
Protein Targeting to Glycogen (PTG): A Promising Player in Glucose and Lipid Metabolism

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure and Characteristics of PP1
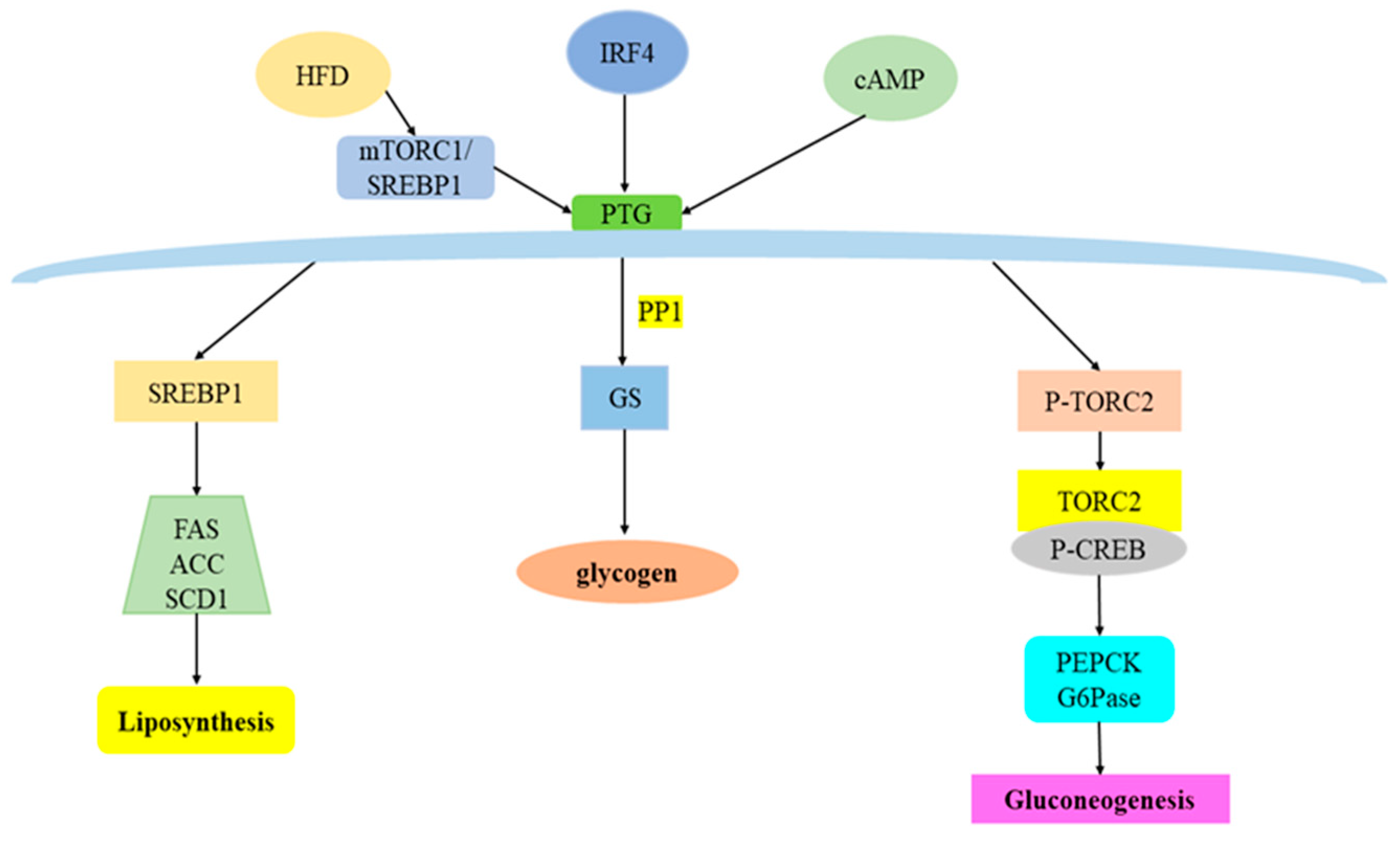
