
Kv7 Channels in Cyclic-Nucleotide Dependent Relaxation of Rat Intra-Pulmonary Artery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Myography
2.2. Kv7 Subunit Expression
2.3. Sources of Drugs
2.4. Data Analysis
3. Results
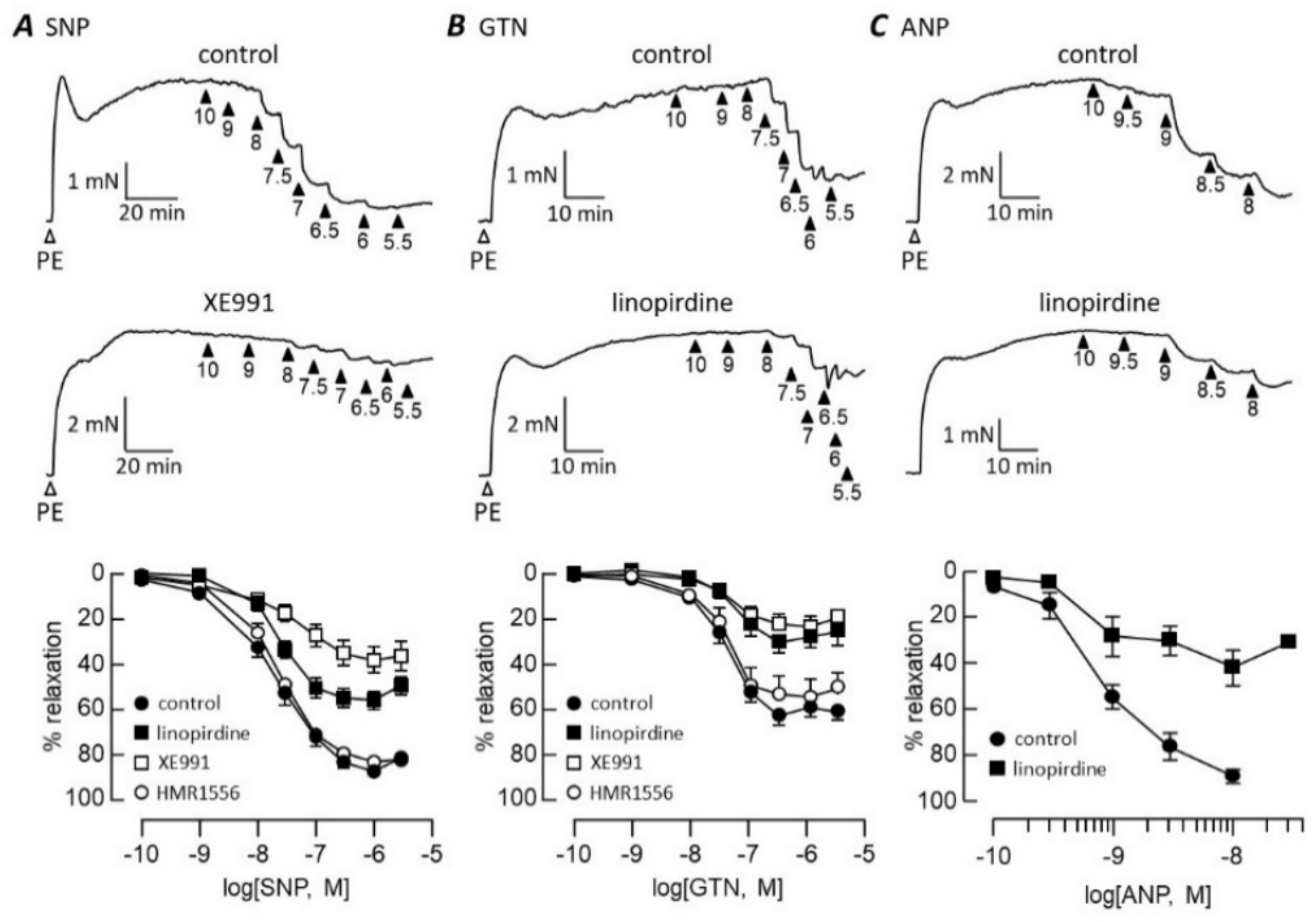
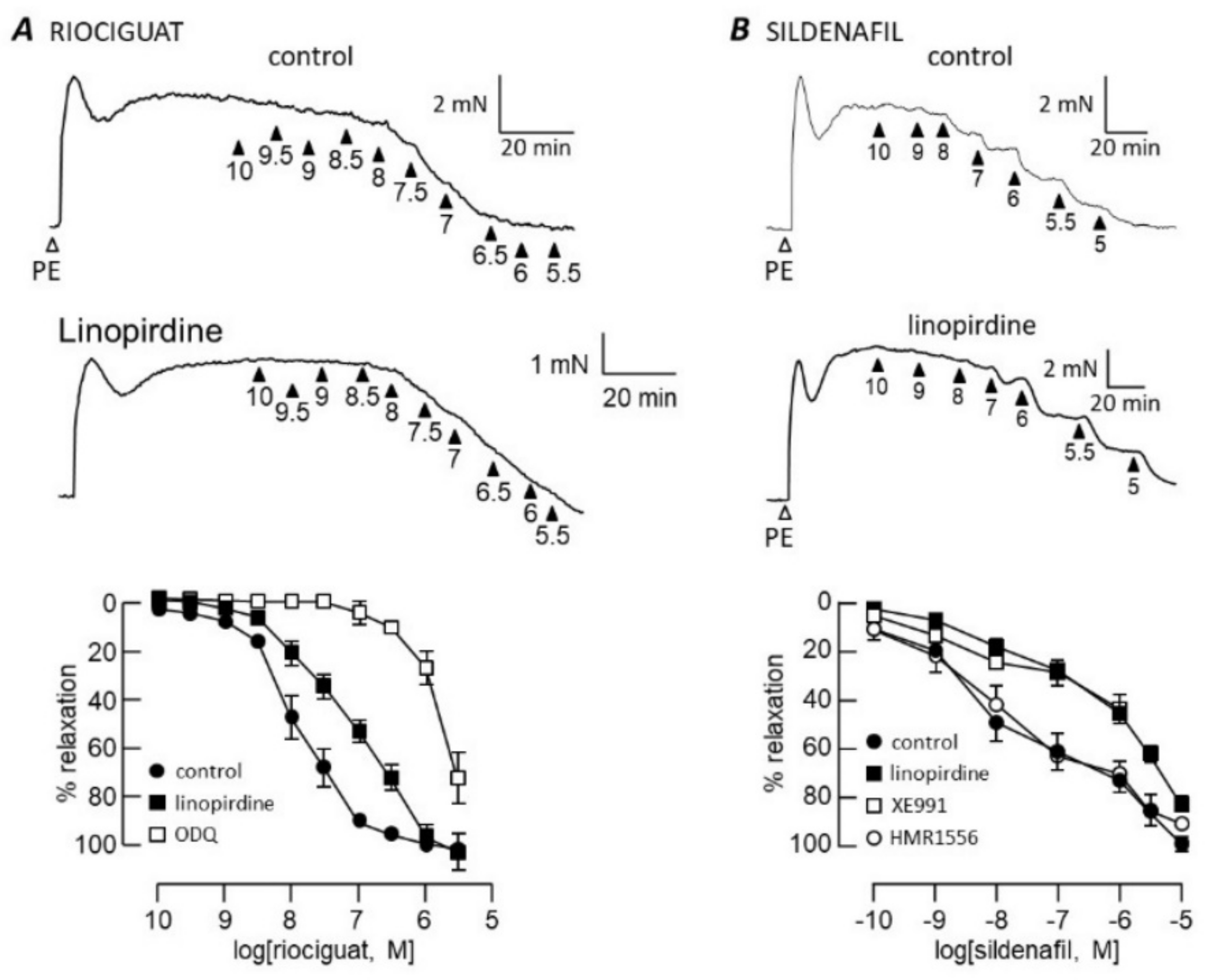
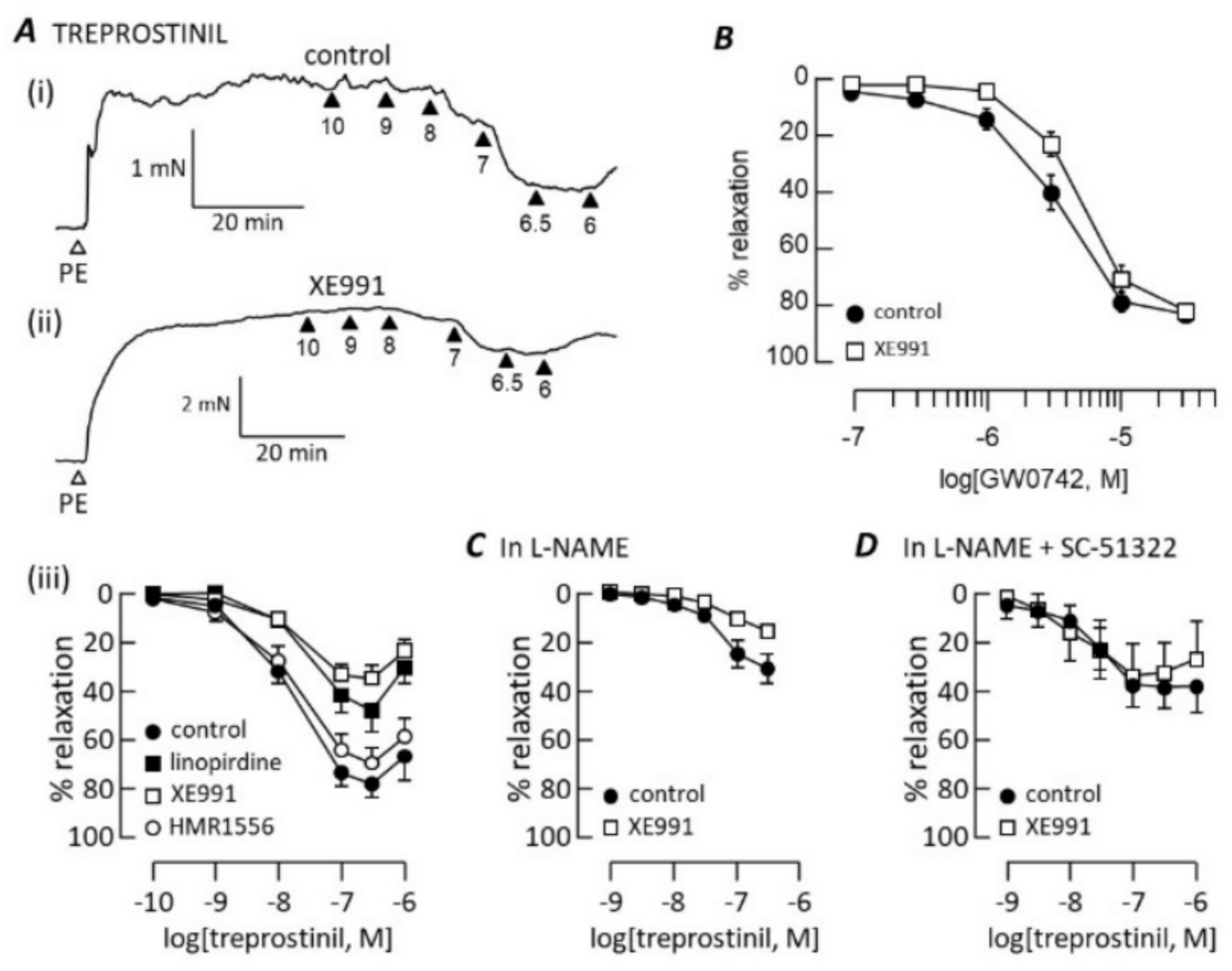
3.1. cGMP-Dependent Vasodilation
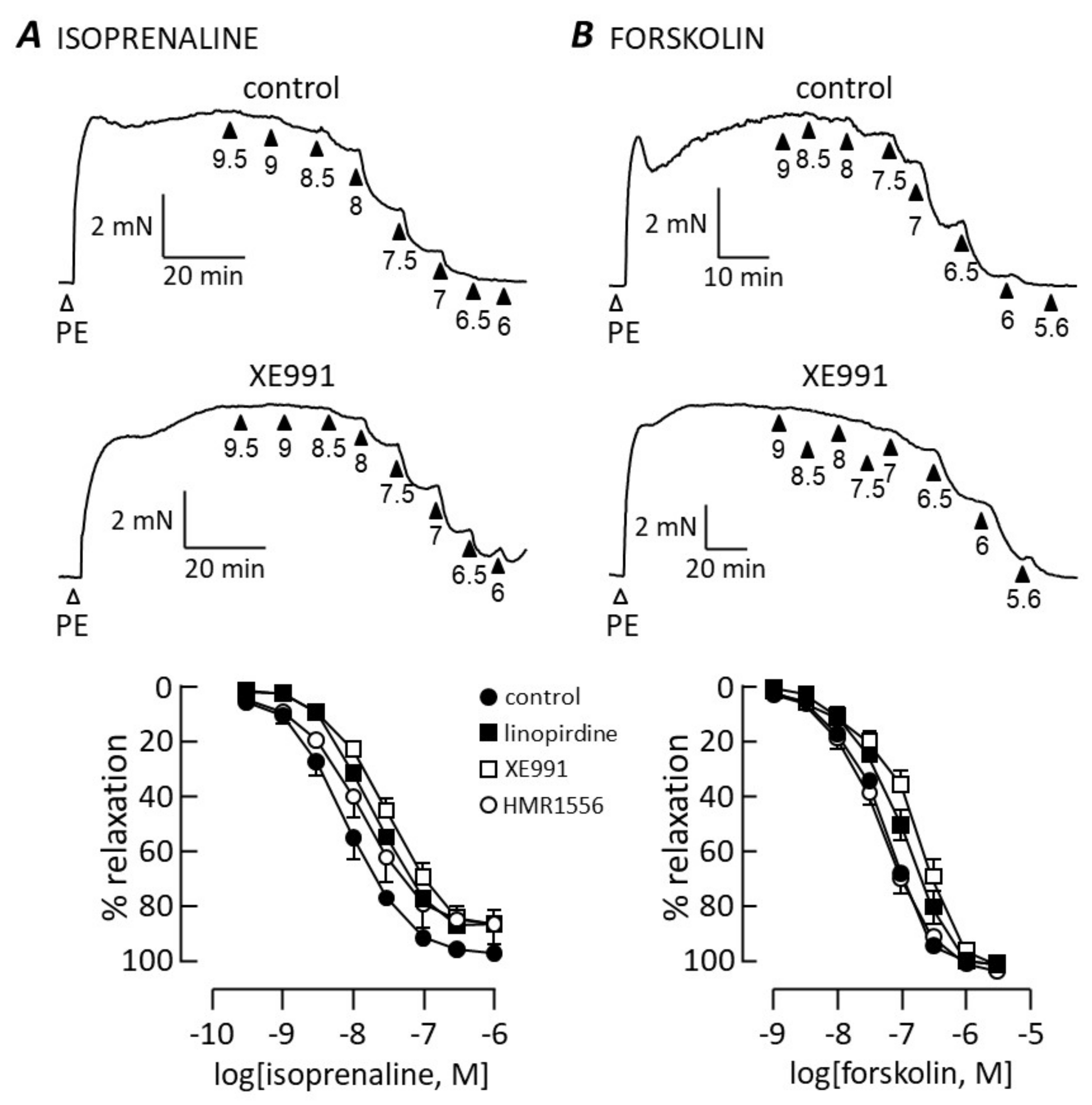
3.2. cAMP-Dependent Vasodilation
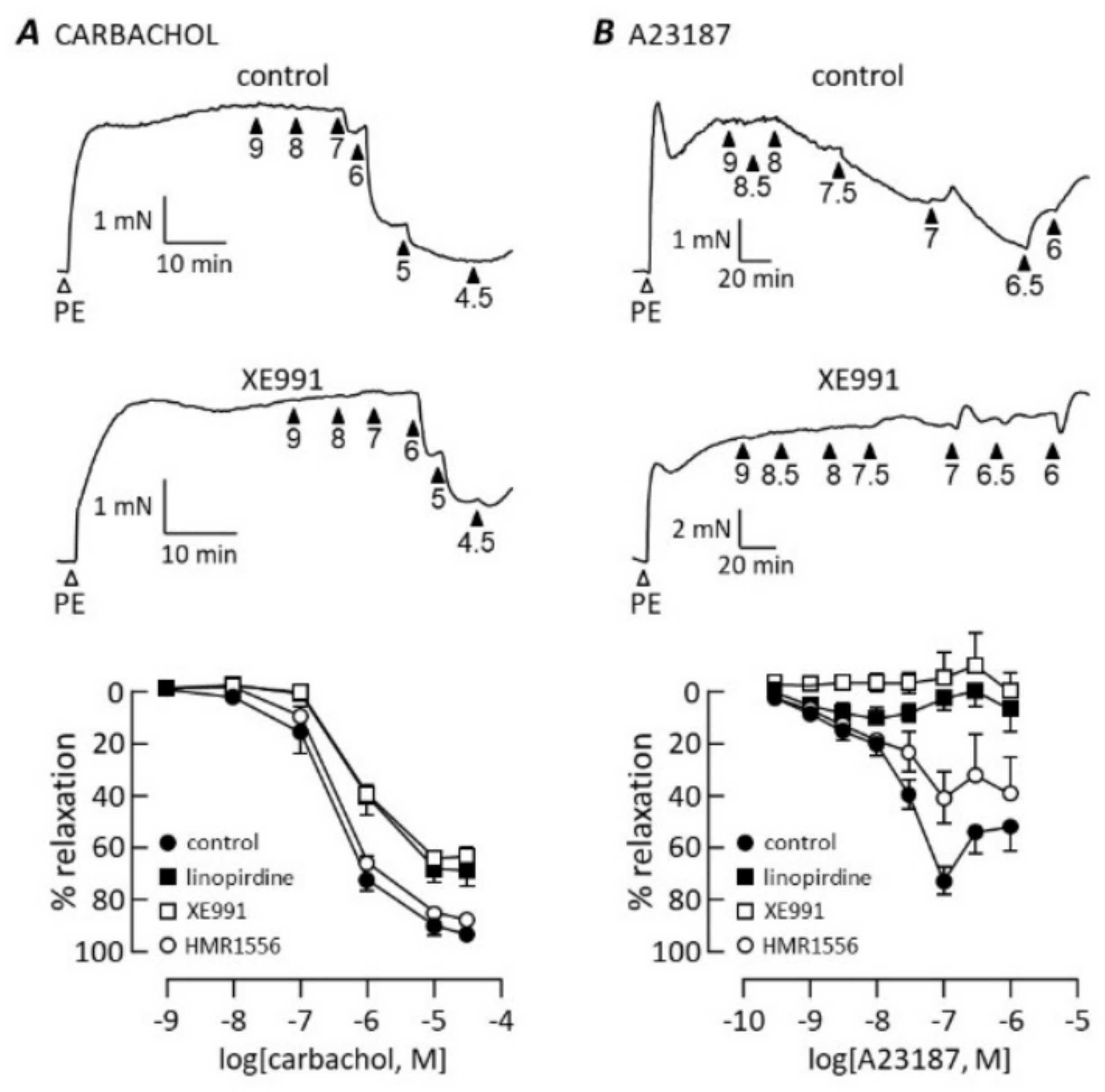
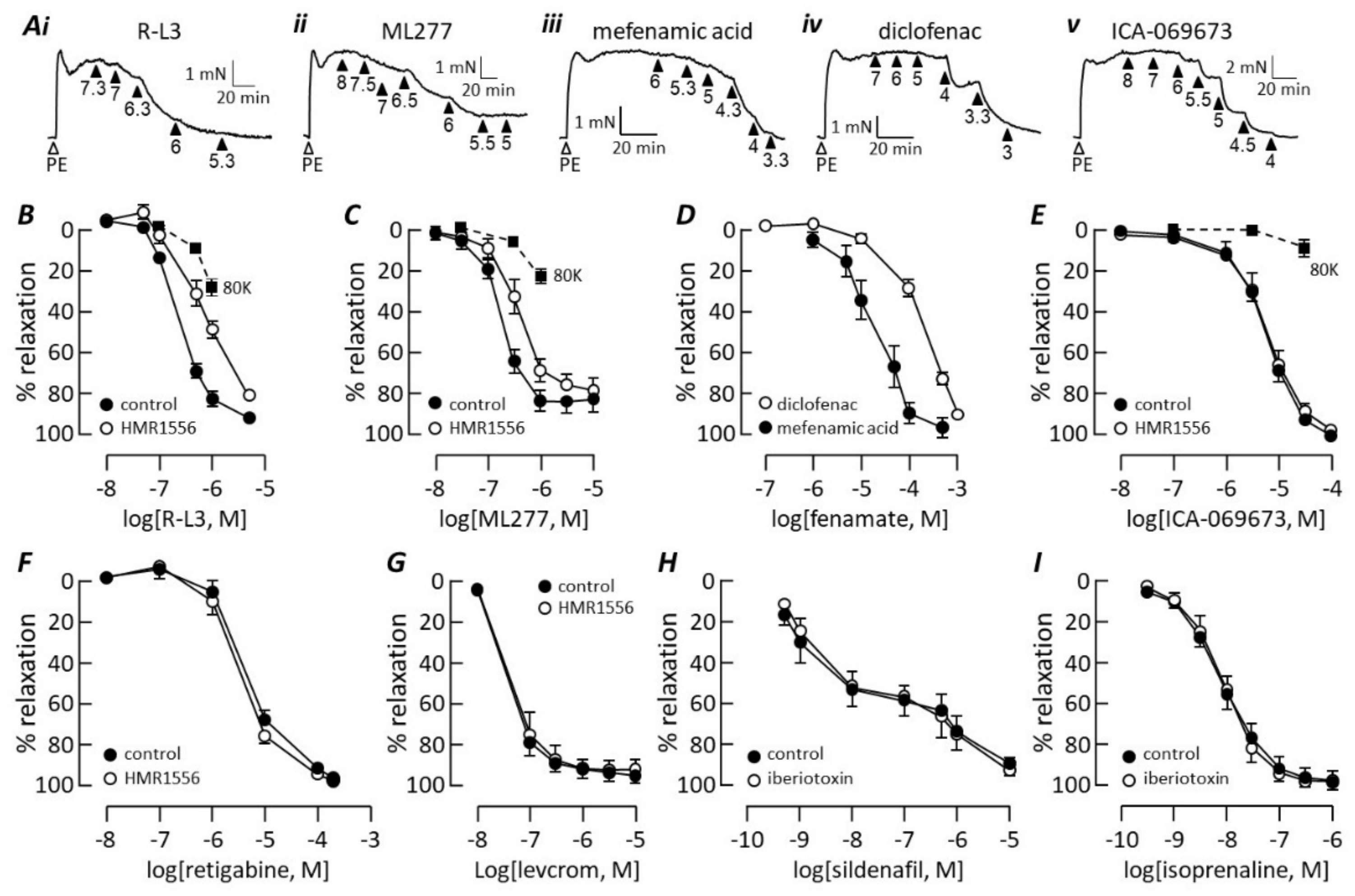
3.3. Kv7 Channel Pharmacology
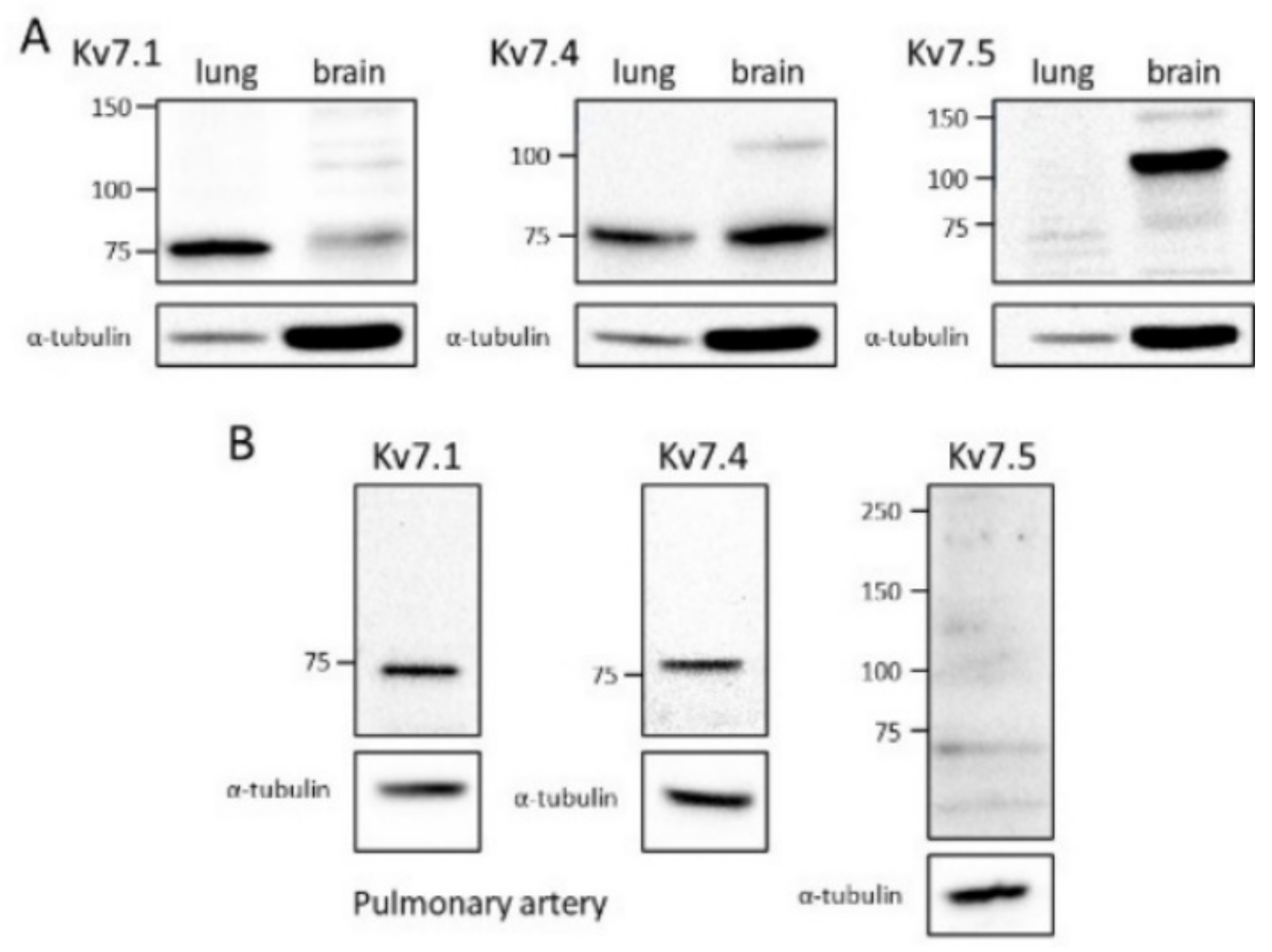
3.4. Kv7 Channel Expression
4. Discussion
4.1. cGMP Signalling
4.2. cAMP Signalling
4.3. Kv7 Channel Subtypes
4.4. Exclusion of Other K Channels
4.5. Implications for PAH
4.6. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montani, D.; Chaumais, M.-C.; Guignabert, C.; Günther, S.; Girerd, B.; Jais, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Aman, J.; Harbaum, L.; Ulrich, A.; Wharton, J.; Rhodes, C. Recent advances in pulmonary arterial hypertension. F1000Research 2018, 7, 1128. [Google Scholar] [CrossRef] [Green Version]
- Nausch, L.W.M.; Ledoux, J.; Bonev, A.D.; Nelson, M.T.; Dostmann, W.R. Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc. Natl. Acad. Sci. USA 2008, 105, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Clapp, L.H.; Gurung, R. The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial hypertension: Role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015, 120, 56–71. [Google Scholar] [CrossRef]
- Chester, A.H.; Yacoub, M.H.; Moncada, S. Nitric oxide and pulmonary arterial hypertension. Glob. Cardiol. Sci. Pr. 2017, 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Balan, P.; Gurney, A.M. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir. Res. 2006, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, S.; Sedivy, V.; Hodyc, D.; Herget, J.; Gurney, A.M. KCNQ Modulators Reveal a Key Role for KCNQ Potassium Channels in Regulating the Tone of Rat Pulmonary Artery Smooth Muscle. J. Pharmacol. Exp. Ther. 2009, 329, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadha, P.S.; Zunke, F.; Davis, A.J.; Jepps, T.A.; Linders, J.T.; Schwake, M.; Towart, R.; Greenwood, I.A. Pharmacological dissection of Kv7.1 channels in systemic and pulmonary arteries. J. Cereb. Blood Flow Metab. 