
Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio


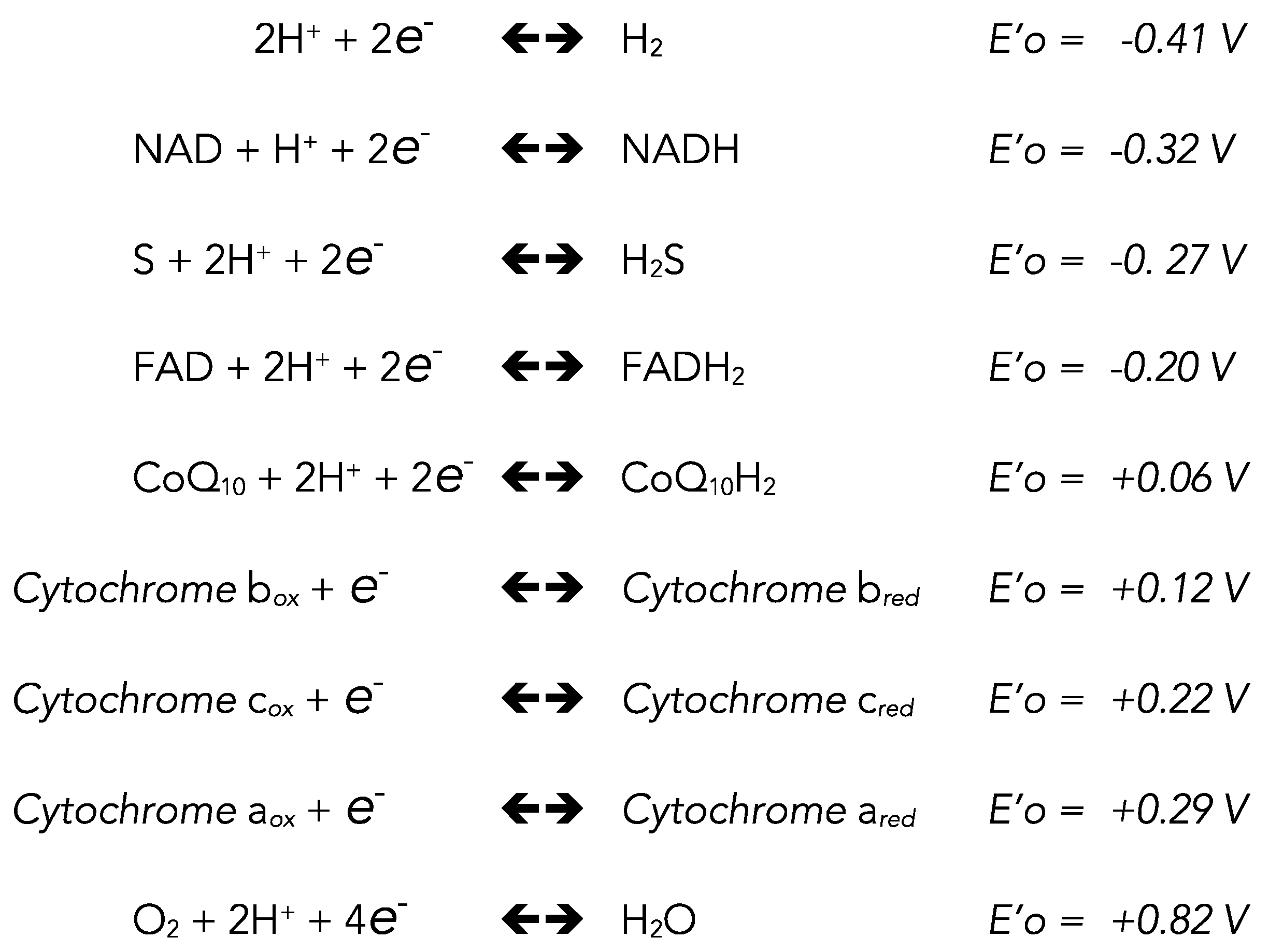{kind=link}
{kind=link}
{kind=link}
{kind=link}
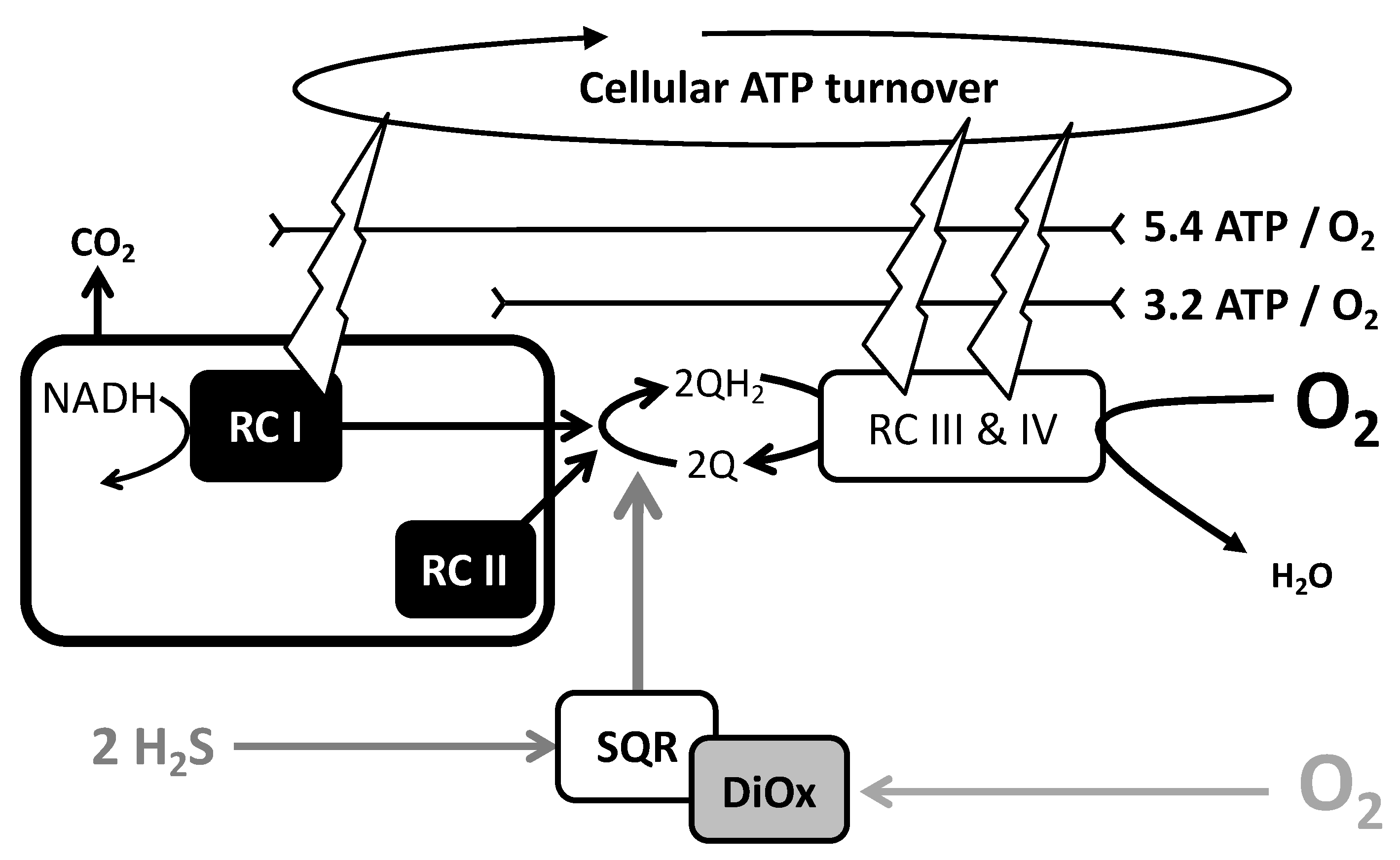{kind=link}
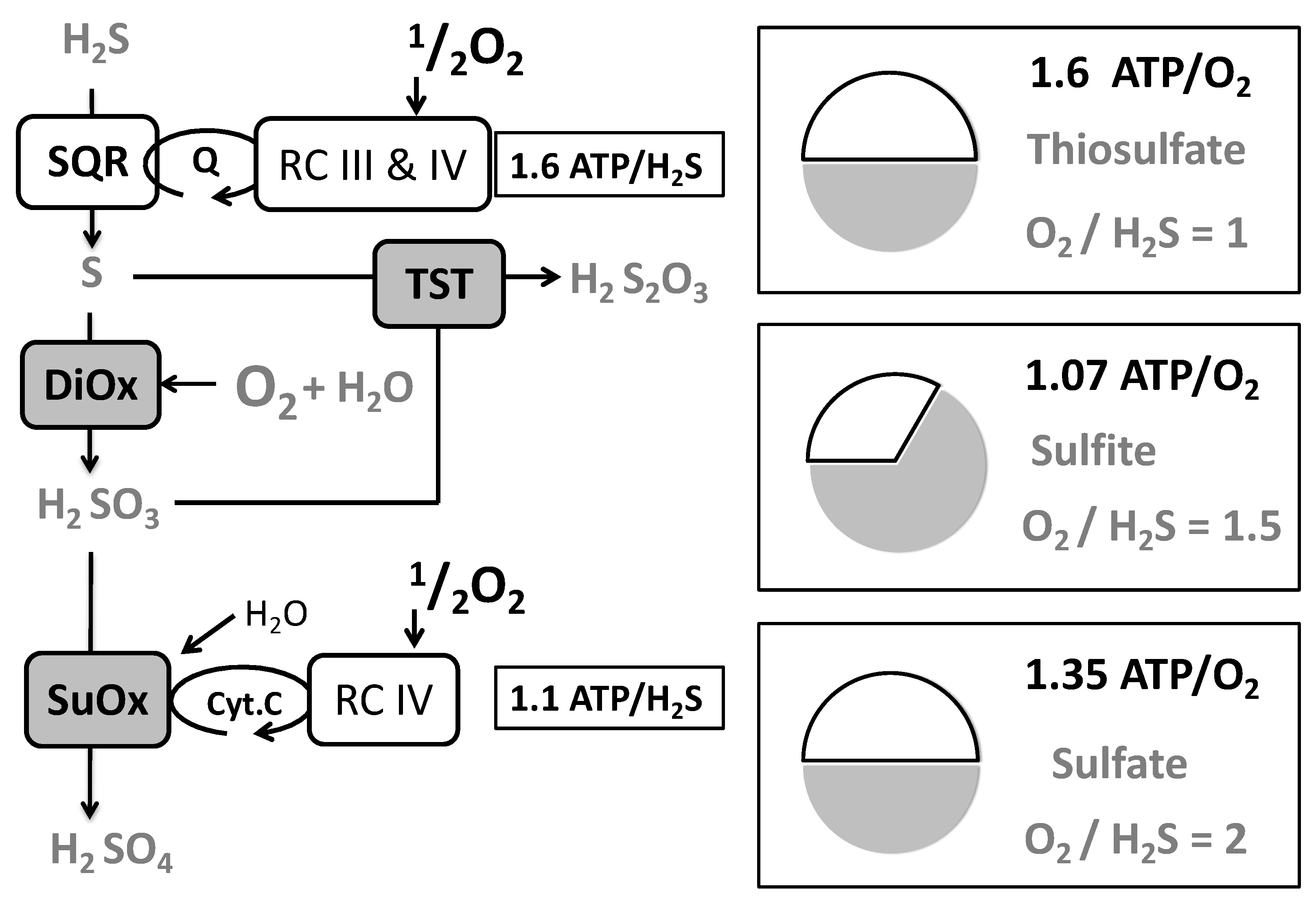{kind=link}
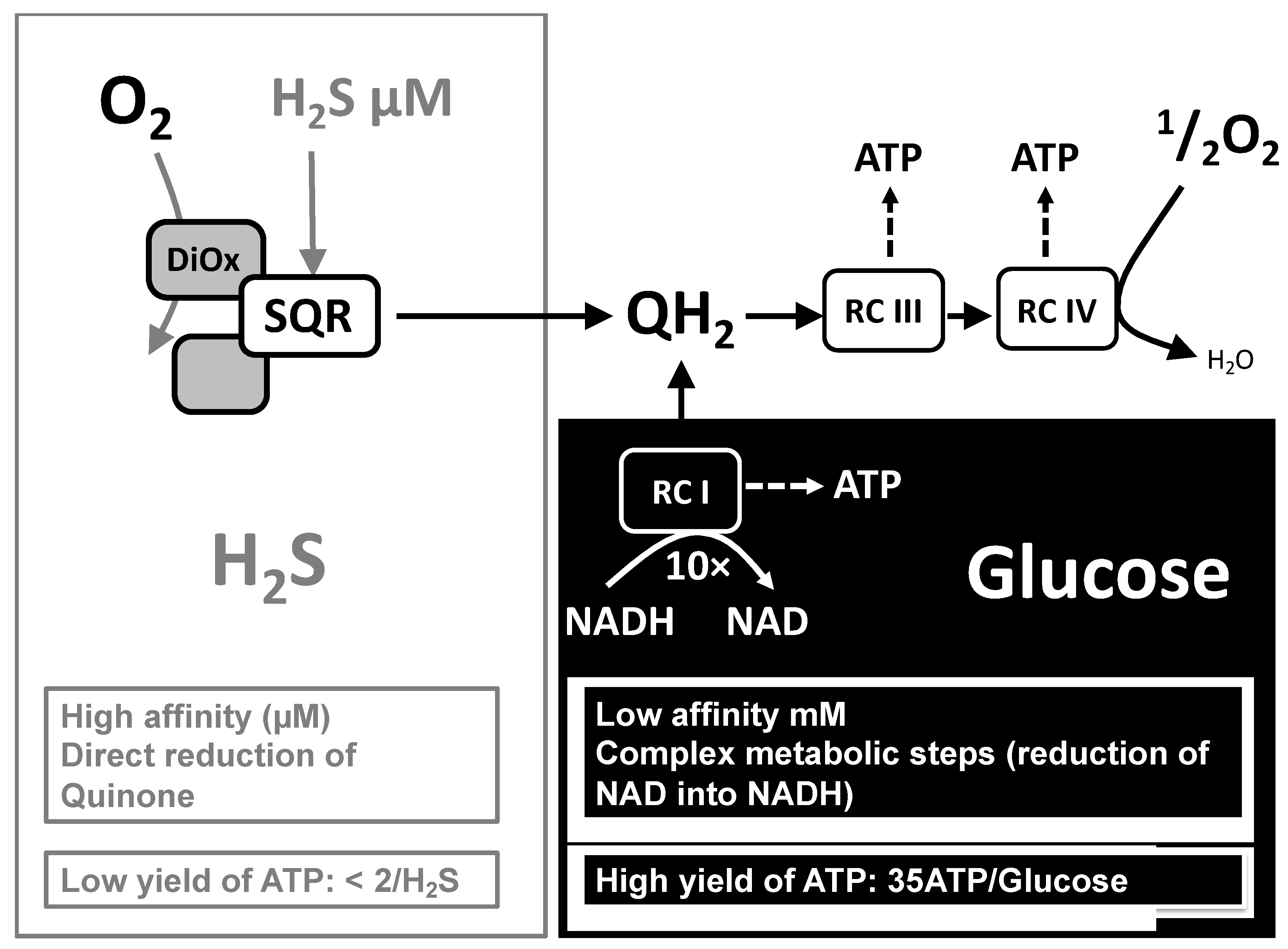{kind=link}
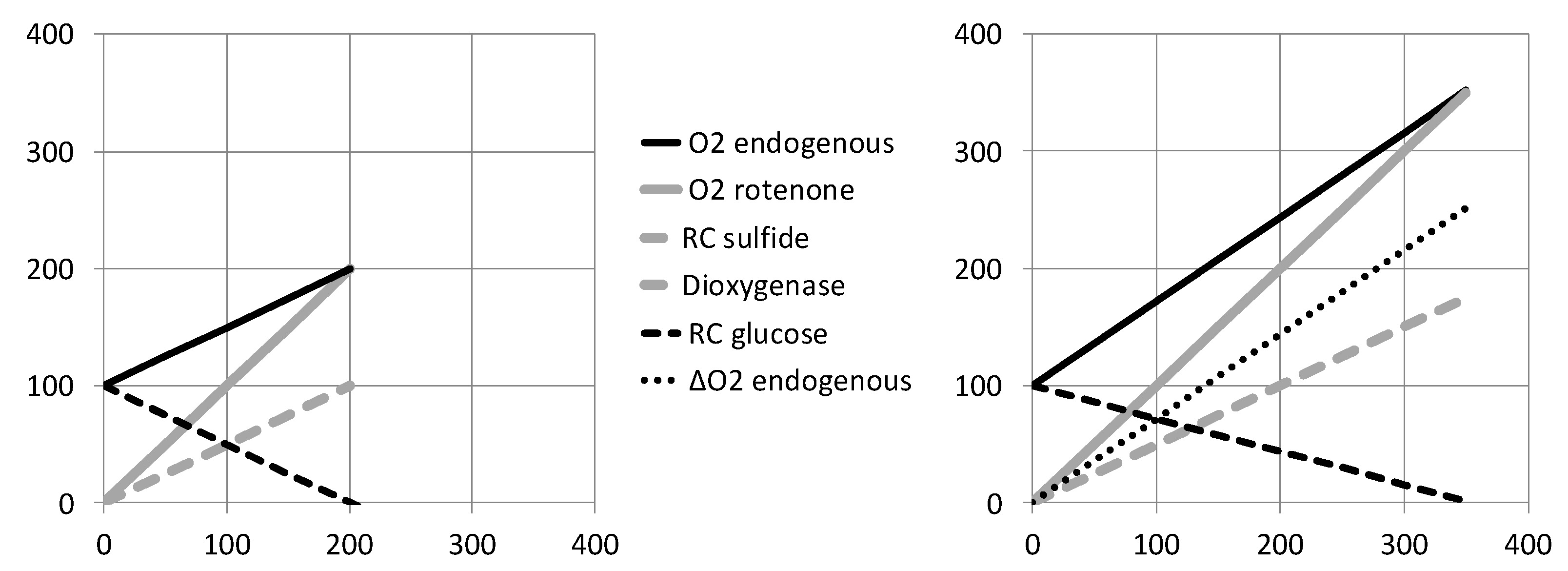{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
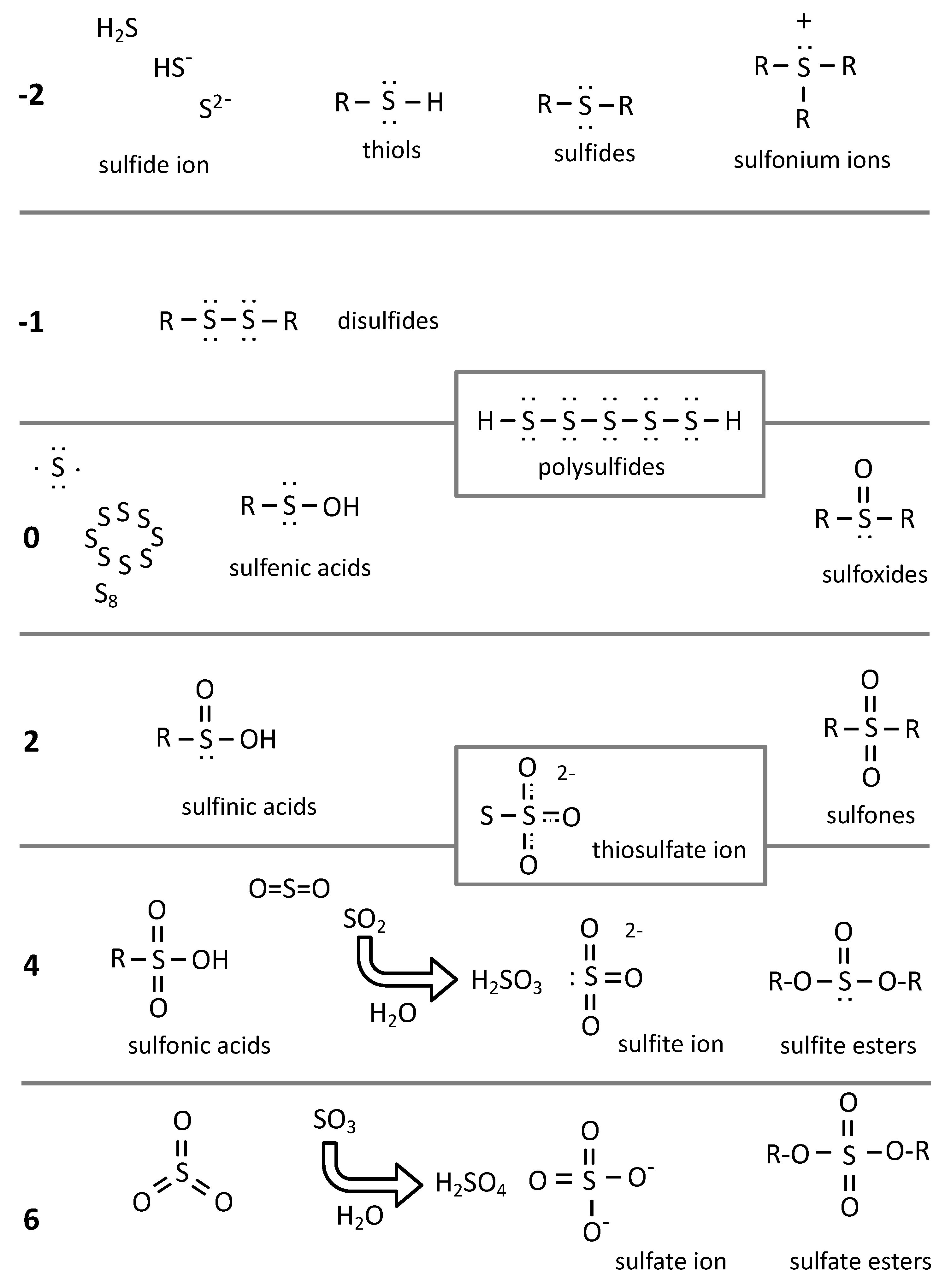
2. Chemistry
3. Oxidation of Sulfide by SQR
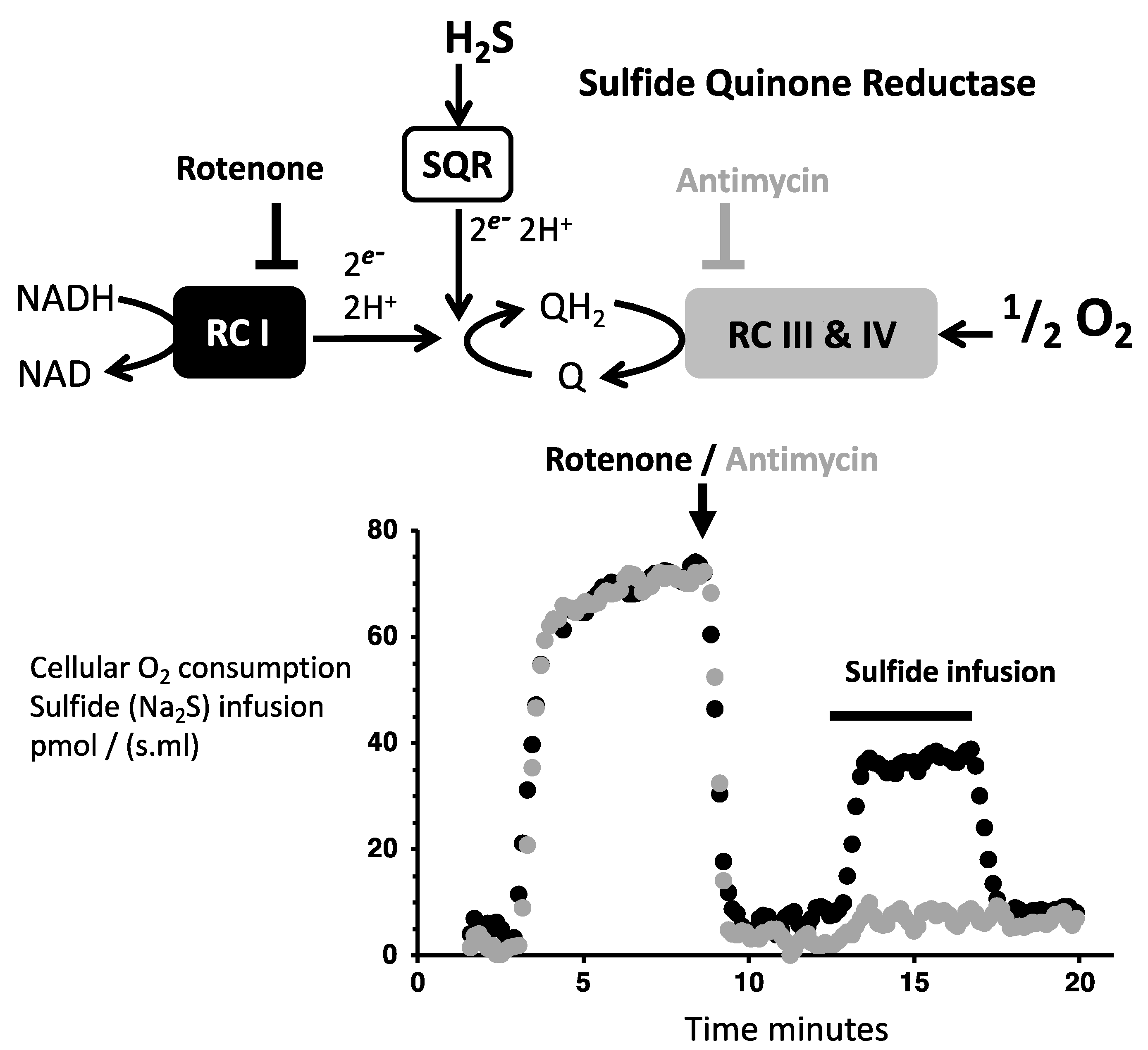
3.1. Evidencing Mitochondrial SQR Activity
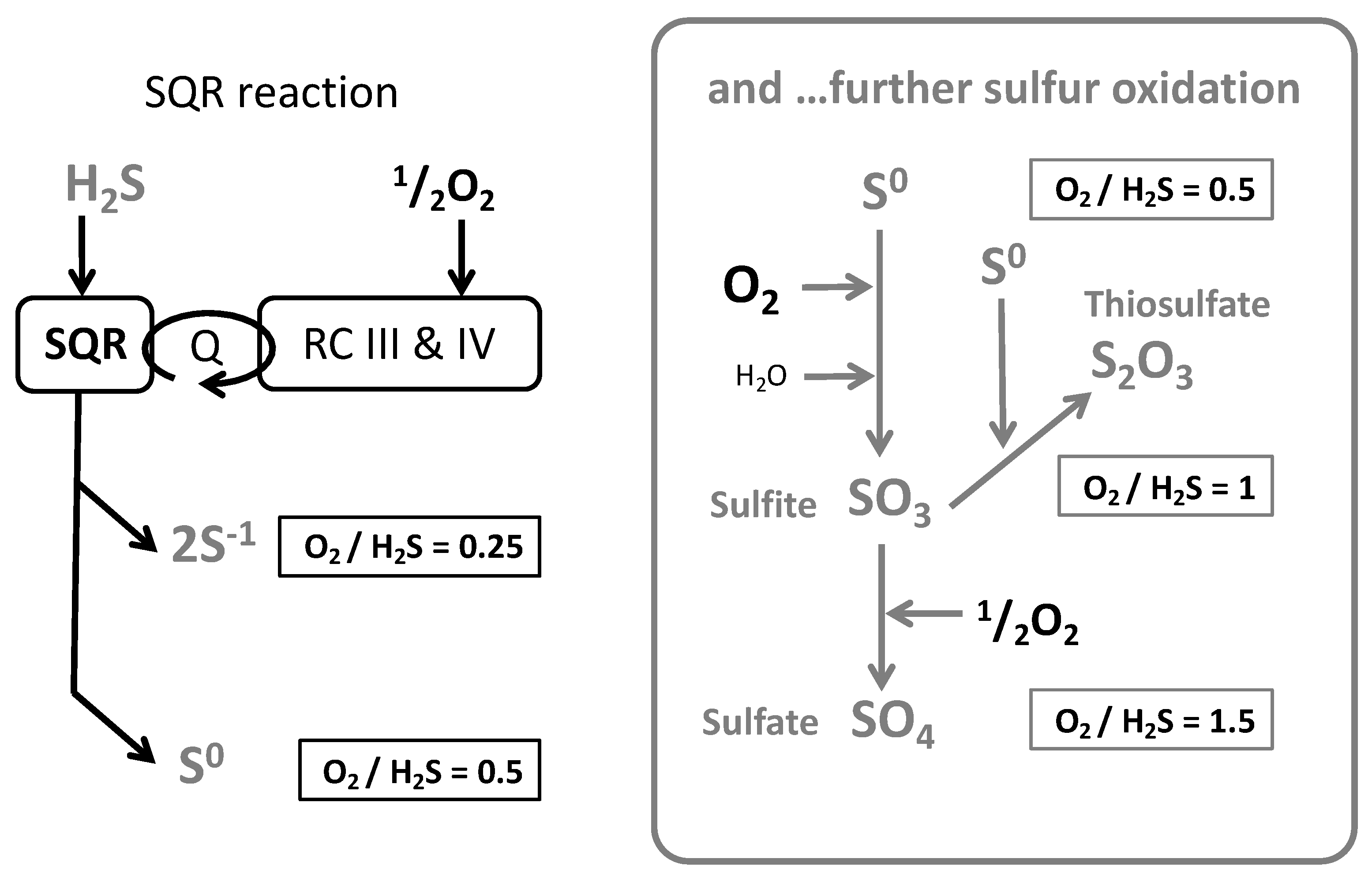
3.2. Oxygen (O2) to Sulfide Ratio
3.3. Infusions in Presence/Absence of Endogenous Respiration
4. A Model for Sulfide Oxidation with Preservation of Cellular ATP Turnover
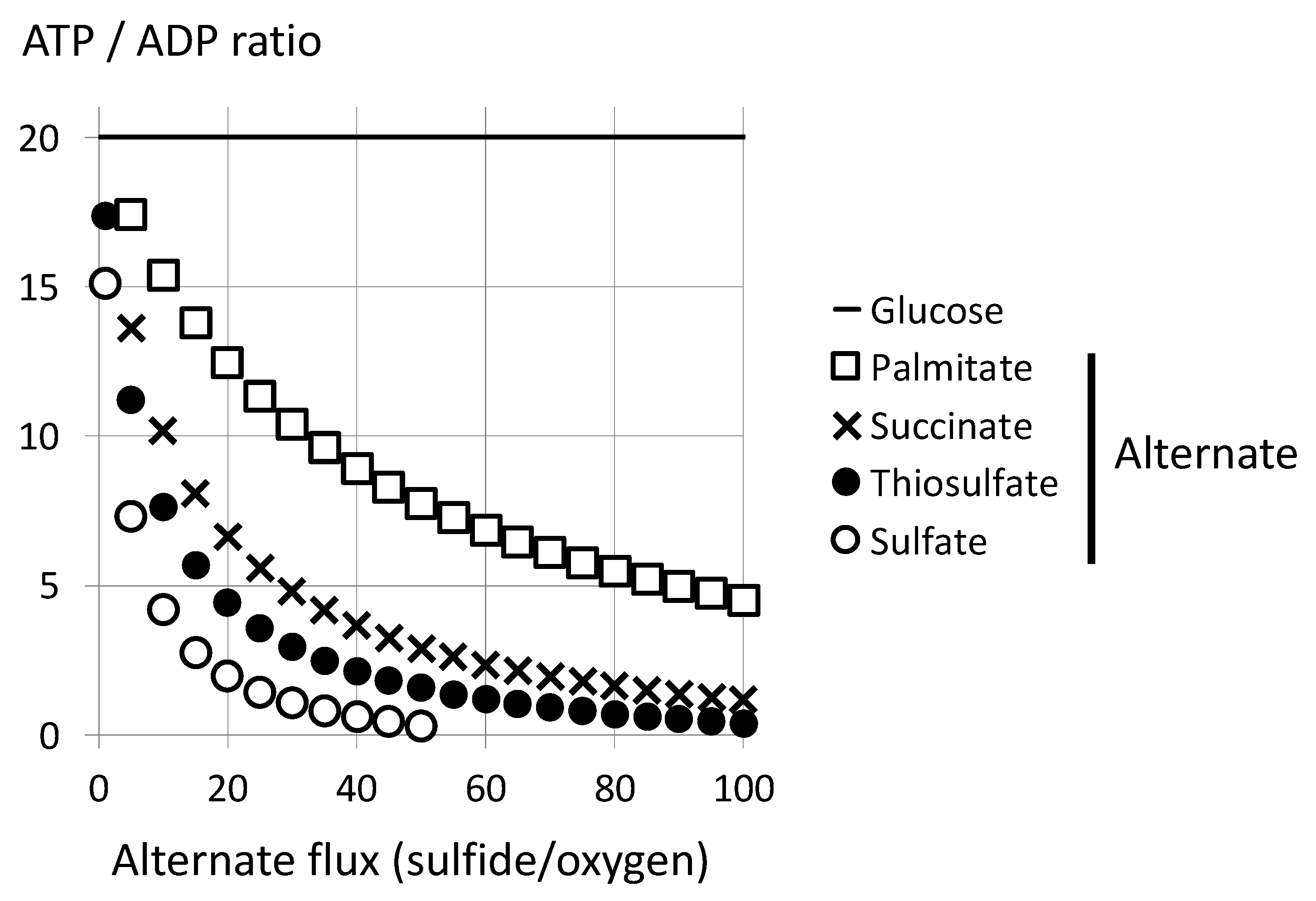
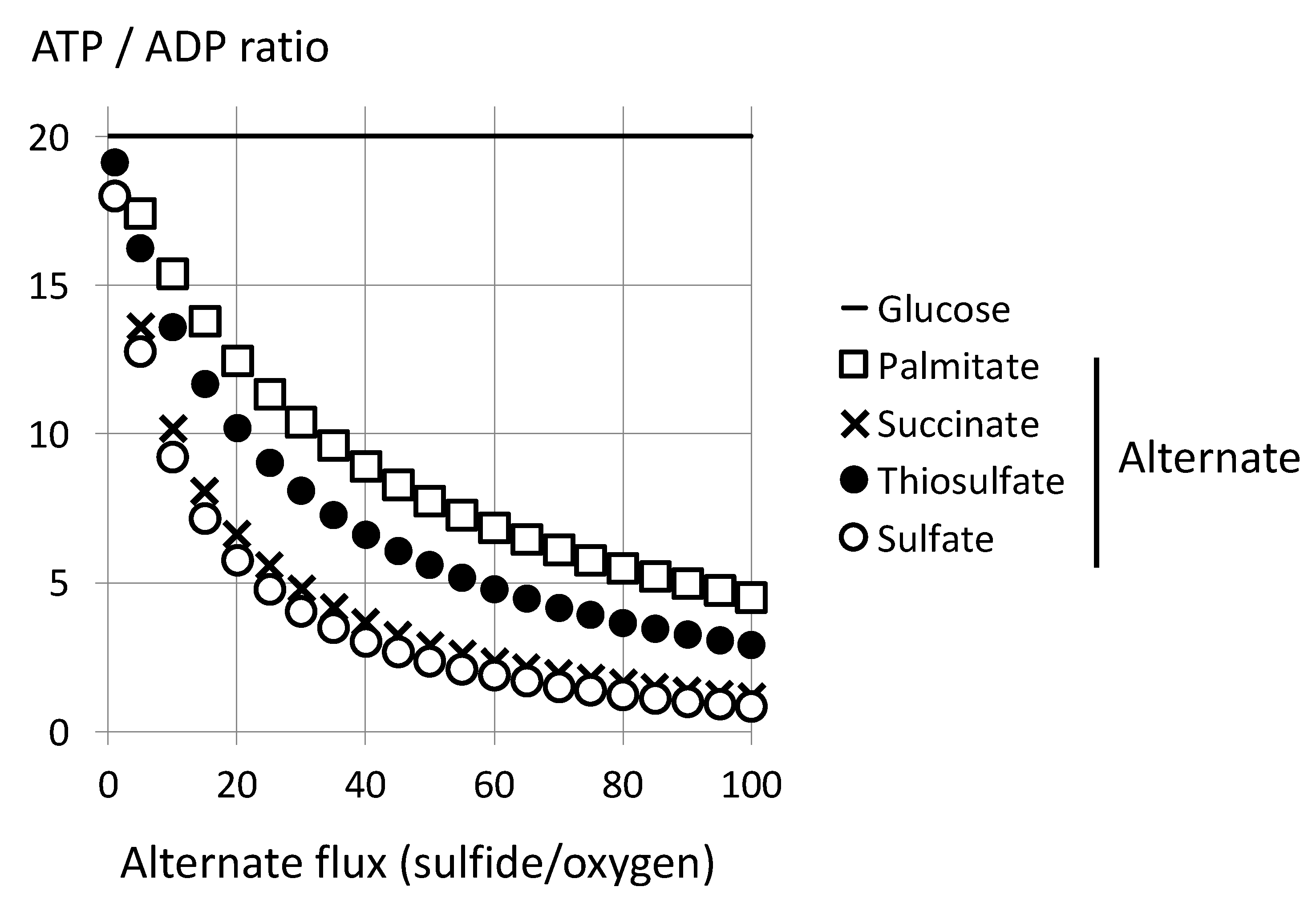
4.1. Limitation by Oxygen Supply: A Marginal Sulfide Flux Impacts on Cellular Bioenergetics
4.2. Limitation by Respiratory Chain Activity
5. Consequences of and Adaptations to Limitation in ATP Regeneration Rate
5.1. Timecourse and Cellular Content in Adenine Nucleotides
5.2. Adaptive Response: Decrease in Cellular ATP Turnover and Its Consequences
5.3. Sustained Sulfide Oxidation Recalls the Effect of Mitochondrial Poisoning
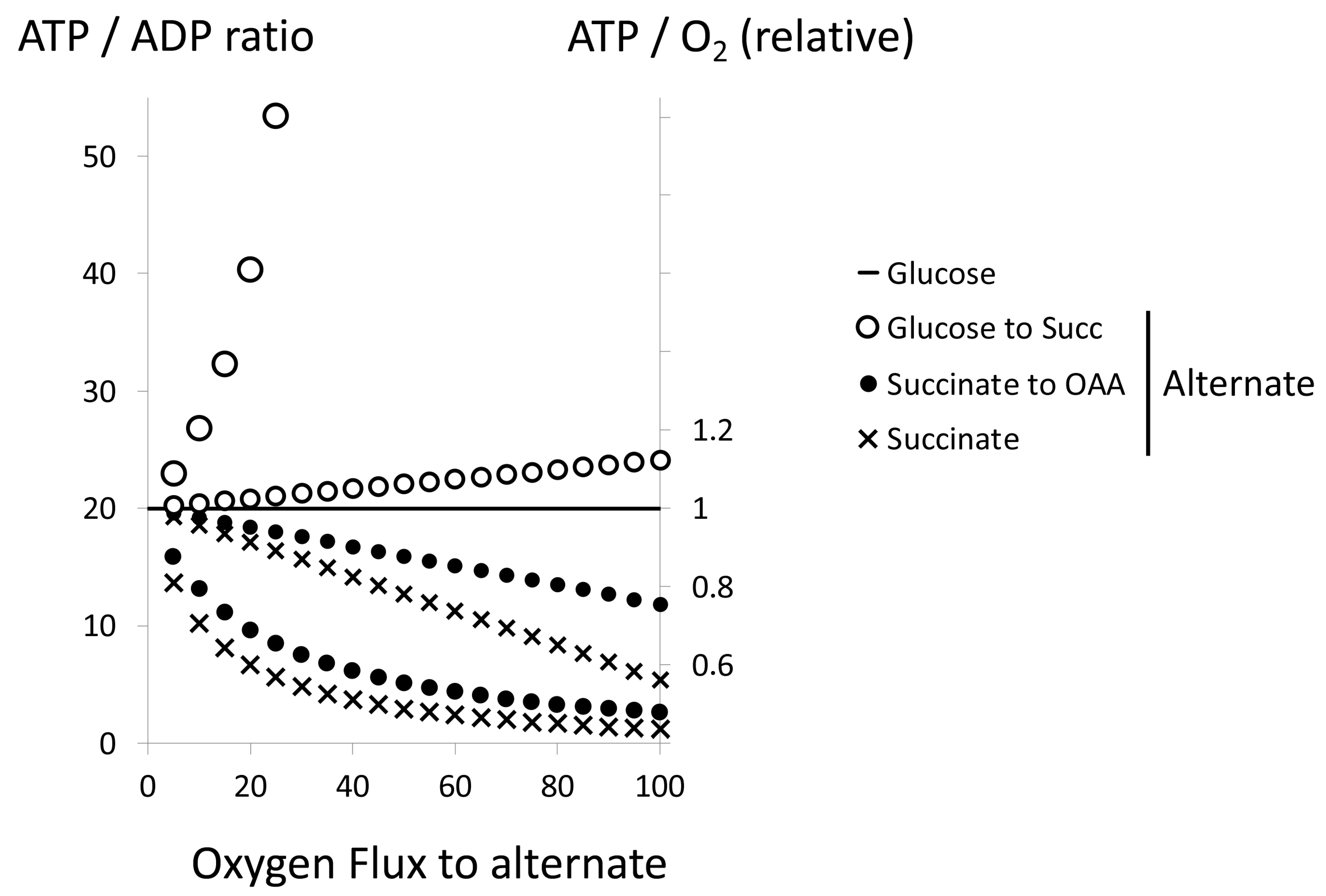
