
VEGFA, B, C: Implications of the C-Terminal Sequence Variations for the Interaction with Neuropilins

 , and
, and
Abstract
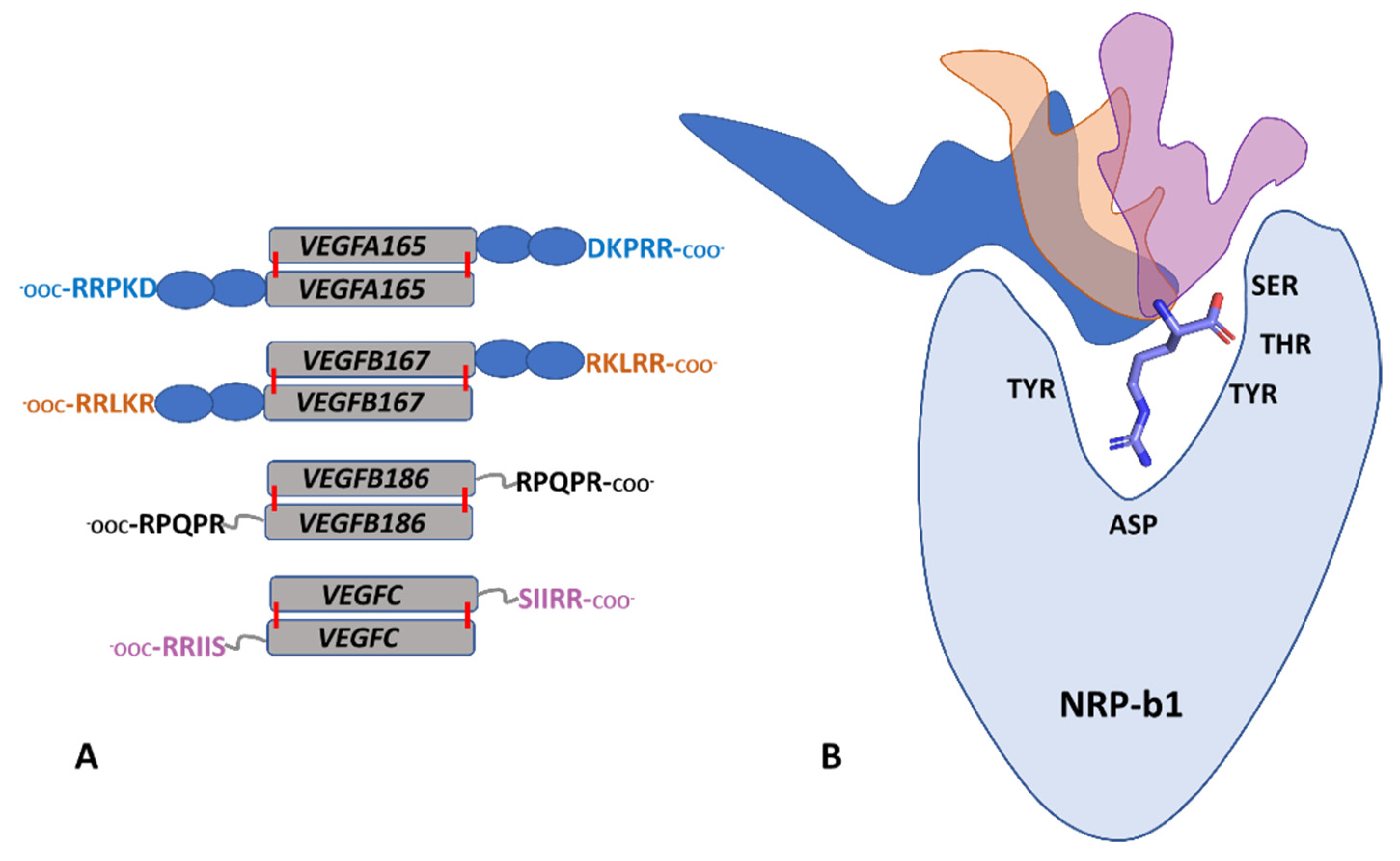
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Peptides
2.3. Surface Plasmon Resonance
2.4. Crystallography
2.5. Thermostability Shift Assays
2.6. Molecular Dynamics Simulations
3. Results
3.1. Interaction with Peptides Increases Thermostability of b1 Domains
3.2. All Peptides Exhibit Binding Affinities in the Micromolar Range by SPR
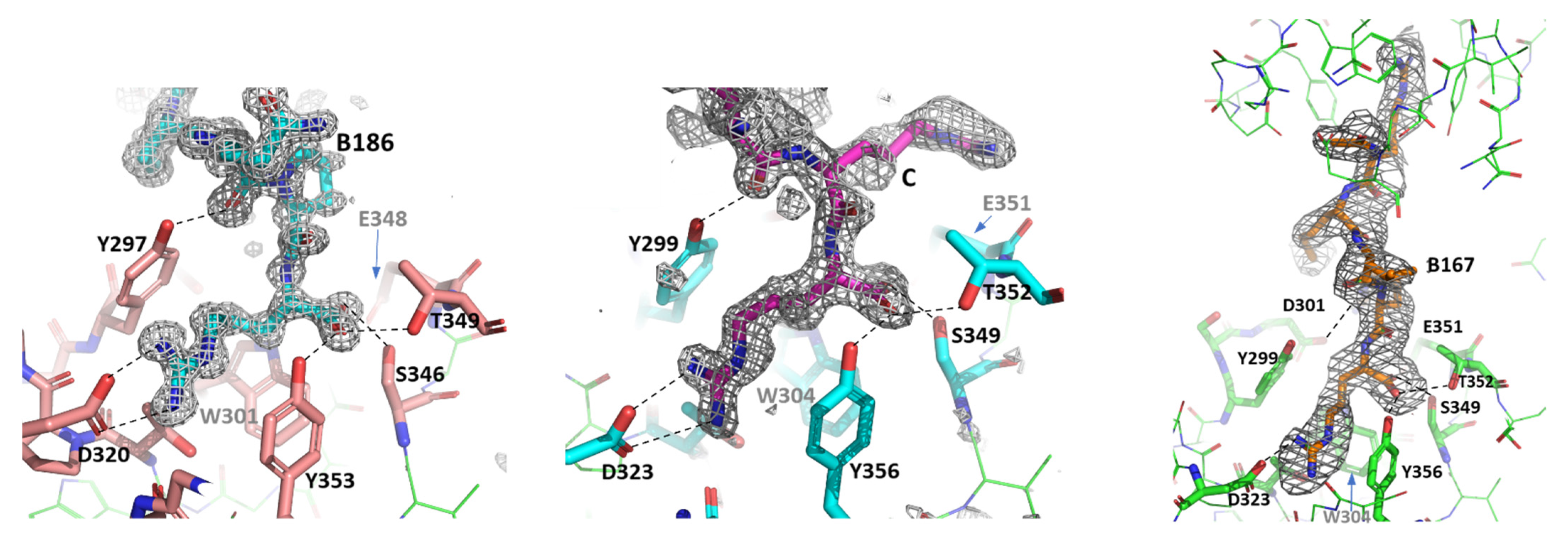
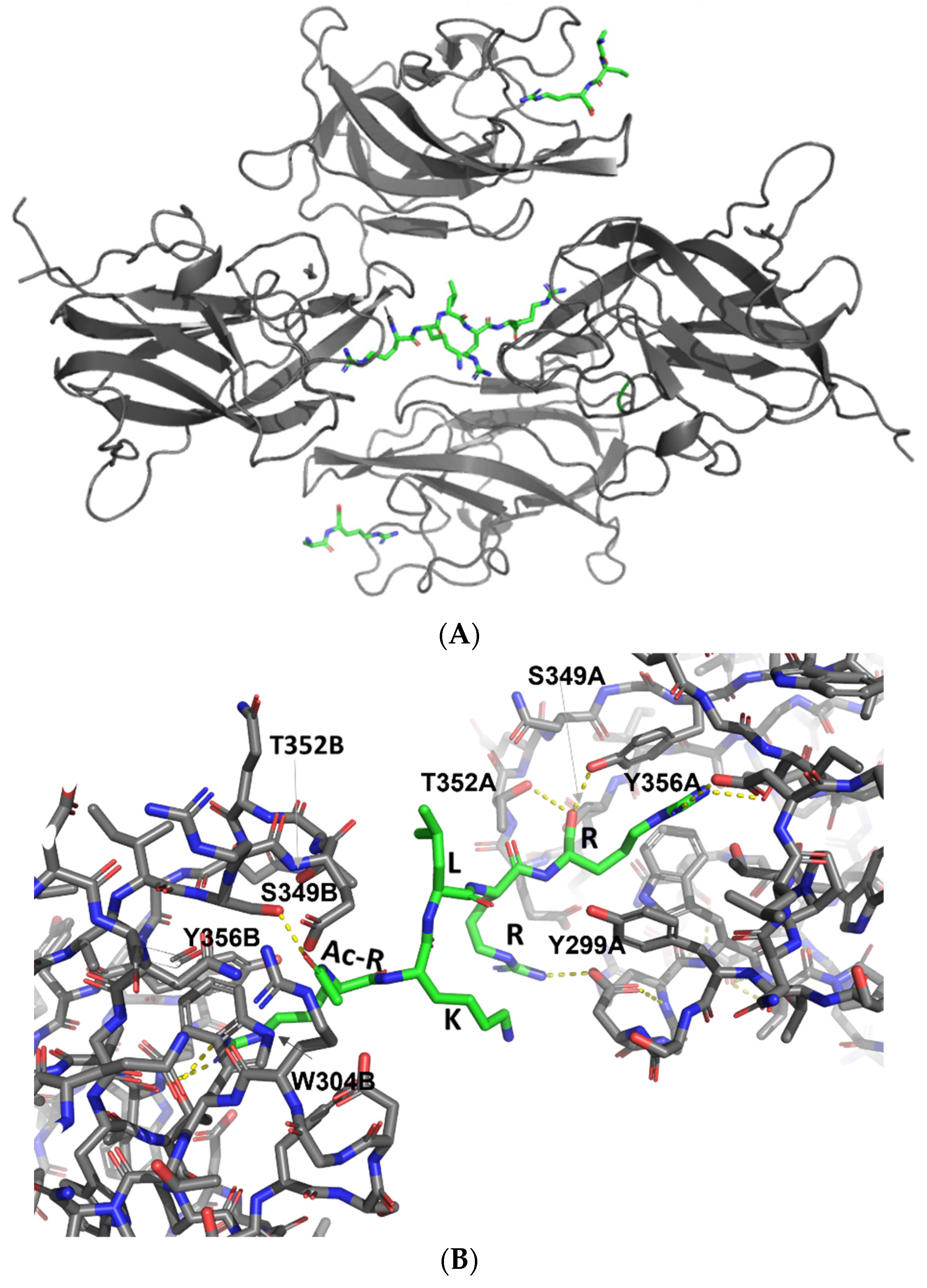
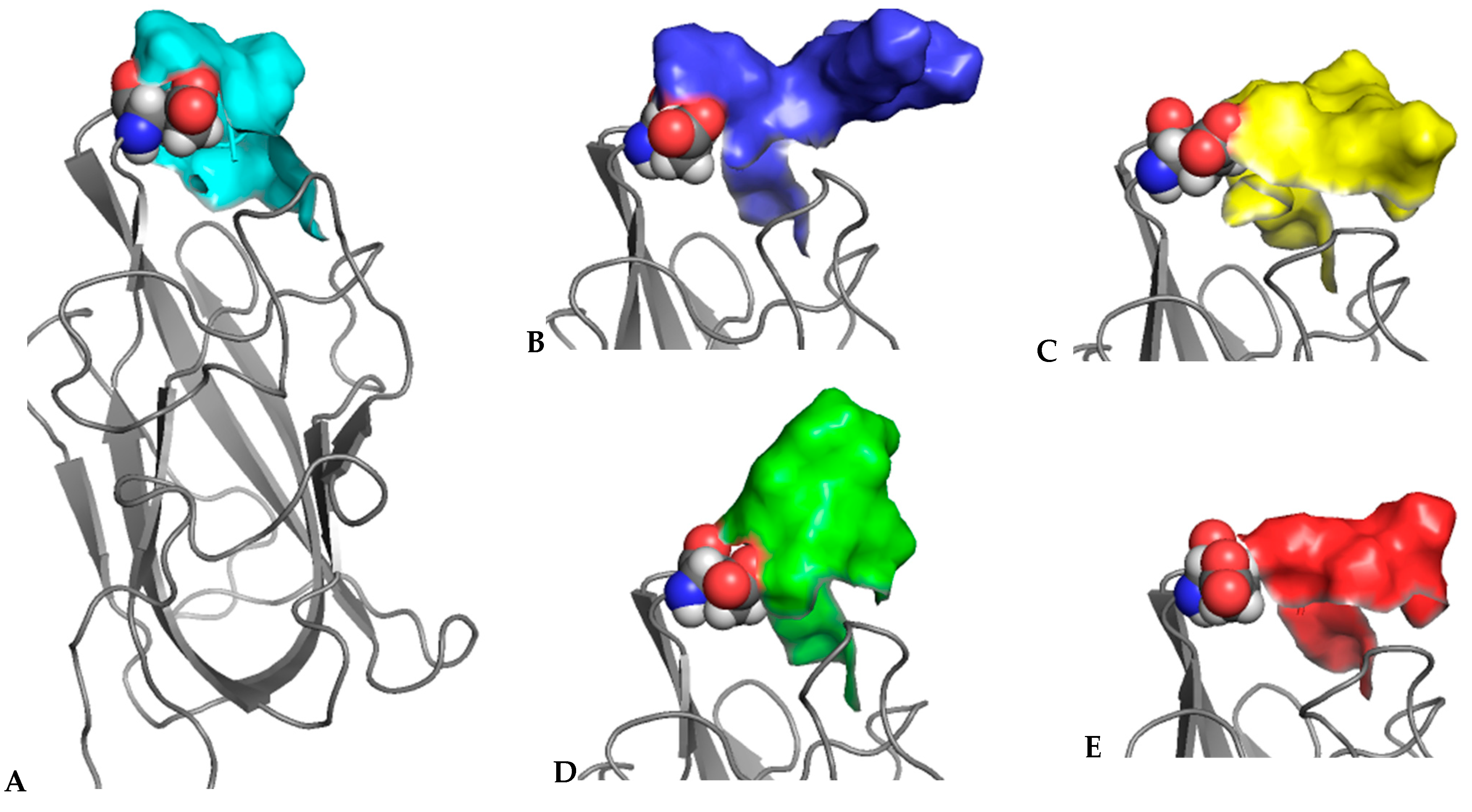
3.3. X-ray Crystal Structures of the Complexes Exhibit Distinct Features
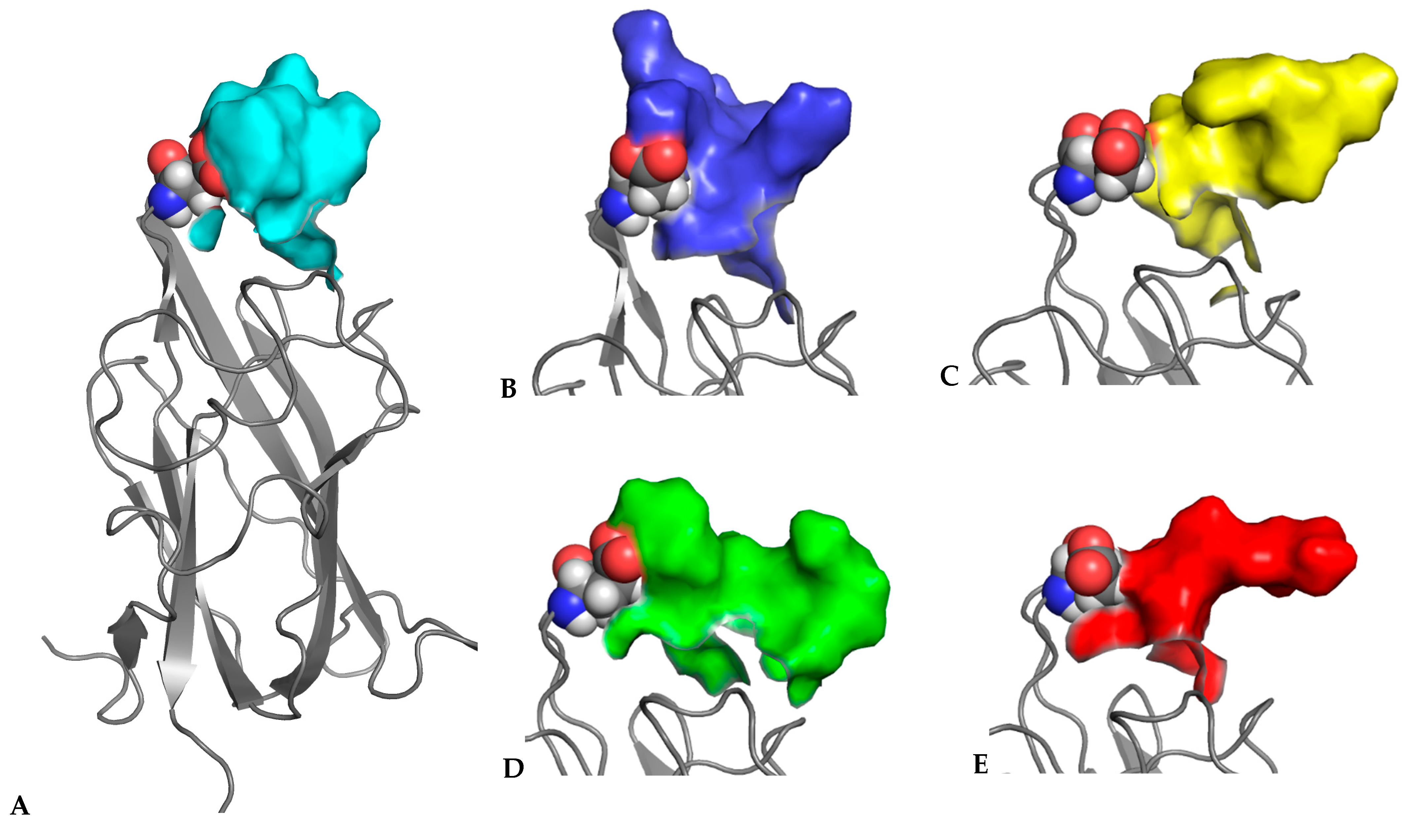
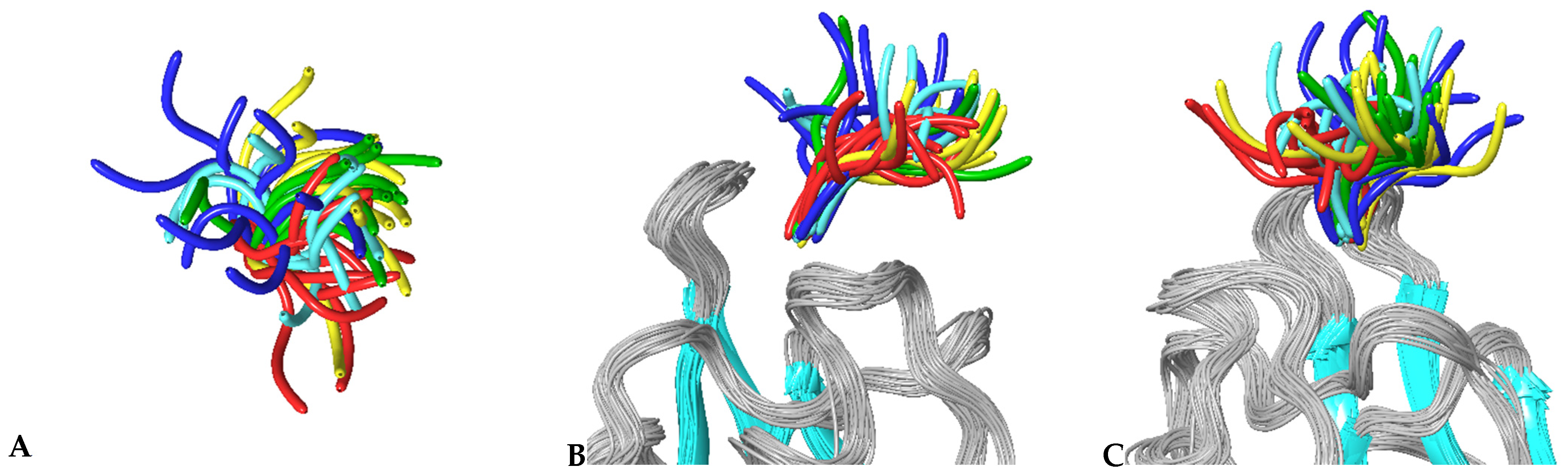
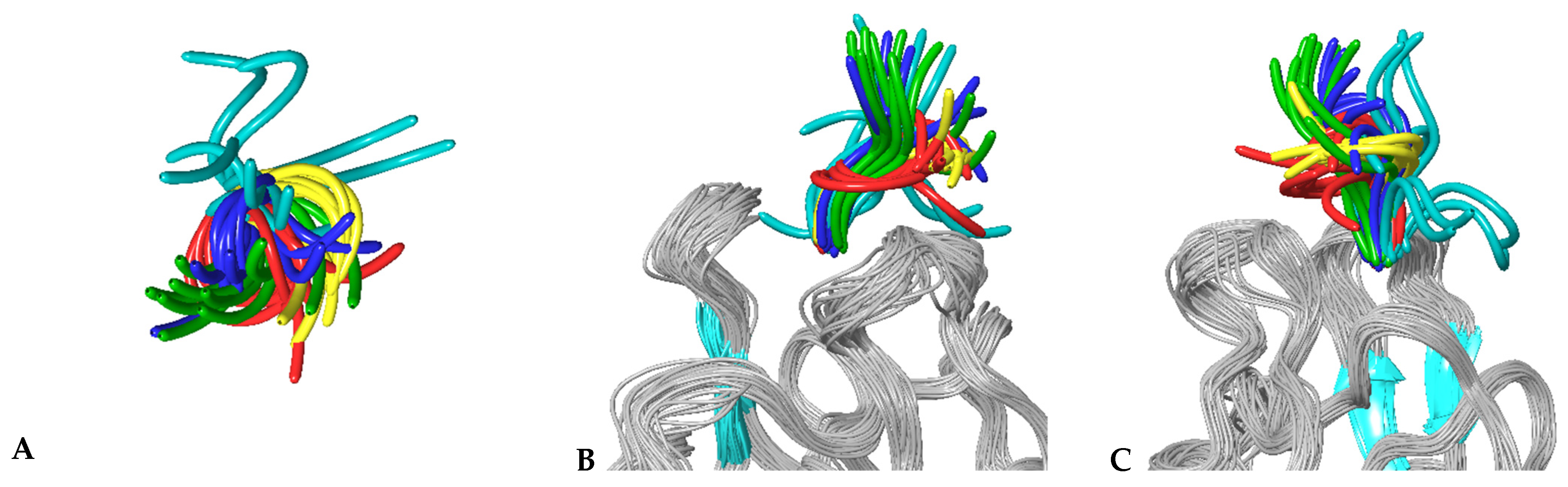
3.4. MD Simulations of Molecular Complexes Reveal Distinct Patterns of Sampling of Conformational Space by the Peptidic Ligands
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Key abbreviations
References
- Parker, M.W.; Guo, H.-F.; Li, X.; Linkugel, A.D.; Vander Kooi, C.W. Function of members of the neuropilin family as essential pleiotropic cell surface receptors. Biochemistry 2012, 51, 9437–9446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev. Cell 2013, 25, 