2012, 166, 1377–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedivy, V.; Joshi, S.; Ghaly, Y.; Mizera, R.; Zaloudikova, M.; Brennan, S.; Novotna, J.; Herget, J.; Gurney, A.M. Role of Kv7 channels in responses of the pulmonary circulation to hypoxia. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L48–L57. [Google Scholar] [CrossRef] [PubMed]
- Van Der Horst, J.; Greenwood, I.A.; Jepps, T.A. Cyclic AMP-Dependent Regulation of Kv7 Voltage-Gated Potassium Channels. Front. Physiol. 2020, 11, 727. [Google Scholar] [CrossRef] [PubMed]
- Stott, J.B.; Barrese, V.; Jepps, T.A.; Leighton, E.V.; Greenwood, I.A. Contribution of Kv7 Channels to Natriuretic Peptide Mediated Vasodilation in Normal and Hypertensive Rats. Hypertension 2015, 65, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Mondéjar-Parreño, G.; Moral-Sanz, J.; Barreira, B.; De la Cruz, A.; Gonzalez, T.; Callejo, M.; Esquivel-Ruiz, S.; Morale-Cano, D.; Moreno, L.; Valenzuela, C.; et al. Activation of Kv7 channels as a novel mechanism for NO/cGMP-induced pulmonary vasodilation. J. Cereb. Blood Flow Metab. 2019, 176, 2131–2145. [Google Scholar] [CrossRef]
- Chadha, P.S.; Zunke, F.; Zhu, H.-L.; Davis, A.J.; Jepps, T.A.; Olesen, S.P.; Cole, W.C.; Moffatt, J.D.; Greenwood, I.A. Reduced KCNQ4-Encoded Voltage-Dependent Potassium Channel Activity Underlies Impaired β-Adrenoceptor–Mediated Relaxation of Renal Arteries in Hypertension. Hypertension 2012, 59, 877–884. [Google Scholar] [CrossRef] [Green Version]
- Chadha, P.S.; Jepps, T.A.; Carr, G.; Stott, J.B.; Zhu, H.-L.; Cole, W.C.; Greenwood, I.A. Contribution of Kv7.4/Kv7.5 Heteromers to Intrinsic and Calcitonin Gene-Related Peptide–Induced Cerebral Reactivity. Arter. Thromb. Vasc. Biol. 2014, 34, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Yang, Y.; Tanner, M.A.; Li, M.; Hill, M. Heterogeneity in Kv7 channel function in the cerebral and coronary circulation. Microcirculation 2015, 22, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Mani, B.; Robakowski, C.; Brueggemann, L.I.; Cribbs, L.L.; Tripathi, A.; Majetschak, M.; Byron, K.L. Kv7.5 Potassium Channel Subunits Are the Primary Targets for PKA-Dependent Enhancement of Vascular Smooth Muscle Kv7 Currents. Mol. Pharmacol. 