6. Sulfide Oxidation Entails Two Oxygen Dependent Steps with Different Affinities
7. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Ranjan, S.; Todd, Z.R.; Sutherland, J.D.; Sasselov, D.D. Sulfidic Anion Concentrations on Early Earth for Surficial Origins-of-Life Chemistry. Astrobiology 2018, 18, 1023–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, C.E.; Brown, G.C. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: Chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 2008, 40, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Katsuki, S.; Arnold, W.; Mittal, C.; Murad, F. Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J. Cyclic. Nucleotide Res. 1977, 3, 23–35. [Google Scholar] [PubMed]
- Ignarro, L.J. Nitric oxide is not just blowing in the wind. Br. J. Pharmacol. 2019, 176, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Suematsu, M.; Goda, N.; Sano, T.; Kashiwagi, S.; Egawa, T.; Shinoda, Y.; Ishimura, Y. Carbon monoxide: An endogenous modulator of sinusoidal tone in the perfused rat liver. J. Clin. Investig. 1995, 96, 2431–2437. [Google Scholar] [CrossRef]
- Davidge, K.S.; Motterlini, R.; Mann, B.E.; Wilson, J.L.; Poole, R.K. Carbon Monoxide in Biology and Microbiology: Surprising Roles for the “Detroit Perfume”. In Advances in Microbial Physiology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 56, pp. 85–167. ISBN 978-0-12-374791-4. [Google Scholar]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid Redox Signal. 