156–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinskey, J.M.; Franks, N.E.; McMellen, A.N.; Giger, R.J.; Allen, B.L. Neuropilin-1 promotes Hedgehog signaling through a novel cytoplasmic motif. J. Biol. Chem. 2017, 292, 15192–15204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinka, Y.; Prud’homme, G.J. Neuropilin-1 is a receptor for transforming growth factor β-1, activates its latent form, and promotes regulatory T cell activity. J. Leukoc. Biol. 2008, 84, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform- specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.-H.; Chen, Y.-L.; Lee, K.-H.; Chang, C.-C.; Cheng, T.-M.; Wu, S.-Y.; Tu, C.-C.; Tsui, W.-L. Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-β- and PDGF-like signals in hepatic stellate cells. Sci. Rep. 2017, 7, 11006. [Google Scholar] [CrossRef] [PubMed]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.; Zachary, I. The VEGF family—Angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeltsch, M.; Kaipainen, A.; Joukov, V.; Meng, X.; Lakso, M.; Rauvala, H.; Swartz, M.; Fukumura, D.; Jain, R.K.; Alitalo, K. Hyperplasia of lymphatic vessels in VEGF-C transgenic mice. Science 1997, 276, 1423–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B.; Pajusola, K.; Kaipainen, A.; von Euler, G.; Joukov, V.; Saksela, O.; Orpana, A.; Pettersson, R.F.; Alitalo, K.; Eriksson, U. Vascular endothelial growth factor B, a novel growth factor for endothelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2576–2581. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, B.; Korpelainen, E.; Pepper, M.S.; Mandriota, S.J.; Aase, K.; Kumar, V.; Gunji, Y.; Jeltsch, M.M.; Shibuya, M.; Alitalo, K.; et al. Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc. Natl. Acad. Sci. USA 1998, 95, 11709–11714. [Google Scholar] [CrossRef] [Green Version]
- Makinen, T.; Olofsson, B.; Karpanen, T.; Hellman, U.; Soker, S.; Klagsbrun, M.; Eriksson, U.; Alitalo, K. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J. Biol. Chem. 1999, 274, 21217–21222. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, C.E.; Mehlem, A.; Falkevall, A.; Muhl, L.; Fam, B.C.; Ortsäter, H.; Scotney, P.; Nyqvist, D.; Samén, E.; Lu, L.; et al. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 2012, 490, 426–430. [Google Scholar] [CrossRef]
- Robciuc, M.R.; Kivelä, R.; Williams, I.M.; de Boer, J.F.; van Dijk, T.H.; Elamaa, H.; Tigistu-Sahle, F.; Molotkov, D.; Leppänen, V.-M.; Käkelä, R.; et al. VEGFB/VEGFR1-Induced Expansion of Adipose Vasculature Counteracts Obesity and Related Metabolic Complications. Cell Metab. 2016, 23, 712–724. [Google Scholar] [CrossRef] [Green Version]
