
Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
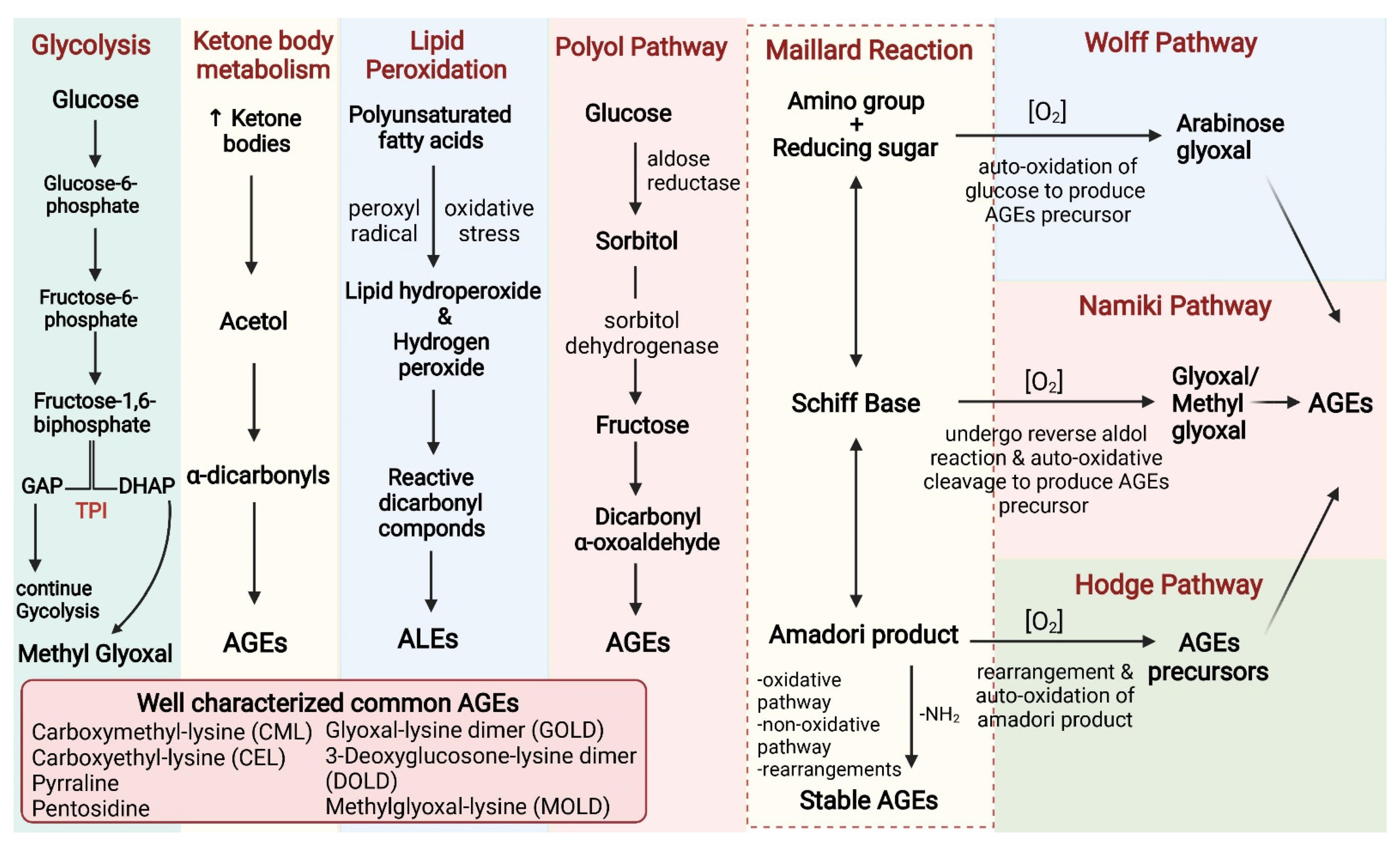
2. Sources of AGEs
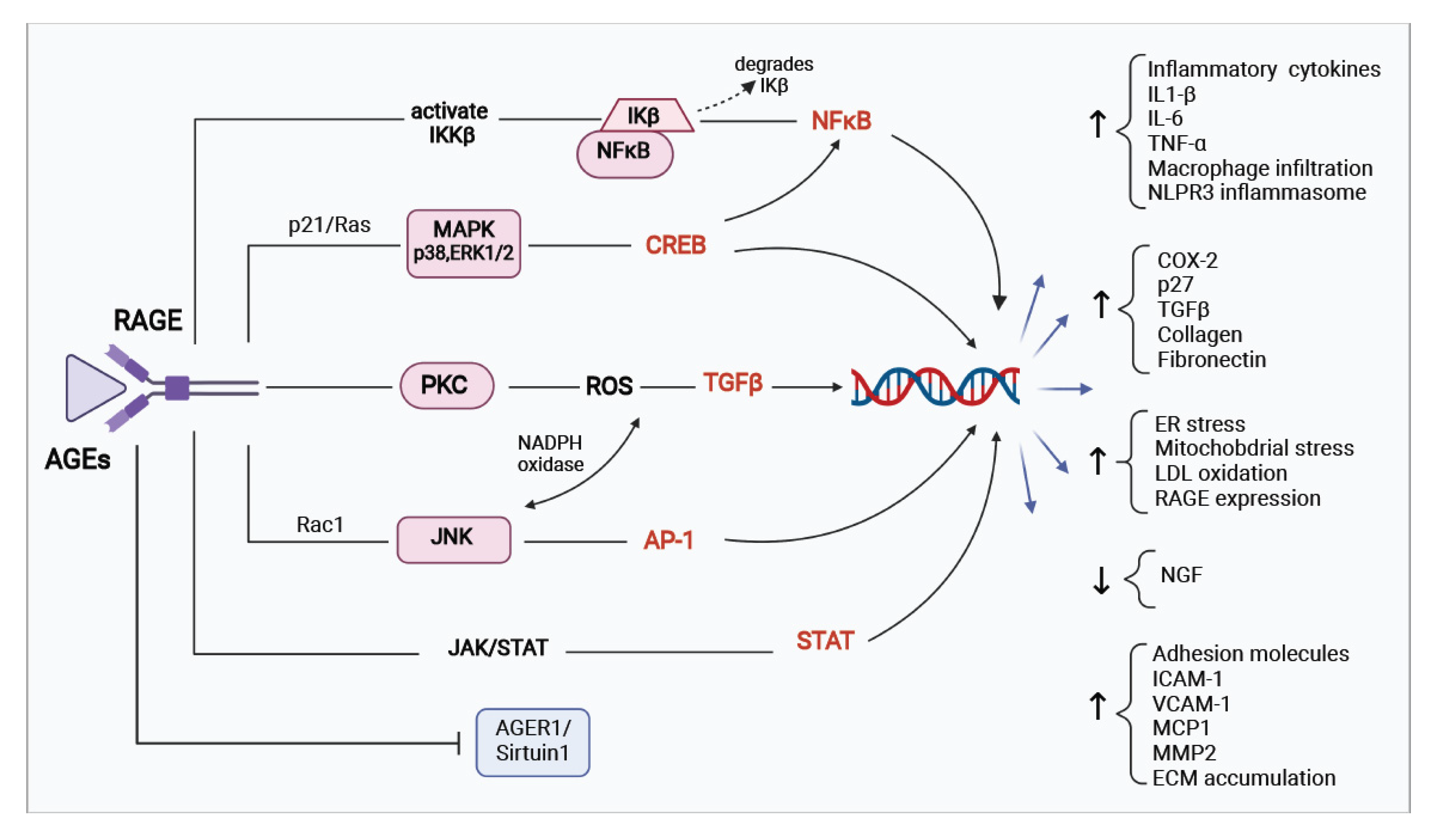
3. Pathophysiology of AGEs/RAGE in Diabetes Mellitus
4. AGEs/RAGE Axis and Pancreatic Beta Cells
5. AGEs/RAGE Axis in Diabetic Complications
6. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced Glycation End Products and Oxidative Stress in Type 2 Diabetes Mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [Green Version]
- Morrish, N.; Wang, S.-L.; Stevens, L.; Fuller, J.; Keen, H. Mortality and Causes of Death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 2001, 44, S14–S21. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.B.; Florez, J.C. Genetics of Diabetes Mellitus and Diabetes Complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Mohammedi, K.; Woodward, M.; Marre, M.; Colagiuri, S.; Cooper, M.; Harrap, S.; Mancia, G.; Poulter, N.; Williams, B.; Zoungas, S. Comparative Effects of Microvascular and Macrovascular Disease on the Risk of Major Outcomes in Patients with Type 2 Diabetes. Cardiovasc. Diabetol. 2017, 16, 95. [Google Scholar] [CrossRef]
- Peppa, M.; Uribarri, J.; Vlassara, H. Glucose, Advanced Glycation End Products, and Diabetes Complications: What Is New and What Works. Clin. Diabetes 2003, 21, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Burillo, S.; Rufián-Henares, J.Á.; Pastoriza, S. Effect of Home Cooking on the Antioxidant Capacity of Vegetables: Relationship with Maillard Reaction Indicators. Food Res. Int. 2019, 121, 514–523. [Google Scholar] [CrossRef]
- Reddy, V.P.; Beyaz, A. Inhibitors of the Maillard Reaction and AGE Breakers as Therapeutics for Multiple Diseases. Drug Discov. Today 2006, 11, 646–654. [Google Scholar] [CrossRef]
- Van Nguyen, C. Toxicity of the AGEs Generated from the Maillard Reaction: On the Relationship of Food-AGEs and Biological-AGEs. Mol. Nutr. Food Res. 2006, 50, 1140–1149. [Google Scholar] [CrossRef]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The Role of Advanced Glycation End-Products in the Development of Coronary Artery Disease in Patients with and without Diabetes Mellitus: A Review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef]
- Nin, J.W.; Jorsal, A.; Ferreira, I.; Schalkwijk, C.G.; Prins, M.H.; Parving, H.-H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D. Higher Plasma Levels of Advanced Glycation End Products Are Associated with Incident Cardiovascular Disease and All-Cause Mortality in Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes Care 2011, 34, 442–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlassara, H.; Striker, G.E. AGE Restriction in Diabetes Mellitus: A Paradigm Shift. Nat. Rev. Endocrinol. 2011, 7, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N. Advanced Glycation Endproducts—Role in Pathology of Diabetic Complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Peppa, M.; Vlassara, H. Advanced Glycation End Products and Diabetic Complications: A General Overview. Hormones 2005, 4, 28–37. [Google Scholar] [CrossRef]
- Wada, R.; Yagihashi, S. Role of Advanced Glycation End Products and Their Receptors in Development of Diabetic Neuropathy. Ann. N. Y. Acad. Sci. 2005, 1043, 598–604. [Google Scholar] [CrossRef]
- Indyk, D.; Bronowicka-Szydełko, A.; Gamian, A.; Kuzan, A. Advanced Glycation End Products and Their Receptors in Serum of Patients with Type 2 Diabetes. Sci. Rep. 2021, 11, 13264. [Google Scholar] [CrossRef]
- Goh, S.-Y.; Cooper, M.E. The Role of Advanced Glycation End Products in Progression and Complications of Diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced Glycation End-Products: A Review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [Green Version]