2010, 13, 157–192. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Cooper, C.E. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994, 356, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Blachier, F.; Davila, A.-M.; Mimoun, S.; Benetti, P.-H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Bouillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2010, 39, 335–347. [Google Scholar] [CrossRef]
- Grieshaber, M.K.; Völkel, S. Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu. Rev. Physiol. 1998, 60, 33–53. [Google Scholar] [CrossRef]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. Production of hydrogen sulfide by the intestinal microbiota and epithelial cells and consequences for the colonic and rectal mucosa. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K. Regulation of cystathionine gamma-lyase/H2S system and its pathological implication. Front. Biosci. 2014, 19, 1355. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular Regulation and Pharmacological Inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H 2 S and polysulfide production and possible SO x production: Role of mercaptopyruvate sulfurtransferase. Br. J. Pharmacol. 2018, 175, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Linden, D.R.; Sha, L.; Mazzone, A.; Stoltz, G.J.; Bernard, C.E.; Furne, J.K.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J. Neurochem. 2008, 106, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [Green Version]
- Friederich, M.W.; Elias, A.F.; Kuster, A.; Laugwitz, L.; Larson, A.A.; Landry, A.P.; Ellwood-Digel, L.; Mirsky, D.M.; Dimmock, D.; Haven, J.; et al. Pathogenic variants in SQOR encoding sulfide:quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis. 2020, 43, 1024–1036. [Google Scholar] [CrossRef] [Green Version]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-β-synthase—Hydrogen sulfide system in Down syndrome. FEBS J. 2020, 287, 3150–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, M.; Ohnishi, T.; Toyoshima, M.; Balan, S.; Maekawa, M.; Shimamoto-Mitsuyama, C.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Ishii, T.; et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 2019, 11, e10695. [Google Scholar] [CrossRef] [PubMed]
- Gaitonde, U.B.; Sellar, R.J.; O’Hare, A.E. Long term exposure to hydrogen sulphide producing subacute encephalopathy in a child. Br. Med. J. (Clin. Res. Ed.) 1987, 294, 614. [Google Scholar] [CrossRef] [Green Version]
- Shibata, H.; Takahashi, M.; Yamaguchi, I.; Kobayashi, S. Sulfide oxidation by gene expressions of sulfide-quinone oxidoreductase and ubiquinone-8 biosynthase in Escherichia coli. J. Biosci Bioeng. 1999, 88, 244–249. [Google Scholar] [CrossRef]