- Arjunan, P.; Lin, X.; Tang, Z.; Du, Y.; Kumar, A.; Liu, L.; Yin, X.; Huang, L.; Chen, W.; Chen, Q.; et al. VEGF-B is a potent antioxidant. Proc. Natl. Acad. Sci. USA 2018, 115, 10351–10356. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C.; Kreusch, A.; McMullan, D.; Ng, K.; Spraggon, G. Crystal structure of the human neuropilin-1 b1 domain. Structure 2003, 11, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-C.I.; Fotinou, C.; Rana, R.; Yelland, T.; Frankel, P.; Zachary, I.; Djordjevic, S. Structural studies of neuropilin-2 reveal a zinc ion binding site remote from the vascular endothelial growth factor binding pocket. FEBS J. 2016, 283, 1921–1934. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Linkugel, A.D.; Goel, H.L.; Wu, T.; Mercurio, A.M.; Vander Kooi, C.W. Structural basis for VEGF-C binding to neuropilin-2 and sequestration by a soluble splice form. Structure 2015, 23, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, A.; Allerston, C.K.; Jia, H.; Herzog, B.; Garza-Garcia, A.; Winfield, N.; Ellard, K.; Aqil, R.; Lynch, R.; Chapman, C.; et al. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J. Med. Chem. 2010, 53, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.; Lobley, C.M.C.; Prince, S.M. Decision making in xia2. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1260–1273. [Google Scholar] [CrossRef] [Green Version]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Dror, R.O.; Shaw, D.E. The midpoint method for parallelization of particle simulations. J. Chem. Phys. 2006, 124, 184109. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 2, 6474–6487. [Google Scholar] [CrossRef]
- Mota, F.; Fotinou, C.; Rana, R.R.; Chan, A.W.E.; Yelland, T.; Arooz, M.T.; O’Leary, A.P.; Hutton, J.; Frankel, P.; Zachary, I.; et al. Architecture and hydration of the arginine-binding site of neuropilin-1. FEBS J. 2018, 285, 1290–1304. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.W.; Xu, P.; Guo, H.F.; Vander Kooi, C.W. Mechanism of Selective VEGF-A Binding by Neuropilin-1 Reveals a Basis for Specific Ligand Inhibition. PLoS ONE 2012, 7, e49177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenker, R.; Jura, N. Receptor tyrosine kinase activation: From the ligand perspective. Curr. Opin. Cell Biol. 2020, 63, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Krilleke, D.; Ng, Y.-S.E.; Shima, D.T. The heparin-binding domain confers diverse functions of VEGF-A in development and disease: A structure-function study. Biochem. Soc. Trans. 2009, 37, 1201–1206. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Peptide Sequence | Name in the Manuscript |
|---|---|---|
