


Functional Implications of Dynamic Structures of Intrinsically Disordered Proteins Revealed by High-Speed AFM Imaging
Abstract
:1. Introduction
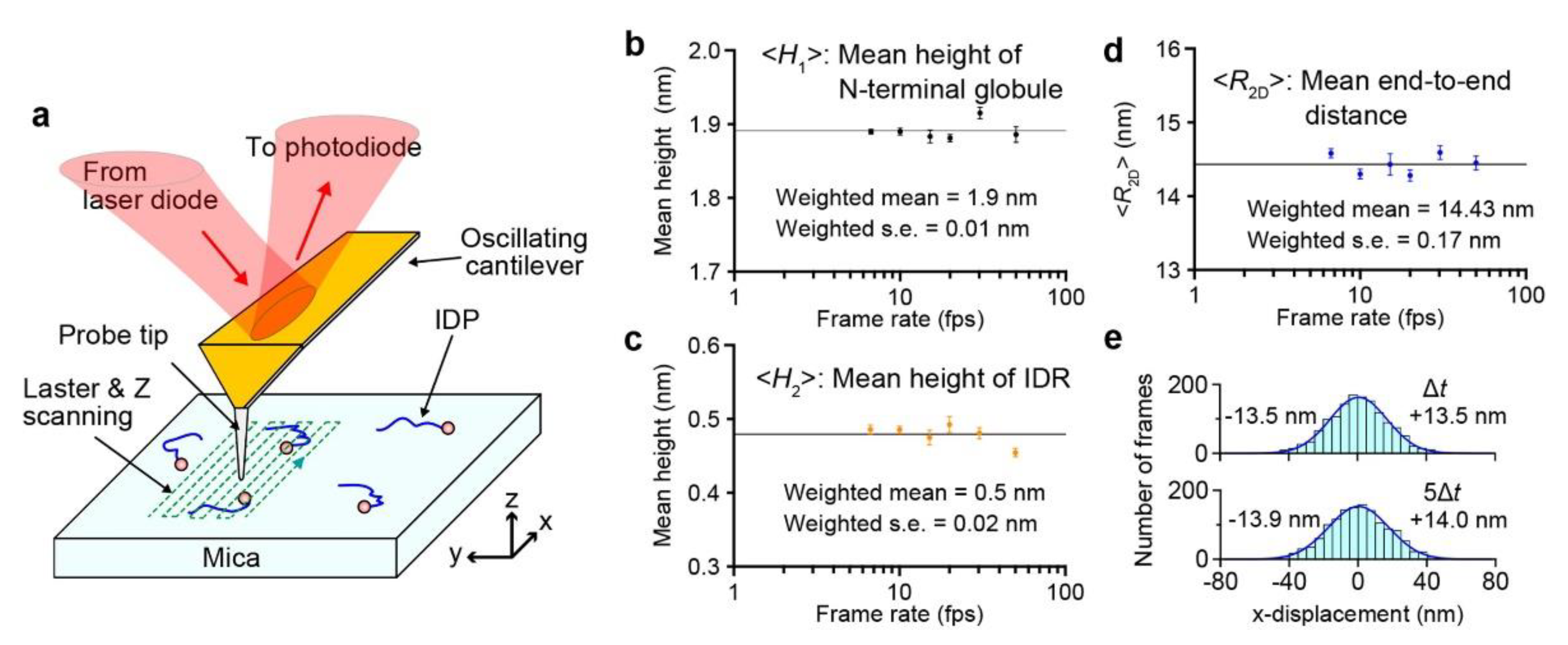
2. HS-AFM Imaging in Amplitude Modulation Mode
3. No Effect of Tip-Sample Contact on the Structure of IDPs
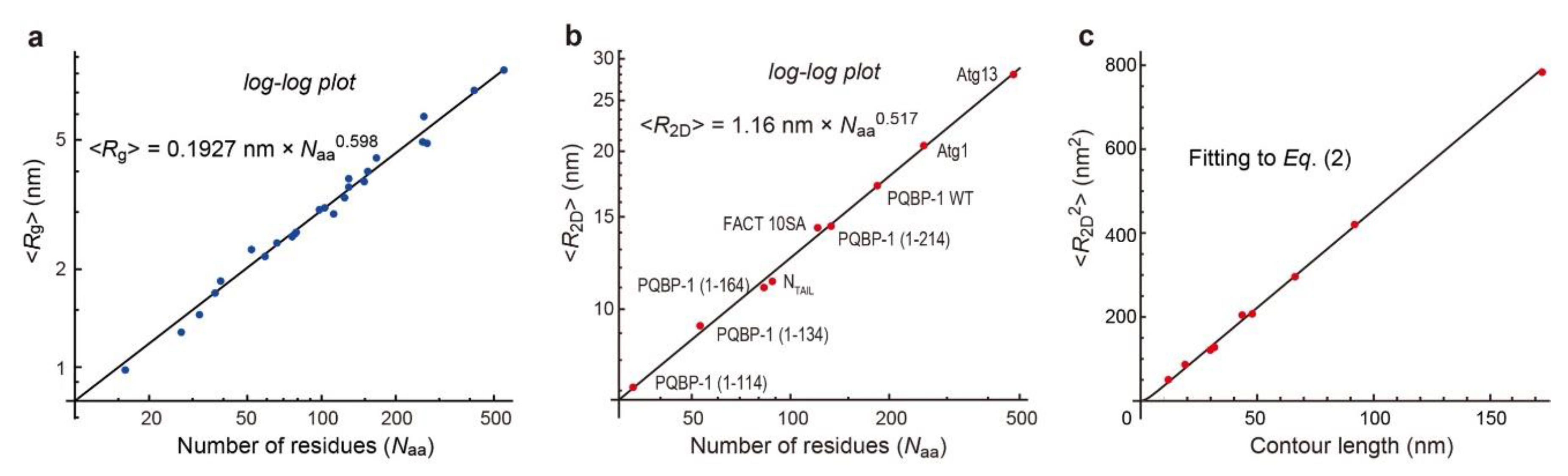
4. Constancy of Flexibility/Rigidity of Fully Disordered Regions
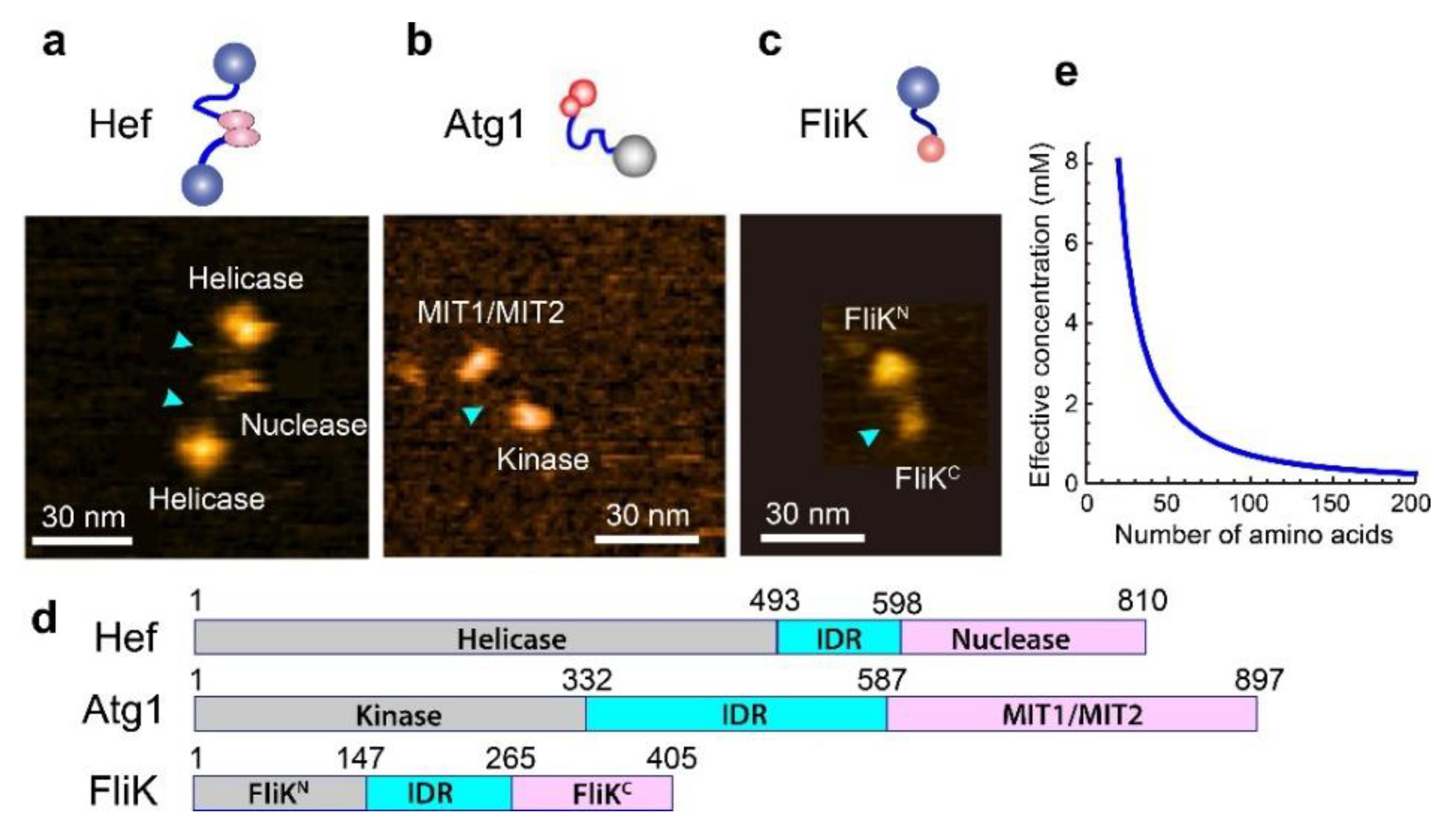
5. IDPs with Dumbbell Shape
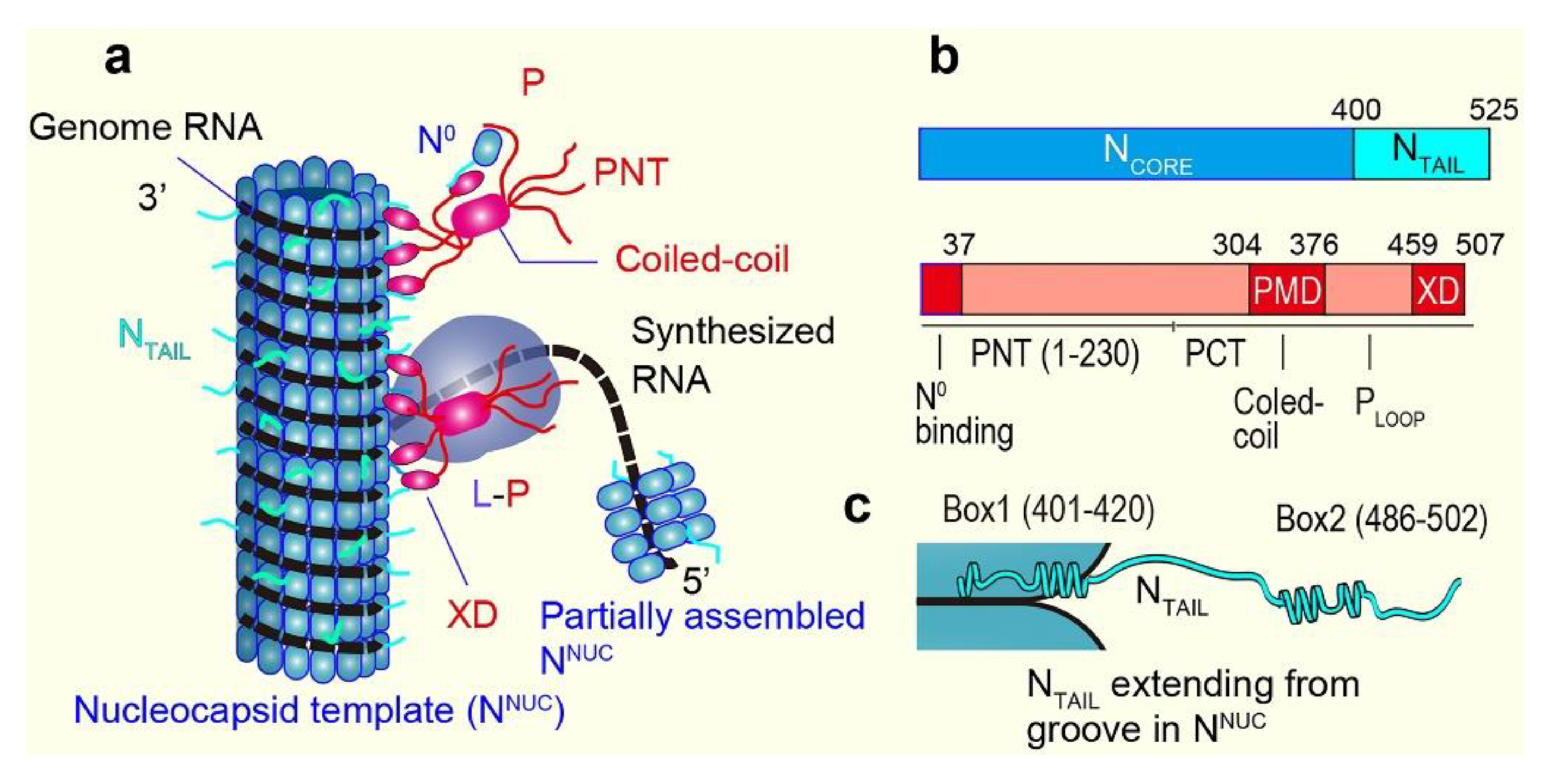
6. Measles Virus IDPs
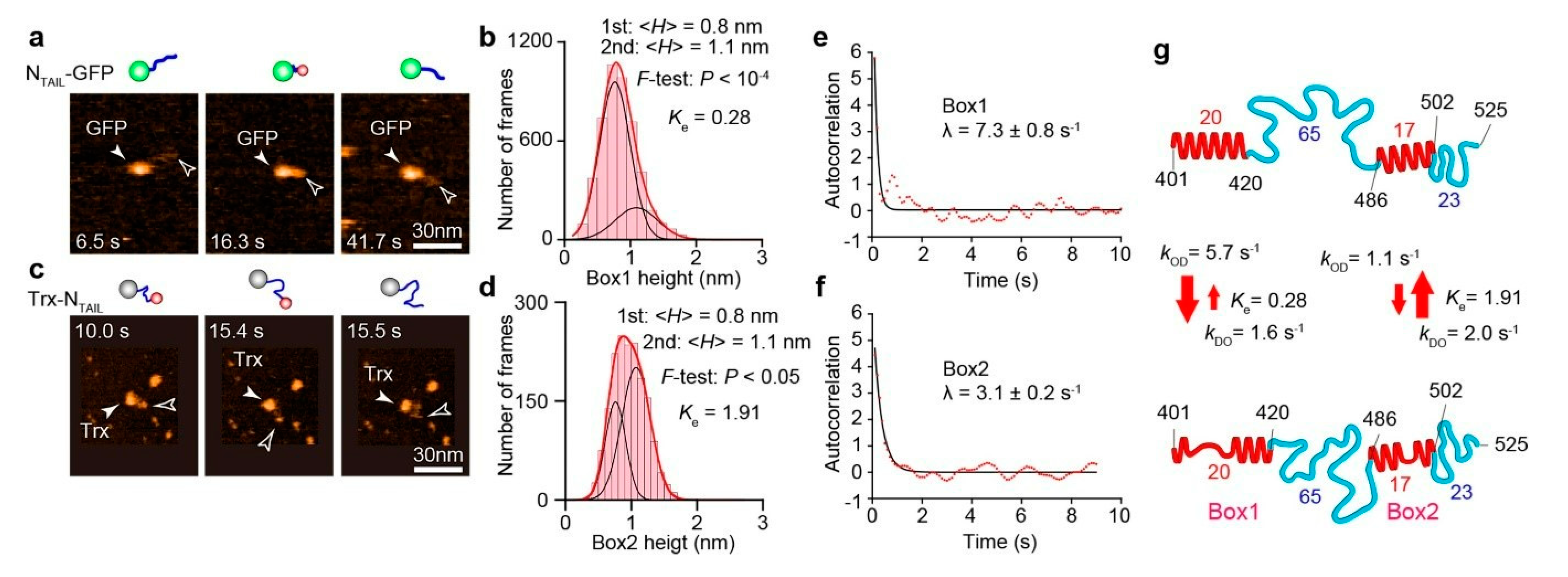
7. HS-AFM Study on NTAIL
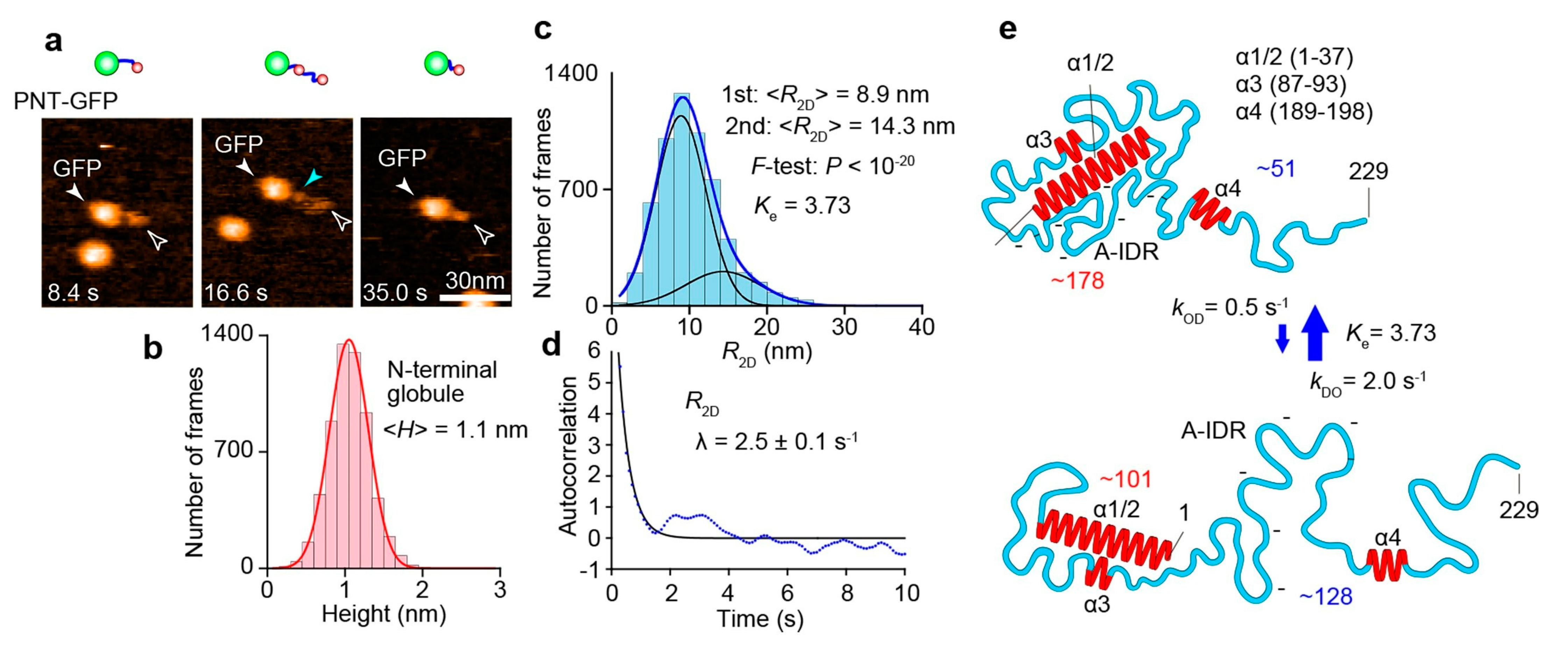
8. HS-AFM Study on PNT
9. Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Longhi, S.; Zhang, K.Y.J.; Bhatia, S. Comprehensive intrinsic disorder analysis of 6108 viral proteomes: From the extent of intrinsic disorder penetrance to functional annotation of disordered viral proteins. J. Proteome Res. 2021, 20, 2704–2713. [Google Scholar] [CrossRef]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 2006, 5, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Karlin, D.; Longhi, S.; Receveur, V.; Canard, B. The N-Terminal Domain of the Phosphoprotein of Morbilliviruses Belongs to the Natively Unfolded Class of Proteins. Virology 2002, 296, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genom. 2008, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Sigalov, A.; Aivazian, D.; Stern, L. Homooligomerization of the cytoplasmic domain of the T cell receptor zeta chain and of other proteins containing the immunoreceptor tyrosine-based activation motif. Biochemistry 2004, 43, 2049–2061. [Google Scholar] [CrossRef]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Schuler, B.; Borgia, A.; Borgia, M.B.; Heidarsson, P.O.; Holmstrom, E.D.; Nettels, D.; Sottini, A. Binding without folding —The biomolecular function of disordered polyelectrolyte complexes. Curr. Opin. Struct. Biol. 2020, 60, 66–76. [Google Scholar] [CrossRef]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Xiang, S.; Kato, M.; Wu, L.C.; Lin, Y.; Ding, M.; Zhang, Y.; Yu, Y.; McKnight, S.L. The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid-like droplets, and nuclei. Cell 2015, 163, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Murthy, A.C.; Dignon, G.L.; Kan, Y.; Zerze, G.H.; Parekh, S.H.; Mittal, J.; Fawzi, N.L. Molecular interactions underlying liquid−liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019, 26, 637–648. [Google Scholar] [CrossRef]
- Fujioka, Y.; Alam, J.M.; Noshiro, D.; Mouri, K.; Ando, T.; Okada, Y.; May, A.I.; Knorr, R.L.; Suzuki, K.; Ohsumi, Y.; et al. Phase separation organizes the site of autophagosome formation. Nature 2020, 678, 301–305. [Google Scholar] [CrossRef]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid–liquid phase separation by intrinsically disordered protein regions of viruses: Roles in viral life cycle and control of virus–host interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Uversky, V. (Ed.) Droplets of Life: Membrane-Less Organelles, Biomolecular Condensates, and Biological Liquid-Liquid Phase Separation, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2022; in press. [Google Scholar]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 539, 2570–2577. [Google Scholar] [CrossRef] [Green Version]