- Griesbeck, C.; Schütz, M.; Schödl, T.; Bathe, S.; Nausch, L.; Mederer, N.; Vielreicher, M.; Hauska, G. Mechanism of sulfide-quinone reductase investigated using site-directed mutagenesis and sulfur analysis. Biochemistry 2002, 41, 11552–11565. [Google Scholar] [CrossRef]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [Green Version]
- Abou-Hamdan, A.; Guedouari-Bounihi, H.; Lenoir, V.; Andriamihaja, M.; Blachier, F.; Bouillaud, F. Oxidation of H2S in mammalian cells and mitochondria. Methods Enzymol. 2015, 554, 201–228. [Google Scholar]
- Abou-Hamdan, A.; Ransy, C.; Roger, T.; Guedouari-Bounihi, H.; Galardon, E.; Bouillaud, F. Positive feedback during sulfide oxidation fine-tunes cellular affinity for oxygen. Biochim. Biophys Acta 2016, 1857, 1464–1472. [Google Scholar] [CrossRef]
- Helmy, N.; Prip-Buus, C.; Vons, C.; Lenoir, V.; Abou-Hamdan, A.; Guedouari-Bounihi, H.; Lombès, A.; Bouillaud, F. Oxidation of hydrogen sulfide by human liver mitochondria. Nitric Oxide 2014, 41, 105–112. [Google Scholar] [CrossRef]
- Cohen, H.J.; Drew, R.T.; Johnson, J.L.; Rajagopalan, K.V. Molecular basis of the biological function of molybdenum: The relationship between sulfite oxidase and the acute toxicity of bisulfite and SO2. Proc. Natl. Acad. Sci. USA 1973, 70, 3655–3659. [Google Scholar] [CrossRef] [Green Version]