| VEGFA165 | Ac-DKPRR | A165 |
| VEGFB167 | Ac-RKLRR | B167 |
| VEGFB186 | Ac-RPQPR | B186 |
| VEGFC | Ac-SIIRR | C |
| Control | Ac-AAAAR | Con |
| Peptide | NRP1-b1 KD (μM) | SE | NRP2-b1 KD (μM) | SE |
|---|---|---|---|---|
| A165 | 7.87 | 0.22 | 30.90 | 0.93 |
| B167 | 4.12 | 0.36 | 13.00 | 0.56 |
| B186 | 10.00 | 0.29 | 24.80 | 0.82 |
| C | 33.20 | 1.30 | 27.10 | 0.96 |
| Con | 33.10 | 2.60 | 92.20 | 13.00 |
| NRP1-b1/B186 | NRP2-b1/B167 | NRP2-b1/C | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PDB Code | 6TKK | 6TDB | 6TJT | ||||||
| Data collection | |||||||||
| Space group | P 21 21 21 | C 1 2 1 | P 1 21 1 | ||||||
| Unit cell | 38.88 | 39.98 | 97.61 | 107.36 | 140.12 | 74.36 | 38.24 | 76.54 | 63.36 |
| 90 | 90 | 90 | 90 | 132.97 | 90 | 90 | 96.06 | 90 | |
| Wavelength, Å | 0.976 | 0.977 | 0.976 | ||||||
| Resolution low, Å | 48.81 | 70.06 | 63.00 | ||||||
| Resolution high, Å | 1.06 | 2.45 | 1.31 | ||||||
| Outer resolution shell, Å | 1.09–1.06 | 2.58–2.45 | 1.38–1.31 | ||||||
| CC half | 0.999 (0.559) * | 0.996 (0.738) | 0.998 (0.905) | ||||||
| Average I/sigma(I) | 29.4 (12.3) | 10.6 (2.3) | 12.6 (2.3) | ||||||
| Redundancy | 5.5 (1.5) | 3.4 (3.4) | 3.1 (2.4) | ||||||
| Completeness, % | 80.2 (12.3) * | 98.9 (99.7) | 98.9 (97.2) | ||||||