- Bernado, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. BioSyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Camacho-Zarco, A.R.; Schnapka, V.; Guseva, S.; Abyzov, A.; Adamski, W.; Milles, S.; Jensen, M.R.; Zidek, L.; Salvi, N.; Blackledge, M. NMR Provides Unique Insight into the Functional Dynamics and Interactions of Intrinsically Disordered Proteins. Chem. Rev. 2022, 122, 9331–9356. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef]
- Pelikan, M.; Hura, G.L.; Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009, 28, 174–189. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.A.; Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–W429. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef]
- Kooshapur, H.; Schwieters, C.D.; Tjandra, N. Conformational ensemble of disordered proteins probed by solvent paramagnetic relaxation enhancement (sPRE). Angew. Chem. Int. Ed. 2018, 57, 13519–13522. [Google Scholar] [CrossRef]
- Kodera, N.; Ando, T. Visualization of intrinsically disordered proteins by high-speed atomic force microscopy. Curr. Opin. Struct. Biol. 2022, 72, 260–266. [Google Scholar] [CrossRef]
- Yamamoto, H.; Fujioka, Y.; Suzuki, S.W.; Noshiro, D.; Suzuki, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ando, T.; Noda, N.N.; et al. The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev. Cell 2016, 38, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Kleckner, I.R.; Foster, M.P. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 2011, 1814, 942–968. [Google Scholar] [CrossRef] [Green Version]
- Vega, S.; Neira, J.L.; Marcuello, C.; Lostao, A.; Abian, O.; Velazquez-Campoy, A. NS3 protease from hepatitis C virus: Biophysical studies on an intrinsically disordered protein domain. Int. J. Mol. Sci. 2013, 14, 13282–13306. [Google Scholar] [CrossRef] [Green Version]
- Bizzarria, A.R.; Agostino, S.D.; Andolfi, L.; Cannistraro, S. A combined atomic force microscopy imaging and docking study to investigate the complex between p53 DNA binding domain and Azurin. J. Mol. Recognit. 2009, 22, 506–515. [Google Scholar] [CrossRef]
- Kodera, N.; Yamamoto, D.; Ishikawa, R.; Ando, T. Video imaging of walking myosin V by high-speed atomic force microscopy. Nature 2010, 468, 72–76. [Google Scholar] [CrossRef]
- Uchihashi, T.; Iino, R.; Ando, T.; Noji, H. High-speed atomic force microscopy reveals rotary catalysis of rotorless F1-ATPase. Science 2011, 333, 755–758. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Uchihashi, T.; Kodera, N. High-speed AFM and applications to biomolecular systems. Annu. Rev. Biophys. 2013, 42, 393–414. [Google Scholar] [CrossRef] [Green Version]
- Ando, T.; Uchihashi, T.; Scheuring, S. Filming biomolecular processes by high-speed atomic force microscopy. Chem. Rev. 2014, 114, 3120–3188. [Google Scholar] [CrossRef]