2016, 89, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Morales-Cano, D.; Moreno, L.; Barreira, B.; Pandolfi, R.; Chamorro, V.; Jimenez, R.; Villamor, E.; Duarte, J.; Perez-Vizcaino, F.; Cogolludo, A. Kv7 channels critically determine coronary artery reactivity: Left-right differences and down-regulation by hyperglycaemia. Cardiovasc. Res. 2015, 106, 98–108. [Google Scholar] [CrossRef]
- Stott, J.; Greenwood, I. Complex role of Kv7 channels in cGMP and cAMP-mediated relaxations. Channels 2015, 9, 117–118. [Google Scholar] [CrossRef] [Green Version]
- Byron, K.L.; Brueggemann, L.I. Kv7 potassium channels as signal transduction intermediates in the control of microvascular tone. Microcirculation 2018, 25, e12419. [Google Scholar] [CrossRef]
- Stott, J.B.; Barrese, V.; Greenwood, I.A. Kv7 Channel Activation Underpins EPAC-Dependent Relaxations of Rat Arteries. Arter. Thromb. Vasc. Biol. 2016, 36, 2404–2411. [Google Scholar] [CrossRef] [Green Version]
- Brueggemann, L.I.; Cribbs, L.L.; Byron, K.L. Heteromeric channels formed from alternating Kv7.4 and Kv7.5 α-subunits display biophysical, regulatory, and pharmacological characteristics of smooth muscle M-currents. Front. Physiol. 2020, 11, 992. [Google Scholar] [CrossRef]
- Melzig, M.F.; Alasbahi, R.H. Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie 2012, 67, 5–13. [Google Scholar]
- Lim, H.; Dey, S.K. Minireview: A Novel Pathway of Prostacyclin Signaling—Hanging Out with Nuclear Receptors. Endocrinology 2002, 143, 3207–3210. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Moreno, L.; Reed, A.; Wort, S.J.; Desvergne, B.; Garland, C.; Zhao, L.; Mitchell, J.A. The PPARβ/δAgonist GW0742 Relaxes Pulmonary Vessels and Limits Right Heart Hypertrophy in Rats with Hypoxia-Induced Pulmonary Hypertension. PLoS ONE 2010, 5, e9526. [Google Scholar] [CrossRef] [Green Version]
- Fuchikami, C.; Murakami, K.; Tajima, K.; Homan, J.; Kosugi, K.; Kuramoto, K.; Oka, M.; Kuwano, K. A comparison of vasodilation mode among selexipag (NS-304; [2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide]), its active metabolite MRE-269 and various prostacyclin receptor agonists in rat, porcine and human pulmonary arteries. Eur. J. Pharmacol. 2017, 795, 75–83. [Google Scholar] [CrossRef]