- Yim, M.B.; Yim, H.; Lee, C.; Kang, S.; Chock, P.B. Protein Glycation: Creation of Catalytic Sites for Free Radical Generation. Ann. N. Y. Acad. Sci. 2001, 928, 48–53. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Asadipooya, K.; Uy, E.M. Advanced Glycation End Products (AGEs), Receptor for AGEs, Diabetes, and Bone: Review of the Literature. J. Endocr. Soc. 2019, 3, 1799–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garay-Sevilla, M.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. [Google Scholar] [CrossRef] [PubMed]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J. Am. Diet. Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Sergi, D.; Boulestin, H.; Campbell, F.M.; Williams, L.M. The Role of Dietary Advanced Glycation End Products in Metabolic Dysfunction. Mol. Nutr. Food Res. 2021, 65, 1900934. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, X.; Li, L.; Li, B.; Yang, Z. The Fate of Dietary Advanced Glycation End Products in the Body: From Oral Intake to Excretion. Crit. Rev. Food Sci. Nutr. 2020, 60, 3475–3491. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Beeri, M.; De la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The Potential Role of Dietary Advanced Glycation Endproducts in the Development of Chronic Non-Infectious Diseases: A Narrative Review. Nutr. Res. Rev. 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Mastrocola, R.; Collotta, D.; Gaudioso, G.; Le Berre, M.; Cento, A.S.; Ferreira Alves, G.; Chiazza, F.; Verta, R.; Bertocchi, I.; Manig, F. Effects of Exogenous Dietary Advanced Glycation End Products on the Cross-Talk Mechanisms Linking Microbiota to Metabolic Inflammation. Nutrients 2020, 12, 2497. [Google Scholar] [CrossRef]
- Gill, V.; Kumar, V.; Singh, K.; Kumar, A.; Kim, J.-J. Advanced Glycation End Products (AGEs) May Be a Striking Link between Modern Diet and Health. Biomolecules 2019, 9, 888. [Google Scholar] [CrossRef] [Green Version]
- Lund, J.; Ouwens, D.M.; Wettergreen, M.; Bakke, S.S.; Thoresen, G.H.; Aas, V. Increased Glycolysis and Higher Lactate Production in Hyperglycemic Myotubes. Cells 2019, 8, 1101. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Mustafiz, A.; Singla-Pareek, S.L.; Shankar Srivastava, P.; Sopory, S.K. Characterization of Stress and Methylglyoxal Inducible Triose Phosphate Isomerase (OscTPI) from Rice. Plant Signal. Behav. 2012, 7, 1337–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipkiss, A. Activity-Induced Deamidation of Triose-Phosphate Isomerase May Explain the Deleterious Effects of Excessive Glucose Consumption. Int. J. Diabetes Clin. Res. 2016, 3, 066. [Google Scholar] [CrossRef]
- Jakuš, V.; Rietbrock, N. Advanced Glycation End-Products and the Progress of Diabetic Vascular Complications. Physiol. Res. 2004, 53, 131–142. [Google Scholar] [PubMed]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of Polyol Pathway to Diabetes-Induced Oxidative Stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, Y.; Araki, N.; Koh, N.; Nakamura, J.; Horiuchi, S.; Hotta, N. Rapid Formation of Advanced Glycation End Products by Intermediate Metabolites of Glycolytic Pathway and Polyol Pathway. Biochem. Biophys. Res. Commun. 1996, 228, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Kuzan, A. Toxicity of Advanced Glycation End Products. Biomed. Rep. 2021, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the Receptor for Advanced Glycation End Products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The Receptor for Advanced Glycation End-Products (RAGE) Is an Important Pattern Recognition Receptor (PRR) for Inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for Advanced Glycation End Products and Its Involvement in Inflammatory Diseases. Int. J. Inflamm. 2013, 2013, 403460. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Du Yan, S.; Yan, S.F.; Stern, D.M. The Biology of the Receptor for Advanced Glycation End Products and Its Ligands. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Prevost, G.; Fajardy, I.; Besmond, C.; Balkau, B.; Tichet, J.; Fontaine, P.; Danze, P.; Marre, M. Polymorphisms of the Receptor of Advanced Glycation Endproducts (RAGE) and the Development of Nephropathy in Type 1 Diabetic Patients. Diabetes Metab. 2005, 31, 35–39. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X.; Tuo, M.; Ma, J.; Xie, A. RAGE and Its Emerging Role in the Pathogenesis of Parkinson’s Disease. Neurosci. Lett. 2018, 672, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, Classification, and Expression of RAGE Gene Splice Variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Keller, J.N. Evaluation of Rage Isoforms, Ligands, and Signaling in the Brain. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2005, 1746, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterenczak, K.A.; Nolte, I.; Escobar, H.M. RAGE Splicing Variants in Mammals. Calcium-Bind. Proteins RAGE 2013, 963, 265–276. [Google Scholar] [CrossRef]
- Scavello, F.; Zeni, F.; Tedesco, C.C.; Mensà, E.; Veglia, F.; Procopio, A.D.; Bonfigli, A.R.; Olivieri, F.; Raucci, A. Modulation of Soluble Receptor for Advanced Glycation End-Products (RAGE) Isoforms and Their Ligands in Healthy Aging. Aging 2019, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Bopp, C.; Bierhaus, A.; Hofer, S.; Bouchon, A.; Nawroth, P.P.; Martin, E.; Weigand, M.A. Bench-to-Bedside Review: The Inflammation-Perpetuating Pattern-Recognition Receptor RAGE as a Therapeutic Target in Sepsis. Crit. Care 2008, 12, 201. [Google Scholar] [CrossRef] [Green Version]
- Stern, D.; Du Yan, S.; Yan, S.F.; Schmidt, A.M. Receptor for Advanced Glycation Endproducts: A Multiligand Receptor Magnifying Cell Stress in Diverse Pathologic Settings. Adv. Drug Deliv. Rev. 2002, 54, 1615–1625. [Google Scholar] [CrossRef]
- Gąsiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Naka, Y.; Schmidt, A.M. Glycation, Inflammation, and RAGE: A Scaffold for the Macrovascular Complications of Diabetes and Beyond. Circ. Res. 2003, 93, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, J.P.; McKinley, B.; Eckel, R.H. The Metabolic Syndrome and Inflammation. Metab. Syndr. Relat. Disord. 2004, 2, 82–104. [Google Scholar] [CrossRef] [PubMed]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein Kinases: Mechanisms and Downstream Targets in Inflammation-Mediated Obesity and Insulin Resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidarala, V.; Kowluru, A. The Regulatory Roles of Mitogen-Activated Protein Kinase (MAPK) Pathways in Health and Diabetes: Lessons Learned from the Pancreatic β-Cell. Recent Pat. Endocr. Metab. Immune Drug Discov. 2016, 10, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Liu, M.; Ji, B.; Bai, B.; Cheng, B.; Wang, C. Role of the Extracellular Signal-Regulated Kinase 1/2 Signaling Pathway in Ischemia-Reperfusion Injury. Front. Physiol. 2019, 10, 1038. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, K.; Awazu, M.; Tamiya, M.; Iwasaki, Y.; Harada, A.; Kugisaki, S.; Tanimura, S.; Kohno, M. Targeting the ERK Signaling Pathway as a Potential Treatment for Insulin Resistance and Type 2 Diabetes. Am. J. Physiol.-Endocrinol. Metab. 2016, 310, E643–E651. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, Z.; Wang, Y.; Tweardy, D.J.; Mitch, W.E. Stat3 Activation Induces Insulin Resistance via a Muscle-Specific E3 Ubiquitin Ligase Fbxo40. Am. J. Physiol.-Endocrinol. Metab. 2020, 318, E625–E635. [Google Scholar] [CrossRef]
- Gabryelska, A.; Karuga, F.F.; Szmyd, B.; Białasiewicz, P. HIF-1α as a Mediator of Insulin Resistance, T2DM, and Its Complications: Potential Links with Obstructive Sleep Apnea. Front. Physiol. 2020, 11, 1035. [Google Scholar] [CrossRef]