- Uchihashi, T.; Watanabe, Y.; Nakazaki, T.; Yamasaki, T.; Watanabe, H.; Maruno, T.; Ishii, K.; Uchiyama, S.; Song, C.; Murata, K.; et al. Dynamic structural states of ClpB involved in its disaggregation function. Nat. Commun. 2018, 9, 2147. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, A.; Tsunaka, Y.; Uchihashi, T.; Mayanagi, K.; Hirose, S.; Morikawa, K.; Ando, T. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. Chem. Phys. Chem. 2008, 9, 1859–1866. [Google Scholar] [CrossRef]
- Hashimoto, M.; Kodera, N.; Tsunaka, Y.; Oda, M.; Tanimoto, M.; Ando, T.; Morikawa, K.; Tate, S. Phosphorylation-coupled intramolecular dynamics of unstructured regions in chromatin remodeler FACT. Biophys. J. 2013, 104, 2222–2234. [Google Scholar] [CrossRef] [Green Version]
- Ishino, S.; Yamagami, T.; Kitamura, M.; Kodera, N.; Mori, T.; Sugiyama, S.; Ando, T.; Goda, N.; Tenno, T.; Hiroaki, H.; et al. Multiple interactions of the intrinsically disordered region between the N-terminal helicase and C-terminal nuclease domains of the archaeal Hef protein. J. Biol. Chem. 2014, 289, 21627–21639. [Google Scholar] [CrossRef] [Green Version]
- Kodera, N.; Uchida, K.; Ando, T.; Aizawa, S. Two-ball structure of the flagellar hook-length control protein FliK as revealed by high-speed atomic force microscopy. J. Mol. Biol. 2015, 427, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Kodera, N.; Noshiro, D.; Dora, S.K.; Mori, T.; Habchi, J.; Blocquel, D.; Gruet, A.; Dosnon, M.; Salladini, E.; Bignon, C.; et al. Structural and dynamics analysis of intrinsically disordered proteins by high-speed atomic force microscopy. Nat. Nanotechnol. 2021, 16, 181–189. [Google Scholar] [CrossRef]
- Terahara, N.; Kodera, N.; Uchihashi, T.; Ando, T.; Namba, K.; Minamino, T. Na+- induced structural transition of MotPS for stator assembly of the Bacillus flagellar motor. Sci. Adv. 2017, 3, eaao4119. [Google Scholar] [CrossRef]
- Watanabe-Nakayama, T.; Nawa, M.; Konno, H.; Kodera, N.; Ando, T.; Teplow, D.B.; Ono, K. Self- and cross-seeding on alpha-synuclein fibril growth kinetics and structure observed by high-speed atomic force microscopy. ACS Nano 2020, 14, 9979–9989. [Google Scholar] [CrossRef]
- Konno, H.; Watanabe-Nakayama, T.; Uchihashi, T.; Okuda, M.; Zhu, L.; Kodera; Kikuchi, Y.; Ando, T.; Taguchi, H. Dynamics of oligomer and amyloid fibril formation by yeast prion Sup35 observed by high-speed atomic force microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 7831–7836. [Google Scholar] [CrossRef]
- Imai, H.; Uchiumi, T.; Kodera, N. Direct visualization of translational GTPase factor pool formed around the archaeal ribosomal P-stalk by high-speed AFM. Proc. Natl. Acad. Sci. USA 2020, 117, 32386–32394. [Google Scholar] [CrossRef]
- Kodera, N.; Ando, T. Guide to studying intrinsically disordered proteins by high-speed atomic force microscopy. Methods 2022, 207, 44–56. [Google Scholar] [CrossRef]
- Ando, T. High-Speed Atomic Force Microscopy in Biology, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–319. [Google Scholar]
- Jang, J.; Schatz, G.C.; Ratner, M.A. Capillary force in atomic force microscopy. J. Chem. Phys. 2004, 120, 1157. [Google Scholar] [CrossRef]