- Claerhout, H.; Witters, P.; Régal, L.; Jansen, K.; Van Hoestenberghe, M.-R.; Breckpot, J.; Vermeersch, P. Isolated sulfite oxidase deficiency. J. Inherit. Metab. Dis. 2018, 41, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Mozo, J.; Ferry, G.; Studeny, A.; Pecqueur, C.; Rodriguez, M.; Boutin, J.A.; Bouillaud, F. Expression of UCP3 in CHO cells does not cause uncoupling, but controls mitochondrial activity in the presence of glucose. Biochem. J. 2006, 393, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillaud, F.; Hammad, N.; Schwartz, L. Warburg Effect, Glutamine, Succinate, Alanine, When Oxygen Matters. Biology 2021, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Mustafa, T. Invertebrate facultative anaerobiosis. Science 1972, 178, 1056–1060. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Hochachka, P.W. Defense strategies against hypoxia and hypothermia. Science 1986, 231, 234–241. [Google Scholar] [CrossRef]
- Boutilier, R.G. Mechanisms of cell survival in hypoxia and hypothermia. J. Exp. Biol. 2001, 204, 3171–3181. [Google Scholar] [CrossRef]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [Green Version]
- Blackstone, E.; Roth, M.B. Suspended animation-like state protects mice from lethal hypoxia. Shock 2007, 27, 370–372. [Google Scholar] [CrossRef]