- Kihira, Y.; Miyake, M.; Hirata, M.; Hoshina, Y.; Kato, K.; Shirakawa, H.; Sakaue, H.; Yamano, N.; Izawa-Ishizawa, Y.; Ishizawa, K. Deletion of Hypoxia-Inducible Factor-1α in Adipocytes Enhances Glucagon-like Peptide-1 Secretion and Reduces Adipose Tissue Inflammation. PLoS ONE 2014, 9, e93856. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The Receptor RAGE: Bridging Inflammation and Cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Tao, M.; Zhao, Y.; Hu, X.; Wang, M. 4′-Methoxyresveratrol Alleviated AGE-Induced Inflammation via RAGE-Mediated NF-ΚB and NLRP3 Inflammasome Pathway. Molecules 2018, 23, 1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheinheimer, J.; de Souza, B.M.; Cardoso, N.S.; Bauer, A.C.; Crispim, D. Current Role of the NLRP3 Inflammasome on Obesity and Insulin Resistance: A Systematic Review. Metabolism 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N. Monounsaturated Fatty Acid–Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome–Mediated IL-1β Secretion and Insulin Resistance despite Obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Fritz, G. RAGE Regulation and Signaling in Inflammation and Beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef]
- Furuya, D.T.; Neri, E.A.; Poletto, A.C.; Anhê, G.F.; Freitas, H.S.; Campello, R.S.; Rebouças, N.A.; Machado, U.F. Identification of Nuclear Factor-ΚB Sites in the Slc2a4 Gene Promoter. Mol. Cell. Endocrinol. 2013, 370, 87–95. [Google Scholar] [CrossRef]
- Khalid, M.; Alkaabi, J.; Khan, M.A.; Adem, A. Insulin Signal Transduction Perturbations in Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 8590. [Google Scholar] [CrossRef]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M. Advanced Glycation End Products-Induced Insulin Resistance Involves Repression of Skeletal Muscle GLUT4 Expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Guerrero-Hernández, A.; Leon-Aparicio, D.; Chavez-Reyes, J.; Olivares-Reyes, J.A.; DeJesus, S. Endoplasmic Reticulum Stress in Insulin Resistance and Diabetes. Cell Calcium 2014, 56, 311–322. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.D.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced Glycation End Products and RAGE: A Common Thread in Aging, Diabetes, Neurodegeneration, and Inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef]
- Hurrle, S.; Hsu, W.H. The Etiology of Oxidative Stress in Insulin Resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, H.J.; Jung, M.H.; Song, J. Coupling Mitochondrial Dysfunction to Endoplasmic Reticulum Stress Response: A Molecular Mechanism Leading to Hepatic Insulin Resistance. Cell. Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaster, M.; Staehr, P.; Beck-Nielsen, H.; Schrøder, H.D.; Handberg, A. GLUT4 Is Reduced in Slow Muscle Fibers of Type 2 Diabetic Patients: Is Insulin Resistance in Type 2 Diabetes a Slow, Type 1 Fiber Disease? Diabetes 2001, 50, 1324–1329. [Google Scholar] [CrossRef] [Green Version]
- Le Bagge, S.; Fotheringham, A.K.; Leung, S.S.; Forbes, J.M. Targeting the Receptor for Advanced Glycation End Products (RAGE) in Type 1 Diabetes. Med. Res. Rev. 2020, 40, 1200–1219. [Google Scholar] [CrossRef]
- Guan, S.-S.; Sheu, M.-L.; Yang, R.-S.; Chan, D.-C.; Wu, C.-T.; Yang, T.-H.; Chiang, C.-K.; Liu, S.-H. The Pathological Role of Advanced Glycation End Products-Downregulated Heat Shock Protein 60 in Islet β-Cell Hypertrophy and Dysfunction. Oncotarget 2016, 7, 23072. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Shu, T.; Lin, Y.; Wang, H.; Yang, J.; Shi, Y.; Han, X. Inhibition of the Receptor for Advanced Glycation Endproducts (RAGE) Protects Pancreatic β-Cells. Biochem. Biophys. Res. Commun. 2011, 404, 159–165. [Google Scholar] [CrossRef]