- Whittle, B.J.; Silverstein, A.M.; Mottola, D.M.; Clapp, L. Binding and activity of the prostacyclin receptor (IP) agonists, treprostinil and iloprost, at human prostanoid receptors: Treprostinil is a potent DP1 and EP2 agonist. Biochem. Pharmacol. 2012, 84, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Corboz, M.R.; Salvail, W.; Gagnon, S.; LaSala, D.; Laurent, C.E.; Salvail, D.; Chen, K.-J.; Cipolla, D.; Perkins, W.R.; Chapman, R.W. Prostanoid receptor subtypes involved in treprostinil-mediated vasodilation of rat pulmonary arteries and in treprostinil-mediated inhibition of collagen gene expression of human lung fibroblasts. Prostaglandins Other Lipid Mediat. 2021, 152, 106486. [Google Scholar] [CrossRef]
- Salata, J.J.; Jurkiewicz, N.K.; Wang, J.; Evans, B.E.; Orme, H.T.; Sanguinetti, M.C. A Novel Benzodiazepine that Activates Cardiac Slow Delayed Rectifier K+ Currents. Mol. Pharmacol. 1998, 54, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Mattmann, M.E.; Yu, H.; Lin, Z.; Xu, K.; Huang, X.; Long, S.; Wu, M.; McManus, O.B.; Engers, D.W.; Le, U.M.; et al. Identification of (R)-N-(4-(4-methoxyphenyl)thiazol-2-yl)-1-tosylpiperidine-2-carboxamide, ML277, as a novel, potent and selective Kv7.1 (KCNQ1) potassium channel activator. Bioorganic Med. Chem. Lett. 2012, 22, 5936–5941. [Google Scholar] [CrossRef] [Green Version]
- Busch, A.E.; Herzer, T.; Wagner, C.A.; Schmidt, F.; Raber, G.; Waldegger, S.; Lang, F. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol. Pharmacol. 1994, 46, 750–753. [Google Scholar]
- Unsöld, B.; Kerst, G.; Brousos, H.; Hübner, M.; Schreiber, R.; Nitschke, R.; Greger, R.; Bleich, M. KCNE1 reverses the response of the human K+ channel KCNQ1 to cytosolic pH changes and alters its pharmacology and sensitivity to temperature. Pflügers Arch. Eur. J. Physiol. 2000, 441, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Peretz, A.; Degani, N.; Nachman, R.; Uziyel, Y.; Gibor, G.; Shabat, D.; Attali, B. Meclofenamic Acid and Diclofenac, Novel Templates of KCNQ2/Q3 Potassium Channel Openers, Depress Cortical Neuron Activity and Exhibit Anticonvulsant Properties. Mol. Pharmacol. 