7 |
| C | 104.3 | 3.8 | 5.29 × 103 | 1.21 × 103 | 5.70 × 10−1 | 1.28 × 10−1 | 107.7 | 1.5 |
| D | 70.0 | 0.7 | 7.06 × 103 | 1.14 × 103 | 5.13 × 10−1 | 8.05 × 10−2 | 72.8 | 0.9 |
| HIV-1 Clade | KD [μM] Equilibrium | ±SD | kon [M−1s−1] | ±SD | koff [s−1] | ±SD | KD [μM] Kinetic/Global | ±SD |
|---|---|---|---|---|---|---|---|---|
| A1 | N.B. | N.B. | N.B. | N.B. | N.B. | N.B. | N.B. | N.B. |
| A2 | 443.3 | 38.1 | 130 × 103 | 3.75 × 102 | 9.60 × 10−1 | 2.13 × 10−1 | 746.3 | 48.5 |
| B (NL4.3) | 380.7 | 34.6 | 1.57 × 103 | 3.60 × 102 | 7.63 × 10−1 | 7.35 × 10−2 | 499.3 | 94.1 |
| C | 503.0 | 46.9 | 9.33 × 102 | 4.31 × 102 | 7.47 × 10−1 | 7.35 × 10−2 | 881.0 | 267.2 |
| D | 308.7 | 42.8 | 1.89 × 103 | 2.91 × 102 | 6.86 × 10−1 | 1.35 × 10−1 | 367.7 | 83.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dick, A.; Meuser, M.E.; Cocklin, S. Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation. Biomolecules 2022, 12, 695. https://doi.org/10.3390/biom12050695
Dick A, Meuser ME, Cocklin S. Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation. Biomolecules. 2022; 12(5):695. https://doi.org/10.3390/biom12050695
Chicago/Turabian StyleDick, Alexej, Megan E. Meuser, and Simon Cocklin. 2022. "Clade-Specific Alterations within the HIV-1 Capsid Protein with Implications for Nuclear Translocation" Biomolecules 12, no. 5: 695. https://doi.org/10.3390/biom12050695