- Raleigh, D.; Zhang, X.; Hastoy, B.; Clark, A. The β-Cell Assassin: IAPP Cytotoxicity. J. Mol. Endocrinol. 2017, 59, R121–R140. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F. RAGE Binds Preamyloid IAPP Intermediates and Mediates Pancreatic β Cell Proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef] [Green Version]
- Abedini, A.; Derk, J.; Schmidt, A.M. The Receptor for Advanced Glycation Endproducts Is a Mediator of Toxicity by IAPP and Other Proteotoxic Aggregates: Establishing and Exploiting Common Ground for Novel Amyloidosis Therapies. Protein Sci. 2018, 27, 1166–1180. [Google Scholar] [CrossRef] [Green Version]
- Bram, Y.; Frydman-Marom, A.; Yanai, I.; Gilead, S.; Shaltiel-Karyo, R.; Amdursky, N.; Gazit, E. Apoptosis Induced by Islet Amyloid Polypeptide Soluble Oligomers Is Neutralized by Diabetes-Associated Specific Antibodies. Sci. Rep. 2014, 4, 4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T. β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition. Am. J. Pathol. 2011, 178, 2632–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceriello, A. The Emerging Challenge in Diabetes: The “Metabolic Memory”. Vasc. Pharmacol. 2012, 57, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Fukami, K.; Matsui, T. Crosstalk between Advanced Glycation End Products (AGEs)-Receptor RAGE Axis and Dipeptidyl Peptidase-4-Incretin System in Diabetic Vascular Complications. Cardiovasc. Diabetol. 2015, 14, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Wu, Y. Metabolic Memory: Mechanisms and Implications for Diabetic Vasculopathies. Sci. China Life Sci. 2014, 57, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, B.; Tang, L. Metabolic Memory: Mechanisms and Implications for Diabetic Retinopathy. Diabetes Res. Clin. Pract. 2012, 96, 286–293. [Google Scholar] [CrossRef]
- Papatheodorou, K.; Papanas, N.; Banach, M.; Papazoglou, D.; Edmonds, M. Complications of Diabetes 2016. J. Diabetes Res. 2016, 2016, 6989453. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. Unlocking the Biology of RAGE in Diabetic Microvascular Complications. Trends Endocrinol. Metab. 2014, 25, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Dal Canto, E.; Ceriello, A.; Rydén, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W. Diabetes as a Cardiovascular Risk Factor: An Overview of Global Trends of Macro and Micro Vascular Complications. Eur. J. Prev. Cardiol. 2019, 26, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Pop-Busui, R.; Pietropaolo, M. Metabolic Syndrome and Inflammation. In Immunoendocrinology: Scientific and Clinical Aspects; Humana Press: Totowa, NJ, USA, 2011; pp. 69–92. [Google Scholar]
- Glovaci, D.; Fan, W.; Wong, N.D. Epidemiology of Diabetes Mellitus and Cardiovascular Disease. Curr. Cardiol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-KB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2014, 13, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Yamagishi, S.; Okuda, S. Role of AGEs-RAGE System in Cardiovascular Disease. Curr. Pharm. Des. 2014, 20, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE Axis: A Fundamental Mechanism Signaling Danger to the Vulnerable Vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betteridge, D.J. What Is Oxidative Stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhao, Z.; Cai, Z.; Dong, N.; Liu, Y. Oxidized Low-Density Lipoprotein Promotes Osteoblastic Differentiation of Valvular Interstitial Cells through RAGE/MAPK. Cardiology 2015, 130, 55–61. [Google Scholar] [CrossRef]
- Sun, L.; Ishida, T.; Yasuda, T.; Kojima, Y.; Honjo, T.; Yamamoto, Y.; Yamamoto, H.; Ishibashi, S.; Hirata, K.; Hayashi, Y. RAGE Mediates Oxidized LDL-Induced pro-Inflammatory Effects and Atherosclerosis in Non-Diabetic LDL Receptor-Deficient Mice. Cardiovasc. Res. 2009, 82, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Matsui, T. Role of Ligands of Receptor for Advanced Glycation End Products (RAGE) in Peripheral Artery Disease. Rejuvenation Res. 2018, 21, 456–463. [Google Scholar] [CrossRef]