2005, 67, 1053–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brueggemann, L.I.; Mackie, A.R.; Martin, J.L.; Cribbs, L.L.; Byron, K.L. Diclofenac Distinguishes among Homomeric and Heteromeric Potassium Channels Composed of KCNQ4 and KCNQ5 Subunits. Mol. Pharmacol. 2011, 79, 10–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, G.; Roeloffs, R.; Rigdon, G.C.; Antonio, B.; Mersch, T.; McNaughton-Smith, G.; Wickenden, A.D.; Fritch, P.; Suto, M.J. N-Pyridyl and Pyrimidine Benzamides as KCNQ2/Q3 Potassium Channel Openers for the Treatment of Epilepsy. ACS Med. Chem. Lett. 2011, 2, 481–484. [Google Scholar] [CrossRef] [Green Version]
- Brueggemann, L.I.; Haick, J.M.; Cribbs, L.L.; Byron, K.L. Differential Activation of Vascular Smooth Muscle Kv7.4, Kv7.5, and Kv7.4/7.5 Channels by ML213 and ICA-069673. Mol. Pharmacol. 2014, 86, 330–341. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.L.; Huang, J.M.; Hampl, V.; Nelson, D.P.; Shultz, P.J.; Weir, E.K. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1994, 91, 7583–7587. [Google Scholar] [CrossRef] [Green Version]
- Barman, S.A.; Zhu, S.; Han, G.; White, R.E. cAMP activates BKCa channels in pulmonary arterial smooth muscle via cGMP-dependent protein kinase. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L1004–L1011. [Google Scholar] [CrossRef]
- Gao, Y.-D.; Garcia, M.L. Interaction of agitoxin2, charybdotoxin, and iberiotoxin with potassium channels: Selectivity between voltage-gated and Maxi-K channels. Proteins Struct. Funct. Bioinform. 2003, 52, 146–154. [Google Scholar] [CrossRef]
- Brown, D.A.; Passmore, G.M. Neural KCNQ (Kv7) channels. J. Cereb. Blood Flow Metab. 2009, 156, 1185–1195. [Google Scholar] [CrossRef]
- Goldman, A.M.; Glasscock, E.; Yoo, J.; Chen, T.T.; Klassen, T.L.; Noebels, J.L. Arrhythmia in Heart and Brain: KCNQ1 Mutations Link Epilepsy and Sudden Unexplained Death. Sci. Transl. Med. 2009, 1, 2ra6. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Feletou, M.; Ku, D.D.; Man, R.Y.K.; Vanhoutte, P.M. The calcium ionophore A23187 induces endothelium-dependent contractions in femoral arteries from rats with streptozotocin-induced diabetes. Br. J. Pharmacol. 