- Kohn, J.E.; Millett, I.S.; Jacob, J.; Zagrovic, B.; Dillon, T.M.; Cingel, N.; Dothager, R.S.; Seifert, S.; Thiyagarajan, P.; Sosnick, T.R.; et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl Acad. Sci. USA 2004, 101, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Pérez, J.; Vachette, P.; Russo, D.; Desmadril, M.; Durand, D. Heat-induced unfolding of neocarzinostatin, a small all-beta protein investigated by small-angle X-ray scattering. J. Mol. Biol. 2001, 308, 721–743. [Google Scholar] [CrossRef]
- Cantor, C.R.; Schimmel, P.R. Configuration statics of polymer chain. In Biophysical Chemistry, Part III: The Behavior of Biological Macromolecules; W. H. Freeman and Company: San Francisco, CA, USA, 1980; pp. 879–1018. [Google Scholar]
- Cordeiro, T.N.; Herranz-Trillo, F.; Urbanek, A.; Estaña, A.; Cortés, J.; Sibille, N.; Bernadó, P. Structural characterization of highly flexible proteins by small-angle scattering. In Biological Small Angle Scattering: Techniques, Strategies and Tips; Chaudhuri, B., Muñoz, I., Qian, S., Urban, V., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2017; Volume 1009, pp. 107–129. [Google Scholar]
- Boze, H.; Marlin, T.; Durand, D.; Pérez, J.; Vernhet, A.; Canon, F.; Sarni-Manchado, P.; Cheynier, V.; Cabane, B. Proline-rich salivary proteins have extended conformations. Biophys. J. 2010, 99, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Nairn, K.M.; Lyons, R.E.; Mulder, R.J.; Mudie, S.T.; Cookson, D.J.; Lesieur, E.; Kim, M.; Lau, D.; Scholes, F.H.; Elvin, C.M. A synthetic resilin is largely unstructured. Biophys. J. 2008, 95, 3358–3365. [Google Scholar] [CrossRef] [Green Version]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR characterization of long-range order in intrinsically disordered proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar] [CrossRef]
- Mylonas, E.; Hascher, A.; Bernadó, P.; Blackledge, M.; Mandelkow, E.; Svergun, D.I. Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry 2008, 47, 10345–10353. [Google Scholar] [CrossRef]
- Rivetti, C.; Guthold, M.; Bustamante, C. Scanning force microscopy of DNA deposited onto mica: Equilibration versus kinetic trapping studied by statistical polymer chain analysis. J. Mol. Biol. 1996, 264, 919–932. [Google Scholar] [CrossRef]
- Baschnagel, J.; Meyer, H.; Wittmer, J.; Kuli´c, I.; Mohrbach, H.; Ziebert, F.; Nam, G.-M.; Lee, N.-K.; Johner, A. Semiflexible chains at surfaces: Worm-like chains and beyond. Polymers 2016, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Kirk, J.; Ilg, P. Chain dynamics in polymer melts at flat surfaces. Macromolecules 2017, 50, 3703–3718. [Google Scholar] [CrossRef] [Green Version]
- Lenton, S.; Seydel, T.; Nylander, T.; Holt, C.; Hartlein, M.; Teixeira, S.; Zaccai, G. Dynamic footprint of sequestration in the molecular fluctuations of osteopontin. J. R. Soc. Interface 2015, 12, 0506. [Google Scholar] [CrossRef]
- Konno, T.; Tanaka, N.; Kataoka, M.; Takano, E.; Maki, M. A circular dichroism study of preferential hydration and alcohol effects on a denatured protein, pig calpastatin domain I. Biochim. Biophys. Acta 1997, 1342, 73–82. [Google Scholar] [CrossRef]