- Hyun, S.P.S.-J.Y.; Shim, J.T.C.Y. RAGE and Cardiovascular Disease. Front. Biosci. 2011, 16, 486–497. [Google Scholar]
- Sanajou, D.; Haghjo, A.G.; Argani, H.; Aslani, S. AGE-RAGE Axis Blockade in Diabetic Nephropathy: Current Status and Future Directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Yoo, H.-J.; Kim, H.; Lee, K.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Choi, D.; Baik, S.-H. Association between Endogenous Secretory RAGE, Inflammatory Markers and Arterial Stiffness. Int. J. Cardiol. 2009, 132, 96–101. [Google Scholar] [CrossRef] [PubMed]
- McNulty, M.; Mahmud, A.; Feely, J. Advanced Glycation End-Products and Arterial Stiffness in Hypertension. Am. J. Hypertens. 2007, 20, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, M.; Marumo, K. Effects of Collagen Crosslinking on Bone Material Properties in Health and Disease. Calcif. Tissue Int. 2015, 97, 242–261. [Google Scholar] [CrossRef] [PubMed]
- London, G.M. Arterial Stiffness in Chronic Kidney Disease and End-Stage Renal Disease. Blood Purif. 2018, 45, 154–158. [Google Scholar] [CrossRef]
- Quinn, U.; Tomlinson, L.A.; Cockcroft, J.R. Arterial Stiffness. JRSM Cardiovasc. Dis. 2012, 1, 1–8. [Google Scholar] [CrossRef]
- Roglic, G. WHO Global Report on Diabetes: A Summary. Int. J. Noncommun. Dis. 2016, 1, 3. [Google Scholar] [CrossRef]
- Jitraknatee, J.; Ruengorn, C.; Nochaiwong, S. Prevalence and Risk Factors of Chronic Kidney Disease among Type 2 Diabetes Patients: A Cross-Sectional Study in Primary Care Practice. Sci. Rep. 2020, 10, 6205. [Google Scholar] [CrossRef] [Green Version]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S. RAGE Drives the Development of Glomerulosclerosis and Implicates Podocyte Activation in the Pathogenesis of Diabetic Nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-Κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.L.; Forbes, J.M.; Cooper, M.E. AGE, RAGE, and ROS in Diabetic Nephropathy; WB Saunders: Philadelphia, PA, USA, 2007; Volume 27, pp. 130–143. [Google Scholar]
- Ilatovskaya, D.V.; Levchenko, V.; Lowing, A.; Shuyskiy, L.S.; Palygin, O.; Staruschenko, A. Podocyte Injury in Diabetic Nephropathy: Implications of Angiotensin II–Dependent Activation of TRPC Channels. Sci. Rep. 2015, 5, 17637. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Receptor for AGE (RAGE): Signaling Mechanisms in the Pathogenesis of Diabetes and Its Complications. Ann. N. Y. Acad. Sci. 2011, 1243, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, H.; Nishizawa, Y. AGEs/RAGE in CKD: Irreversible Metabolic Memory Road toward CVD? Eur. J. Clin. Investig. 2010, 40, 623–635. [Google Scholar] [CrossRef]
- An, X.; Zhang, L.; Yao, Q.; Li, L.; Wang, B.; Zhang, J.; He, M.; Zhang, J. The Receptor for Advanced Glycation Endproducts Mediates Podocyte Heparanase Expression through NF-ΚB Signaling Pathway. Mol. Cell. Endocrinol. 2018, 470, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A. Metabolic Memory Phenomenon in Diabetes Mellitus: Achieving and Perspectives. Diabetes Metab. Syndr. Clin. Res. Rev. 2016, 10, S176–S183. [Google Scholar] [CrossRef] [PubMed]