2007, 150, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Feil, R.; Mülsch, A.; Lohmann, S.M.; Hofmann, F.; Walter, U. Physiology and Pathophysiology of Vascular Signaling Controlled by Cyclic Guanosine 3′,5′-Cyclic Monophosphate–Dependent Protein Kinase. Circulation 2003, 108, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Pauvert, O.; Lugnier, C.; Keravis, T.; Marthan, R.; Rousseau, E.; Savineau, J.P. Effect of sildenafil on cyclic nucleotide phosphodiesterase activity, vascular tone and calcium signaling in rat pulmonary artery. J. Cereb. Blood Flow Metab. 2003, 139, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Moreland, R.B.; Goldstein, I.; Traish, A. Sildenafil, a novel inhibitor of phosphodiesterase type 5 in human corpus cavernosum smooth muscle cells. Life Sci. 1998, 62, PL309–PL318. [Google Scholar] [CrossRef]
- Sandner, P.; Zimmer, D.P.; Milne, G.T.; Follmann, M.; Hobbs, A.; Stasch, J.-P. Soluble Guanylate Cyclase Stimulators and Activators. In Reactive Oxygen Species; Springer: Cham, Switzerland, 2018; Volume 264, pp. 355–394. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Brandish, P.E.; DiValentin, M.; Schelvis, J.P.M.; Babcock, G.T.; Marletta, M.A. Inhibition of Soluble Guanylate Cyclase by ODQ. Biochemistry 2000, 39, 10848–10854. [Google Scholar] [CrossRef]
- Garthwaite, J.; Southam, E.; Boulton, C.L.; Nielsen, E.B.; Schmidt, K.; Mayer, B. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 1995, 48, 184–188. [Google Scholar]
- Stasch, J.-P.; Becker, E.M.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Feurer, A.; Gerzer, R.; Minuth, T.; Perzborn, E.; Pleiß, U.; et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 2001, 410, 212–215. [Google Scholar] [CrossRef]
- Dhanakoti, S.N.; Gao, Y.; Nguyen, M.Q.; Raj, J.U. Involvement of cGMP-dependent protein kinase in the relaxation of ovine pulmonary arteries to cGMP and cAMP. J. Appl. Physiol. 2000, 88, 1637–1642. [Google Scholar] [CrossRef]
- Shimokawa, H.; Flavahan, N.A.; Lorenz, R.R.; Vanhoutte, P.M. Prostacyclin releases endothelium-derived relaxing factor and potentiates its action in coronary arteries of the pig. J. Cereb. Blood Flow Metab. 1988, 95, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Norel, X.; de Montpreville, V.; Brink, C. Vasoconstriction induced by activation of EP1 and EP3 receptors in human lung: Effects of ONO-AE-248, ONO-DI-004, ONO-8711 or ONO-8713. Prostaglandins Other Lipid Mediat. 2004, 74, 101–112. [Google Scholar] [CrossRef]