- Hemelrijk, S.D.; Dirkes, M.C.; van Velzen, M.H.N.; Bezemer, R.; van Gulik, T.M.; Heger, M. Exogenous hydrogen sulfide gas does not induce hypothermia in normoxic mice. Sci. Rep. 2018, 8, 3855. [Google Scholar] [CrossRef]
- Borregaard, N.; Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Auré, K.; Dubourg, O.; Jardel, C.; Clarysse, L.; Sternberg, D.; Fournier, E.; Laforêt, P.; Streichenberger, N.; Petiot, P.; Gervais-Bernard, H.; et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 2013, 81, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Buckler, K.J. Effects of exogenous hydrogen sulphide on calcium signalling, background (TASK) K channel activity and mitochondrial function in chemoreceptor cells. Pflugers Arch. 2012, 463, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabil, O.; Motl, N.; Strack, M.; Seravalli, J.; Metzler-Nolte, N.; Banerjee, R. Mechanism-based inhibition of human persulfide dioxygenase by γ-glutamyl-homocysteinyl-glycine. J. Biol. Chem. 2018, 293, 12429–12439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E.; Lassnig, B.; Kuznetsov, A.; Rieger, G.; Margreiter, R. Mitochondrial oxygen affinity, respiratory flux control and excess capacity of cytochrome c oxidase. J. Exp. Biol. 1998, 201, 1129–1139. [Google Scholar] [CrossRef]