- Gazi, A.D.; Bastaki, M.; Charova, S.N.; Gkougkoulia, E.A.; Kapellios, E.A.; Panopoulos, N.J.; Kokkinidis, M. Evidence for a coiled-coil interaction mode of disordered proteins from bacterial type III secretion systems. J. Biol. Chem. 2008, 283, 34062–34068. [Google Scholar] [CrossRef] [Green Version]
- Kjaergaard, M.; Nørholm, A.B.; Hendus-Altenburger, R.; Pedersen, S.F.; Poulsen, F.M.; Kragelund, B.B. Temperature-dependent structural changes in intrinsically disordered proteins: Formation of alpha-helices or loss of polyproline II? Protein Sci. 2010, 19, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Paz, A.; Zeev-Ben-Mordehai, T.; Lundqvist, M.; Sherman, E.; Mylonas, E.; Weiner, L.; Haran, G.; Svergun, D.I.; Mulder, F.A.A.; Sussman, J.L.; et al. Biophysical characterization of the unstructured cytoplasmic domain of the human neuronal adhesion protein neuroligin 3. Biophys. J. 2008, 95, 1928–1944. [Google Scholar] [CrossRef] [Green Version]
- Ruskamo, S.; Chukhlieb, M.; Vahokoski, J.; Bhargav, S.P.; Liang, F.; Kursula, I.; Kursula, P. Juxtanodin is an intrinsically disordered F-actin-binding protein. Sci. Rep. 2012, 2, 899. [Google Scholar] [CrossRef]
- Wells, M.; Tidow, H.; Rutherford, T.J.; Markwick, P.; Jensen, M.R.; Mylonas, E.; Svergun, D.I.; Blackledge, M.; Fersht, A.R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 2008, 105, 5762–5767. [Google Scholar] [CrossRef] [Green Version]
- Cragnell, C.; Durand, D.; Cabane, B.; Skepö, M. Coarse-grained modelling of the intrinsically disordered protein Histatin 5 in solution. Monte Carlo simulations in combination with SAXS. Proteins 2016, 84, 777–791. [Google Scholar] [CrossRef]
- Fuertes, G.; Banterle, N.; Ruff, K.M.; Chowdhury, A.; Mercadante, D.; Koehler, C.; Kachala, M.; Girona, G.E.; Milles, S.; Mishra, A.; et al. Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proc. Natl. Acad. Sci. USA 2017, 114, E6342–E6351. [Google Scholar] [CrossRef] [Green Version]
- Borgia, A.; Zheng, W.; Buholzer, K.; Borgia, M.B.; Schüler, A.; Hofmann, H.; Soranno, A.; Nettels, D.; Gast, K.; Grishaev, A.; et al. Consistent view of polypeptide chain expansion in chemical denaturants from multiple experimental methods. J. Am. Chem. Soc. 2016, 138, 11714–11726. [Google Scholar] [CrossRef] [Green Version]
- Moncoq, K.; Broutin, I.; Craescu, C.T.; Vachette, P.; Ducruix, A.; Durand, D. SAXS study of the PIR domain from the Grb14 molecular Adaptor: A natively unfolded protein with a transient structure primer? Biophy. J. 2004, 87, 4056–4064. [Google Scholar] [CrossRef] [Green Version]
- Holehouse, A.S.; Garai, K.; Lyle, N.; Vitalis, A.; Pappu, R.V. Quantitative assessments of the distinct contributions of polypeptide backbone amides versus side chain groups to chain expansion via chemical denaturation. J. Am. Chem. Soc. 2015, 137, 2984–2995. [Google Scholar] [CrossRef] [Green Version]
- Vigil, D.; Blumenthal, D.K.; Heller, W.T.; Brown, S.; Canaves, J.M.; Taylor, S.S.; Trewhella, J. Conformational differences among solution structures of the type Ia, IIa and IIb protein kinase A regulatory subunit homodimers: Role of the linker regions. J. Mol. Biol. 2004, 337, 1183–1194. [Google Scholar] [CrossRef]