| No. unique reflections measured | 55,937 (645) | 29,170 (4278) | 86,786 (12,438) | ||||||
| Refinement | |||||||||
| Resolution low, Å | 48.805 | 70.06 | 48.64 | ||||||
| Resolution high, Å | 1.06 | 2.45 | 1.31 | ||||||
| Unique reflections work/free | 53,084/2785 | 27,731/1428 | 82,426/4267 | ||||||
| R value work/free | 0.1361/0.1495 | 0.1827/0.2497 | 0.1533/0.1790 | ||||||
| No. of atoms | 1543 | 5471 | 2955 | ||||||
| Ramachandran plot, favoured/allowed/outliers, % | 97.48/2.52/0.00 | 92.66/6.70/0.64 | 95.87/4.13/0.00 | ||||||
| RMSD bond lengths, Å | 0.012 | 0.004 | 0.008 | ||||||
| RMSD bond angles, ° | 1.299 | 0.691 | 1.095 | ||||||
| Fraction Time of Interaction Occurrence (%) | ||||||
|---|---|---|---|---|---|---|
| Residue | Binding Mode | A165 | B167 | B186 | C | Control |
| Y297 | 51 | 40 | 88 | 52 | 38 | |
| N300 | WB | 51 | 52 | 45 | ||
| W301 | 96 | 72 | 98 | 93 | 68 | |
| N313 | WB | 52 | 33 | 43 | 51 | |
| T316 | 96 | 94 | 74 | 95 | 96 | |
| D320 | BF | 99 | 99 | 99 | 99 | 99 |
| S346 | 98 | 81 | 99 | 97 | 63 | |
| K347 | WB | 32 | 60 | 47 | 33 | 30 |
| E348 | 57 | 93 | 53 | |||
| WB | 58 | 52 | ||||
| T349 | 47 | 52 | ||||
| K351 | WB | 32 | ||||
| Y353 | 35 | 67 | ||||
| Fraction Time of Interaction Occurrence (%) | ||||||
|---|---|---|---|---|---|---|
| Residue | Binding Mode | A165 | B167 | B186 | C | Control |