- Hirai, D.M.; Colburn, T.D.; Craig, J.C.; Hotta, K.; Kano, Y.; Musch, T.I.; Poole, D.C. Skeletal muscle interstitial O 2 pressures: Bridging the gap between the capillary and myocyte. Microcirculation 2019, 26, e12497. [Google Scholar] [CrossRef]
- Olson, K.R. A Case for Hydrogen Sulfide Metabolism as an Oxygen Sensing Mechanism. Antioxidants 2021, 10, 1650. [Google Scholar] [CrossRef]
- Olson, K.R.; Dombkowski, R.A.; Russell, M.J.; Doellman, M.M.; Head, S.K.; Whitfield, N.L.; Madden, J.A. Hydrogen sulfide as an oxygen sensor/transducer in vertebrate hypoxic vasoconstriction and hypoxic vasodilation. J. Exp. Biol. 2006, 209, 4011–4023. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R.; Biscoe, T.J. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J. Physiol. 1992, 450, 13–31. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouillaud, F. Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio. Biomolecules 2022, 12, 361. https://doi.org/10.3390/biom12030361
Bouillaud F. Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio. Biomolecules. 2022; 12(3):361. https://doi.org/10.3390/biom12030361
Chicago/Turabian StyleBouillaud, Frédéric. 2022. "Sulfide Oxidation Evidences the Immediate Cellular Response to a Decrease in the Mitochondrial ATP/O2 Ratio" Biomolecules 12, no. 3: 361. https://doi.org/10.3390/biom12030361