- Abbott, M.B.; Gaponenko, V.; Abusamhadneh, E.; Finley, N.; Li, G.; Dvoretsky, A.; Rance, M.; Solaro, R.J.; Rosevear, P.R. Regulatory domain conformational exchange and linker region flexibility in cardiac troponin C bound to cardiac troponin I. J. Biol. Chem. 2000, 275, 20610–20617. [Google Scholar] [CrossRef] [Green Version]
- Morton, C.R.; Rzechorzek, N.J.; Maman, J.D.; Kuramochi, M.; Sekiguchi, H.; Rambo, R.; Sasaki, Y.C.; Davies, O.R.; Pellegrini, L. Structural basis for the coiled-coil architecture of human CtIP. Open Biol. 2021, 11, 210060. [Google Scholar] [CrossRef]
- Hesgrove, C.S.; Nguyen, K.H.; Biswas, S.; Childs, C.A.; Shraddha, K.C.; Medina, B.X.; Alvarado, V.; Sukenik, S.; Yu, F.; Malferrari, M.; et al. Molecular Swiss army knives: Tardigrade CAHS proteins mediate desiccation tolerance through multiple mechanisms. BioRxiv 2021. [Google Scholar] [CrossRef]
- Jaiswal, S.; He, Y.; Lu, H.P. Probing functional conformation-state fluctuation dynamics in recognition binding between calmodulin and target peptide. J. Chem. Phys. 2022, 156, 055102. [Google Scholar] [CrossRef]
- Komori, K.; Fujikane, R.; Shinagawa, H.; Ishino, Y. Novel endonuclease in Archaea cleaving DNA with various branched structure. Genes Genet. Syst. 2002, 77, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Komori, K.; Hidaka, M.; Horiuchi, T.; Fujikane, R.; Shinagawa, H.; Ishino, Y. Cooperation of the N-terminal helicase and C-terminal endonuclease activities of archaeal Hef protein in processing stalled replication forks. J. Biol. Chem. 2004, 279, 53175–53185. [Google Scholar] [CrossRef] [Green Version]
- Fujikane, R.; Ishino, S.; Ishino, Y.; Forterre, P. Genetic analysis of DNA repair in the hyperthermophilic archaeon, Thermococcus kodakaraensis. Genes Genet. Syst. 2010, 85, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Gonzalez-Pedrajo, B.; Yamaguchi, K.; Aizawa, S.; Macnab, R.M. FliK, the protein responsible for flagellar hook Two-Ball Structure of FliK length control in Salmonella, is exported during hook assembly. Mol. Microbiol. 1999, 34, 295–304. [Google Scholar] [CrossRef]
- Mizuno, S.; Amida, H.; Kobayashi, N.; Aizawa, S.; Tate, S. The NMR structure of FliK, the trigger for the switch of substrate specificity in the flagellar type III secretion apparatus. J. Mol. Biol. 2011, 409, 558–573. [Google Scholar] [CrossRef]
- Yao, W.; Li, Y.; Chen, Y.; Chen, Y.; Xie, Y.; Ye, M.; Zhang, Y.; Chen, X.; Wu, X.; Feng, Y.; et al. Atg1-mediated Atg11 phosphorylation is required for selective autophagy by regulating its association with receptor proteins. Autophagy 2022, in press. [Google Scholar] [CrossRef]
- Kira, S.; Noguchi, M.; Araki, Y.; Oikawa, Y.; Yoshimori, T.; Miyahara, A.; Noda, T. Vacuolar protein Tag1 and Atg1–Atg13 regulate autophagy termination during persistent starvation in S. cerevisiae. J. Cell Sci. 2021, 134, jcs253682. [Google Scholar] [CrossRef]
- Suzuki, S.W.; Yamamoto, H.; Oikawa, Y.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Ohsumi, Y. Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc. Natl. Acad. Sci. USA 2015, 112, 3350–3355. [Google Scholar] [CrossRef] [Green Version]
