
Did Homocysteine Take Part in the Start of the Synthesis of Peptides on the Early Earth?



Abstract
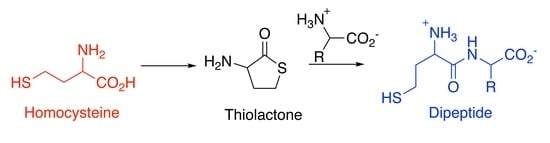
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
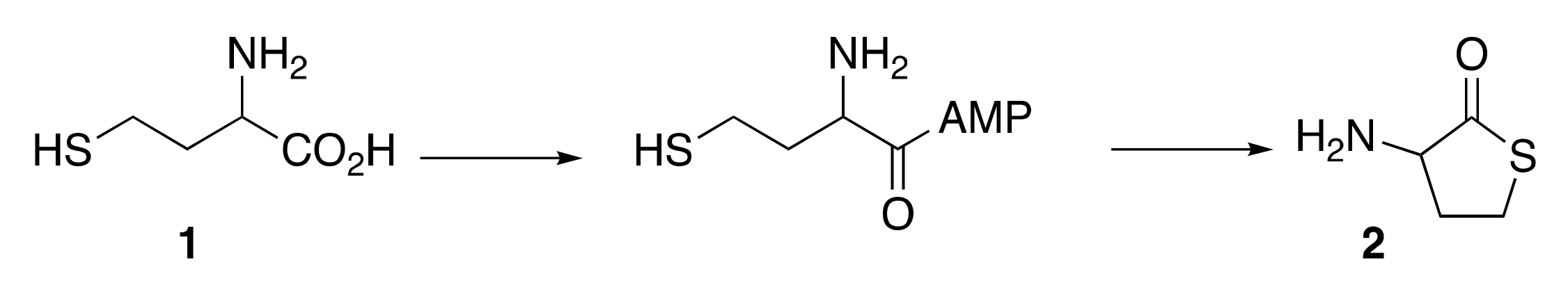{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
3.1. Cyclisation of Homocysteine: Experimental Results
3.2. Formation of Dipeptides from Homocysteine Thiolactone and Its N-formyl and N-acetyl Derivatives
4. Discussion
4.1. Cyclisation of Homocysteine: Theoretical Calculations
4.2. Hypothesis: Hcy Was the First StartResidue
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krishnamurthy, R. Giving Rise to Life: Transition from Prebiotic Chemistry to Protobiology. Acc. Chem. Res. 2017, 50, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Zahnle, K.; Schaefer, L.; Fegley, B. Earth’s Earliest Atmospheres. Cold Spring Harb. Perspect. Biol. 2010, 2, a004895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muchowska, K.B.; Varma, S.J.; Moran, J. Nonenzymatic Metabolic Reactions and Life’s Origins. Chem. Rev. 2020, 120, 7708–7744. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.D. The Origin of Life—Out of the Blue. Angew. Chem. Int. Ed. 2016, 55, 104–121. [Google Scholar] [CrossRef]
- Marchi, S.; Bottke, W.F.; Elkins-Tanton, L.T.; Bierhaus, M.; Wuennemann, K.; Morbidelli, A.; Kring, D.A. Widespread mixing and burial of Earth’s Hadean Crust by asteroid impacts. Nature 2014, 511, 578–582. [Google Scholar] [CrossRef]
- Youssef-Saliba, S.; Vallée, Y. Sulfur Amino Acids: From Prebiotic Chemistry to Biology and Vice Versa. Synthesis 2021, 53, 2798–2808. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Editing, Thioester Chemistry, Coenzyme A, and the Origin of Coded Peptide Synthesis. Life 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of Homocysteine Toxicity in Humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef]
- Parker, E.T.; Cleaves, H.J.; Callahan, M.P.; Dworkin, J.P.; Glavin, D.P.; Lazcano, A.; Bada, J.L. Prebiotic Synthesis of Methionine and Other Sulfur-Containing Organic Compounds on the Primitive Earth: A Contemporary Reassessment Based on an Unpublished 1958 Stanley Miller Experiment. Orig. Life Evol. Biosph. 2011, 41, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Shalayel, I.; Vallée, Y. Chemistry of Homocysteine Thiolactone in A Prebiotic Perspective. Life 2019, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Deepak, R.N.V.K.; Chandrakar, B.; Sankararamakrishnan, R. Comparison of Metal Binding Strength between Methionine and Cysteine Residues: Implications for the Design of Metal-Binding Motifs in Proteins. Biophysic. Chem. 2017, 224, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P. Proteome-pl: Proteome Isoelectric Point Database. Nucleic Acids Res. 2017, 45, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N. The Triplet Code from First Principles. J. Biomol. Struct. Dyn. 2004, 22, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Blattner, F.R.; Plunkett, G., III; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belinky, F.; Rogozin, I.B.; Koonin, E.V. Selection on Start Codons in Prokaryotes and Potential Compensatory Nucleotide Substitutions. Sci. Rep. 2017, 7, 1242. [Google Scholar] [CrossRef] [Green Version]
- De Duve, C. A Research Proposal on the Origin of Life. Orig. Life Evol. Biosph. 2003, 33, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Risse, C. Homocysteinthiolacton-Abkömmlinge und Verfahren zu deren Herstellung. German Patent DE1972-2213028, 21 November 1985. [Google Scholar]
- Abelson, P.H. Chemical Events on the Primitive Earth. Proc. Natl. Acad. Sci. USA 1966, 55, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Mendham, A.P.; Spencer, J.; Chowdhry, B.Z.; Dines, T.J.; Mujahid, M.; Palmer, R.A.; Tizzard, G.J.; Coles, S.J. X-Ray Crystallographic Structure and Absolute Configuration of the Cyclic Di-amino Acid Peptide: Cyclo(L-HomoCySH-L-HomoCySH). J. Chem. Crystallogr. 2011, 41, 1328–1334. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solavation Model Based on Solute Electron Density and on Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tension. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A Post-Hartree-Fock Model of Intermolecular Interactions: Inclusion of Higher-Order Corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [Green Version]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An Electronic Structure and Molecular Dynamics Software Package—Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- Pinti, D.L. The Origin and Evolution of the Oceans. Lect. Astrobiol. 2005, 1, 83–112. [Google Scholar] [CrossRef]