| Y299 | 31 | 64 | 86 | 32 | 85 | |
| D301 | WB | 88 | 100 | 97 | 93 | |
| R303 | 30 | 100 | ||||
| WB | 42 | 93 | ||||
| W304 | 98 | 99 | 78 | 41 | 98 | |
| T319 | 96 | 75 | 80 | 92 | 71 | |
| D323 | BF | 99 | 99 | 99 | 99 | 98 |
| S349 | 89 | 74 | 67 | 67 | 54 | |
| R350 | WB | 61 | ||||
| E351 | 37 | 74 | 63 | 55 | ||
| BF | 48 | 30 | 30 | |||
| T352 | 56 | 99 | 78 | 81 | 60 | |
| Y356 | 99 | 98 | 94 | 99 | ||
| I418 | 57 | 50 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eldrid, C.; Zloh, M.; Fotinou, C.; Yelland, T.; Yu, L.; Mota, F.; Selwood, D.L.; Djordjevic, S. VEGFA, B, C: Implications of the C-Terminal Sequence Variations for the Interaction with Neuropilins. Biomolecules 2022, 12, 372. https://doi.org/10.3390/biom12030372
Eldrid C, Zloh M, Fotinou C, Yelland T, Yu L, Mota F, Selwood DL, Djordjevic S. VEGFA, B, C: Implications of the C-Terminal Sequence Variations for the Interaction with Neuropilins. Biomolecules. 2022; 12(3):372. https://doi.org/10.3390/biom12030372
Chicago/Turabian StyleEldrid, Charles, Mire Zloh, Constantina Fotinou, Tamas Yelland, Lefan Yu, Filipa Mota, David L. Selwood, and Snezana Djordjevic. 2022. "VEGFA, B, C: Implications of the C-Terminal Sequence Variations for the Interaction with Neuropilins" Biomolecules 12, no. 3: 372. https://doi.org/10.3390/biom12030372