- Moriya, N.; Minamino, T.; Hughes, K.T.; Macnab, R.M.; Namba, K. The type III flagellar export specificity switch is dependent on FliK ruler and a molecular clock. J. Mol. Biol. 2006, 359, 466–477. [Google Scholar] [CrossRef]
- Longhi, S. Nucleocapsid Structure and Function. In Measles. Current Topics in Microbiology and Immunology; Griffin, D.E., Oldstone, M.B.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; Volume 329, pp. 103–128. [Google Scholar]
- Bourhis, J.-M.; Canard, B.; Longhi, S. Structural disorder within the replicative complex of measles virus: Functional implications. Virology 2006, 344, 94–110. [Google Scholar] [CrossRef] [Green Version]
- Bignon, C.; Troilo, F.; Gianni, S.; Longhi, S. Modulation of measles virus NTAIL interactions through fuzziness and sequence features of disordered binding sites. Biomolecules 2019, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Guseva, S.; Milles, S.; Blackledge, M.; Ruigrok, R.W.H. The nucleoprotein and phosphoprotein of measles virus. Front. Microbiol. 2019, 10, 1832. [Google Scholar] [CrossRef] [Green Version]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Structural virology. Near-atomic Cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef]
- Vreede, F.T.; Jung, T.E.; Brownlee, G.G. Model suggesting that replication of influenza virus Is regulated by stabilization of replicative intermediates. J. Virol. 2004, 78, 9568–9572. [Google Scholar] [CrossRef] [Green Version]
- Bloyet, L.M.; Brunel, J.; Dosnon, M.; Hamon, V.; Erales, J.; Gruet, A.; Lazert, C.; Bignon, C.; Roche, P.; Longhi, S.; et al. Modulation of re-initiation of measles virus transcription at intergenic regions by PXD to Ntail binding strength. PLoS Pathog. 2016, 12, e1006058. [Google Scholar] [CrossRef] [Green Version]
- Mavrakis, M.; Iseni, F.; Mazza, C.; Schoehn, G.; Ebel, C.; Gentzel, M.; Franz, T.; Ruigrok, R.W. Isolation and characterisation of the rabies virus N0-P complex produced in insect cells. Virology 2003, 305, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Communie, G.; Ribeiro, E.A., Jr.; Martinez, N.; Desfosses, A.; Salmon, L.; Mollica, L.; Gabel, F.; Jamin, M.; Longhi, S.; et al. Intrinsic disorder in measles virus nucleocapsids. Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Gely, S.; Lowry, D.F.; Bernard, C.; Jensen, M.R.; Blackledge, M.; Costanzo, S.; Bourhis, J.-M.; Darbon, H.; Daughdrill, G.; Longhi, S. Solution structure of the C-terminal X domain of the measles virus phosphoprotein and interaction with the intrinsically disordered C-terminal domain of the nucleoprotein. J. Mol. Recognit. 2010, 23, 435–447. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Romero, P.; Uversky, V.N.; Dunker, A.K. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef]
- Morin, B.; Bourhis, J.-M.; Belle, V.; Woudstra, M.; Carrière, F.; Guigliarelli, B.; Fournel, A.; Longhi, S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. J. Phys. Chem. B. 2006, 110, 20596–20608. [Google Scholar] [CrossRef]
- Belle, V.; Rouger, S.; Costanzo, S.; Liquière, E.; Strancar, J.; Guigliarelli, B.; Fournel, A.; Longhi, S. Mapping α-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins 2008, 73, 973–988. [Google Scholar] [CrossRef]