- Chwatko, G.; Jakubowsky, H. The Determination of Homocysteine-thiolactone in human plasma. Anal. Biochem. 2005, 337, 271–277. [Google Scholar] [CrossRef]
- Velázquez-Ríos, I.O.; Rincón-Rosales, R.; Gutiérrez-Miceli, F.A.; Alcántara-Hernández, R.J.; Ruíz-Valdiviezo, V.E. Prokariotic diversity across a pH gradient in the « El Chichón » crater-lake: A naturally thermo-acidic environment. Extremophiles 2022, 26, 8. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.B.; Nesbitt, R.W. Trace element distributions in the chalcopyrite wall of a black smoker chimney: Insights from laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). Earth Planet. Sci. Lett. 1999, 167, 335–345. [Google Scholar] [CrossRef]
- Keil, L.M.R.; Möller, F.M.; Kieß, M.; Kudella, P.W.; Mast, C.B. Proton gradient and pH oscillations emerge from heat flow at the microscale. Nat. Commun. 2017, 8, 1897. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.; de Graaf, R.; Strandoo Rodin, M.; Ohno, A.; Lane, N.; McGlynn, S.E.; Yamada, Y.M.A.; Nakamura, R.; Barge, L.M.; Braun, D.; et al. CO2 reduction driven by a pH gradient. Proc. Natl. Acad. Sci. USA 2020, 117, 22873–22879. [Google Scholar] [CrossRef]
- Lane, N. Proton gradients at the origin of life. Bioessays 2017, 39, 1600217. [Google Scholar] [CrossRef] [Green Version]
- Holm, N.G.; Hennet, R.J.C. Hydrothermal systems: Their varieties, dynamics, and suitability for prebiotic chemistry. Orig. Life Evol. Biosph. 1992, 22, 15–31. [Google Scholar] [CrossRef]
- Foden, S.; Islam, C.; Fernández-Garcia, C.; Maugeri, L.; Sheppard, T.D.; Powner, M.W. Prebiotic synthesis of cysteine peptides that catalyze peptide ligation in neutral water. Science 2020, 370, 865–869. [Google Scholar] [CrossRef]
- Canavelli, P.; Islam, S.; Powner, M.W. Peptide ligation by chemoselective aminonitrile coupling in water. Nature 2019, 571, 546–549. [Google Scholar] [CrossRef]
- Sauer, F.; Haas, M.; Sydow, C.; Siegle, A.F.; Lauer, C.A.; Trapp, O. From amino acid mixtures to peptides in liquid sulphur dioxide on early Earth. Nat. Commun. 2021, 12, 7182. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine modification in protein structure/function and in human disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Fucks, E.; Huber, L.; Schinkel, T.; Trapp, O. Investigation of straightforward, photoinduced alkylations of electron-rich heterocompounds with electron-deficient alkyl bromides in the sole presence of 2,6-lutidine. Eur. J. Org. Chem. 2020, 2020, 6192–6198. [Google Scholar] [CrossRef]
- Closs, C.; Bechtel, M.; Trapp, O. Dynamic exchange of substituents in a prebiotic organocatalyst: Initial steps towards an evolutionary system. Angew. Chem. Int. Ed. 2022, 61, e202112563. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. Cp2k: Atomistic Simulations of Condensed Matter Systems. WIREs Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Van de Vondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef-Saliba, S.; Milet, A.; Vallée, Y. Did Homocysteine Take Part in the Start of the Synthesis of Peptides on the Early Earth? Biomolecules 2022, 12, 555. https://doi.org/10.3390/biom12040555
Youssef-Saliba S, Milet A, Vallée Y. Did Homocysteine Take Part in the Start of the Synthesis of Peptides on the Early Earth? Biomolecules. 2022; 12(4):555. https://doi.org/10.3390/biom12040555
Chicago/Turabian StyleYoussef-Saliba, Sparta, Anne Milet, and Yannick Vallée. 2022. "Did Homocysteine Take Part in the Start of the Synthesis of Peptides on the Early Earth?" Biomolecules 12, no. 4: 555. https://doi.org/10.3390/biom12040555