- Al-Haddad, R.; Karnib, N.; Abi Assaad, R.; Bilen, Y.; Emmanuel, N.; Ghanem, A.; Younes, J.; Zibara, V.; Stephan, J.S.; Sleiman, S.F. Epigenetic Changes in Diabetes. Neurosci. Lett. 2016, 625, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Balcerczyk, A.; Okabe, J.; El-Osta, A. Epigenetic Phenomena Linked to Diabetic Complications. Nat. Rev. Endocrinol. 2010, 6, 665–675. [Google Scholar] [CrossRef]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic Mechanisms in Diabetic Complications and Metabolic Memory. Diabetologia 2015, 58, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Shiu, S.; Chow, W.; Leng, L.; Bucala, R.; Betteridge, D. Association between Serum Levels of Soluble Receptor for Advanced Glycation End Products and Circulating Advanced Glycation End Products in Type 2 Diabetes. Diabetologia 2006, 49, 2756–2762. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of Advanced Glycation End Products in Cellular Signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [Green Version]
- Younessi, P.; Yoonessi, A. Advanced Glycation End-Products and Their Receptor-Mediated Roles: Inflammation and Oxidative Stress. Iran. J. Med. Sci. 2011, 36, 154. [Google Scholar] [PubMed]
- Bucciarelli, L.G.; Wendt, T.; Qu, W.; Lu, Y.; Lalla, E.; Rong, L.L.; Goova, M.T.; Moser, B.; Kislinger, T.; Lee, D.C. RAGE Blockade Stabilizes Established Atherosclerosis in Diabetic Apolipoprotein E–Null Mice. Circulation 2002, 106, 2827–2835. [Google Scholar] [CrossRef] [PubMed]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M. Deletion of the Receptor for Advanced Glycation End Products Reduces Glomerulosclerosis and Preserves Renal Function in the Diabetic OVE26 Mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christaki, E.; Opal, S.M.; Keith Jr, J.C.; Kessimian, N.; Palardy, J.E.; Parejo, N.A.; Tan, X.Y.; Piche-Nicholas, N.; Tchistiakova, L.; Vlasuk, G.P. A Monoclonal Antibody against RAGE Alters Gene Expression and Is Protective in Experimental Models of Sepsis and Pneumococcal Pneumonia. Shock 2011, 35, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xu, H.; Wu, X.; Xie, Y.; Lu, X.; Wang, L. Design, Synthesis and Anti-TNBC Activity of Azeliragon Triazole Analogues. Bioorg. Med. Chem. Lett. 2021, 54, 128444. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, T.; Yang, Y.; Lu, D.; Xu, A.; Li, K. FPS-ZM1 Alleviates Neuroinflammation in Focal Cerebral Ischemia Rats via Blocking Ligand/RAGE/DIAPH1 Pathway. ACS Chem. Neurosci. 2020, 12, 63–78. [Google Scholar] [CrossRef]
- Hong, Y.; Shen, C.; Yin, Q.; Sun, M.; Ma, Y.; Liu, X. Effects of RAGE-Specific Inhibitor FPS-ZM1 on Amyloid-β Metabolism and AGEs-Induced Inflammation and Oxidative Stress in Rat Hippocampus. Neurochem. Res. 2016, 41, 1192–1199. [Google Scholar] [CrossRef]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral Advanced Glycation Endproducts (AGEs) Promote Insulin Resistance and Diabetes by Depleting the Antioxidant Defenses AGE Receptor-1 and Sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of Advanced Glycation End Products Improves Insulin Resistance in Human Type 2 Diabetes: Potential Role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A. Oral AGE Restriction Ameliorates Insulin Resistance in Obese Individuals with the Metabolic Syndrome: A Randomised Controlled Trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef]
- Chen, J.-H.; Lin, X.; Bu, C.; Zhang, X. Role of Advanced Glycation End Products in Mobility and Considerations in Possible Dietary and Nutritional Intervention Strategies. Nutr. Metab. 2018, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Cai, W.; Mitsuhashi, T.; Vlassara, H.; Bucala, R. Lysozyme Enhances Renal Excretion of Advanced Glycation Endproducts in Vivo and Suppresses Adverse Age-Mediated Cellular Effects in Vitro: A Potential AGE Sequestration Therapy for Diabetic Nephropathy? Mol. Med. 2001, 7, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.G.; Tan, G.; Binger, K.J.; Pickering, R.J.; Thomas, M.C.; Nagaraj, R.H.; Cooper, M.E.; Wilkinson-Berka, J.L. Candesartan Attenuates Diabetic Retinal Vascular Pathology by Restoring Glyoxalase-I Function. Diabetes 2010, 59, 3208–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Padiya, R.; Adela, R.; Putcha, U.K.; Reddy, G.S.; Reddy, B.R.; Kumar, K.P.; Chakravarty, S.; Banerjee, S.K. Garlic and Resveratrol Attenuate Diabetic Complications, Loss of β-Cells, Pancreatic and Hepatic Oxidative Stress in Streptozotocin-Induced Diabetic Rats. Front. Pharmacol. 2016, 7, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin Lowers Methylglyoxal Dependent Protein Glycation and Limits the Complications of Diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef] [PubMed]