- Kozłowska, H.; Baranowska-Kuczko, M.; Schlicker, E.; Kozłowski, M.; Zakrzeska, A.; Grzęda, E.; Malinowska, B. EP3 receptor-mediated contraction of human pulmonary arteries and inhibition of neurogenic tachycardia in pithed rats. Pharmacol. Rep. 2012, 64, 1526–1536. [Google Scholar] [CrossRef]
- Tatulian, L.; Delmas, P.; Abogadie, F.C.; Brown, D.A. Activation of Expressed KCNQ Potassium Currents and Native Neuronal M-Type Potassium Currents by the Anti-Convulsant Drug Retigabine. J. Neurosci. 2001, 21, 5535–5545. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, D.; Kaßmann, M.; Tano, J.-Y.; Chen, L.; Schleifenbaum, J.; Voelkl, J.; Lang, F.; Huang, Y.; Gollasch, M. Do KV7.1 channels contribute to control of arterial vascular tone? J. Cereb. Blood Flow Metab. 2017, 174, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.C.; Hechenberger, M.; Weinreich, F.; Kubisch, C.; Jentsch, T.J. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 2000, 275, 24089–24095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-S.; Ran, Y.-J.; Zhang, D.-D.; Li, S.-Z.; Zhu, D. MicroRNA-190 Regulates Hypoxic Pulmonary Vasoconstriction by Targeting a Voltage-Gated K+ Channel in Arterial Smooth Muscle Cells. J. Cell. Biochem. 2014, 115, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Mondéjar-Parreño, G.; Barreira, B.; Callejo, M.; Morales-Cano, D.; Barrese, V.; Esquivel-Ruiz, S.; Olivencia, M.A.; Macías, M.; Moreno, L.; Greenwood, I.A.; et al. Uncovered Contribution of Kv7 Channels to Pulmonary Vascular Tone in Pulmonary Arterial Hypertension. Hypertension 2020, 76, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.W.; Yang, R.; Kurata, H.T. Sequence determinants of subtype-specific actions of KCNQ channel openers. J. Physiol. 2017, 595, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Hampl, V.; Huang, J.M.; Weir, E.K.; Archer, S.L. Activation of the cGMP-dependent protein kinase mimics the stimulatory effect of nitric oxide and cGMP on calcium-gated potassium channels. Physiol. Res. 1995, 44, 39–44. [Google Scholar]