- Longhi, S.; Belle, V.; Fournel, A.; Guigliarelli, B.; Carrière, F. Probing structural transitions in both structured and disordered proteins using site-directed spin-labeling EPR spectroscopy. J. Pept. Sci. 2011, 17, 315–328. [Google Scholar] [CrossRef]
- Kingston, R.L.; Hamel, D.J.; Gay, L.S.; Dahlquist, F.W.; Matthews, B.W. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [Google Scholar] [CrossRef] [Green Version]
- Bourhis, J.M.; Receveur- Bréchot, V.; Oglesbee, M.; Zhang, X.; Buccellato, M.; Darbon, H.; Canard, B.; Finet, S.; Longhi, S. The intrinsically disordered C-terminal domain of the measles virus nucleoprotein interacts with the C-terminal domain of the phosphoprotein via two distinct sites and remains predominantly unfolded. Protein Sci. 2005, 14, 1975–1992. [Google Scholar] [CrossRef]
- Plumet, S.; Duprex, W.P.; Gerlier, D. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 2005, 79, 6900–6908. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Houben, K.; Lescop, E.; Blanchard, L.; Ruigrok, R.W.H.; Blackledge, M. Quantitative conformational analysis of partially folded proteins from residual dipolar couplings: Application to the molecular recognition element of Sendai virus nucleoprotein. J. Am. Chem. Soc. 2008, 130, 8055–8061. [Google Scholar] [CrossRef]
- Habchi, J.; Mamelli, L.; Darbon, H.; Longhi, S. Structural disorder within henipavirus nucleoprotein and phosphoprotein: From predictions to experimental assessment. PLoS ONE 2010, 5, e11684. [Google Scholar] [CrossRef]
- Guryanov, S.G.; Liljeroos, L.; Kasaragod, P.; Kajander, T.; Butcher, S.J. Crystal structure of the measles virus nucleoprotein core in complex with an N-terminal region of phosphoprotein. J. Virol. 2016, 90, 2849–2857. [Google Scholar] [CrossRef]
- Milles, S.; Jensen, M.R.; Lazert, C.; Guseva, S.; Ivashchenko, S.; Communie, G.; Maurin, D.; Gerlier, D.; Ruigrok, R.W.H.; Blackledge, M. An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 2018, 4, eaat7778. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection Time | Sample Amount | Spatial Resolution | Dynamics | Molecular Weight limitation | |
|---|---|---|---|---|---|
| NMR (a) | days–month | 2–100 mg | ~0.2 nm | ps–ms (b) | <40 kDa |
| SAXS | s–h (c) | 0.1–10 mg | 1–10 nm (d) | s-min | none |
| HS-AFM | ~10 s | 0.1 μg | 2–3 nm (XY) 0.1 nm (Z) | 20–100 ms (e) | none |
| AFM | min | 0.1 μg | 2–3 nm (XY) 0.1 nm (Z) | >30 s | none |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, T. Functional Implications of Dynamic Structures of Intrinsically Disordered Proteins Revealed by High-Speed AFM Imaging. Biomolecules 2022, 12, 1876. https://doi.org/10.3390/biom12121876
Ando T. Functional Implications of Dynamic Structures of Intrinsically Disordered Proteins Revealed by High-Speed AFM Imaging. Biomolecules. 2022; 12(12):1876. https://doi.org/10.3390/biom12121876
Chicago/Turabian StyleAndo, Toshio. 2022. "Functional Implications of Dynamic Structures of Intrinsically Disordered Proteins Revealed by High-Speed AFM Imaging" Biomolecules 12, no. 12: 1876. https://doi.org/10.3390/biom12121876