- Ruhee, R.T.; Suzuki, K. The Integrative Role of Sulforaphane in Preventing Inflammation, Oxidative Stress and Fatigue: A Review of a Potential Protective Phytochemical. Antioxidants 2020, 9, 521. [Google Scholar] [CrossRef]
- Suantawee, T.; Thilavech, T.; Cheng, H.; Adisakwattana, S. Cyanidin Attenuates Methylglyoxal-Induced Oxidative Stress and Apoptosis in INS-1 Pancreatic β-Cells by Increasing Glyoxalase-1 Activity. Nutrients 2020, 12, 1319. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of Insulin Resistance in Overweight and Obese Subjects by Trans-Resveratrol and Hesperetin Combination—Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic Targeting of Advanced Glycation End-Products in Age-Related Diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef]
- Machado, A.P.; Pinto, R.S.; Moysés, Z.P.; Nakandakare, E.R.; Quintão, E.C.; Passarelli, M. Aminoguanidine and Metformin Prevent the Reduced Rate of HDL-Mediated Cell Cholesterol Efflux Induced by Formation of Advanced Glycation End Products. Int. J. Biochem. Cell Biol. 2006, 38, 392–403. [Google Scholar] [CrossRef]
- Harvey, A.; Martorana, R.; Colangeli, J.; Ahmed, H.; Delgado, D.; Kim, A.; Sandhu, R.; Barsotti, R.J.; Young, L.H.; Chen, Q. The Effects of Metformin, Aminoguanidine, and Pyridoxamine on Methylglyoxal Induced Cardiac Myocyte Injury; The Digital Commons@PCOM: Philadelphia, PA, USA, 2019. [Google Scholar]
- Lima, T.F.O.; Costa, M.C.; Figueiredo, I.D.; Inácio, M.D.; Rodrigues, M.R.; Assis, R.P.; Baviera, A.M.; Brunetti, I.L. Curcumin, Alone or in Combination with Aminoguanidine, Increases Antioxidant Defenses and Glycation Product Detoxification in Streptozotocin-Diabetic Rats: A Therapeutic Strategy to Mitigate Glycoxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 1036360. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.; Ruggiero-Lopez, D. Metformin Inhibition of Glycation Processes. Diabetes Metab. 2003, 29, S95–S96. [Google Scholar] [CrossRef]
- Solís-Calero, C.; Ortega-Castro, J.; Frau, J.; Muñoz, F. Scavenger Mechanism of Methylglyoxal by Metformin. A DFT Study; Springer: Berlin/Heidelberg, Germany, 2016; pp. 191–204. [Google Scholar]
- Dariya, B.; Nagaraju, G.P. Advanced Glycation End Products in Diabetes, Cancer and Phytochemical Therapy. Drug Discov. Today 2020, 25, 1614–1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yagiz, Y.; Buran, T.J.; do Nascimento Nunes, C.; Gu, L. Phytochemicals from Berries and Grapes Inhibited the Formation of Advanced Glycation End-products by Scavenging Reactive Carbonyls. Food Res. Int. 2011, 44, 2666–2673. [Google Scholar] [CrossRef]
- Martínez, Y.; Más, D.; Betancur, C.; Gebeyew, K.; Adebowale, T.; Hussain, T.; Lan, W.; Ding, X. Role of the Phytochemical Compounds like Modulators in Gut Microbiota and Oxidative Stress. Curr. Pharm. Des. 2020, 26, 2642–2656. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. https://doi.org/10.3390/biom12040542
Khalid M, Petroianu G, Adem A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules. 2022; 12(4):542. https://doi.org/10.3390/biom12040542
Chicago/Turabian StyleKhalid, Mariyam, Georg Petroianu, and Abdu Adem. 2022. "Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives" Biomolecules 12, no. 4: 542. https://doi.org/10.3390/biom12040542