- Klinger, J.R.; Kadowitz, P.J. The Nitric Oxide Pathway in Pulmonary Vascular Disease. Am. J. Cardiol. 2017, 120, S71–S79. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Vanderpool, R.R.; Wang, J.; Yuan, J.X.-J. Targeting L-arginine-nitric oxide-cGMP pathway in pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 569–571. [Google Scholar] [CrossRef] [Green Version]
- García, M.L.; Knaus, H.G.; Munujos, P.; Slaughter, R.S.; Kaczorowski, G.J. Charybdotoxin and its effects on potassium channels. Am. J. Physiol. Physiol. 1995, 269, C1–C10. [Google Scholar] [CrossRef]
- Garcia, M.L.; Garcia-Calvo, M.; Hidalgo, P.; Lee, A.; MacKinnon, R. Purification and Characterization of Three Inhibitors of Voltage-Dependent K+ Channels from Leiurus Quinquestriatus var. Hebraeus Venom. Biochemistry 1994, 33, 6834–6839. [Google Scholar] [CrossRef]
- Candia, S.; Garcia, M.; Latorre, R. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca2+-activated K+ channel. Biophys. J. 1992, 63, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Perez-Vizcaino, F.; Cogolludo, A.; Mondejar-Parreño, G. Transcriptomic profile of cationic channels in human pulmonary arterial hypertension. Sci. Rep. 2021, 11, 15829. [Google Scholar] [CrossRef]
- Mandegar, M.; Yuan, J.X.-J. Role of K+ channels in pulmonary hypertension. Vasc. Pharmacol. 2002, 38, 25–33. [Google Scholar] [CrossRef]
- Morecroft, I.; Murray, A.; Nilsen, M.; Gurney, A.; Lean, M. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. J. Cereb. Blood Flow Metab. 2009, 157, 1241–1249. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Chawishly, M.; Loveland, O.; Gurney, A.M. Kv7 Channels in Cyclic-Nucleotide Dependent Relaxation of Rat Intra-Pulmonary Artery. Biomolecules 2022, 12, 429. https://doi.org/10.3390/biom12030429
Al-Chawishly M, Loveland O, Gurney AM. Kv7 Channels in Cyclic-Nucleotide Dependent Relaxation of Rat Intra-Pulmonary Artery. Biomolecules. 2022; 12(3):429. https://doi.org/10.3390/biom12030429
Chicago/Turabian StyleAl-Chawishly, Mohammed, Oliver Loveland, and Alison M. Gurney. 2022. "Kv7 Channels in Cyclic-Nucleotide Dependent Relaxation of Rat Intra-Pulmonary Artery" Biomolecules 12, no. 3: 429. https://doi.org/10.3390/biom12030429