3. Overview of PTG and Its Regulatory Factors
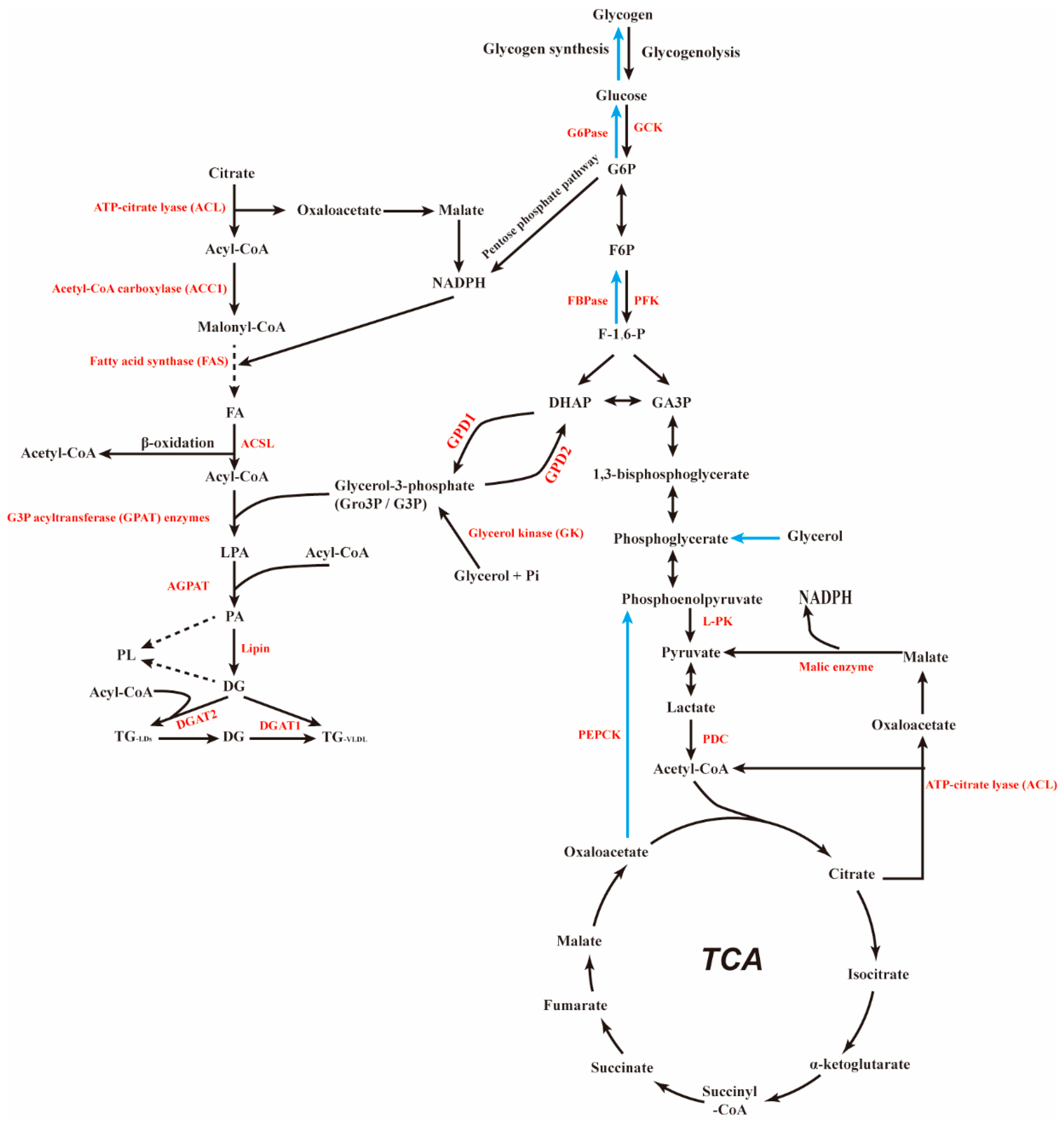
4. PTG and Glucose Metabolism
4.1. PTG and Glycogen Synthesis
4.2. PTG and Gluconeogenesis
5. PTG and Lipid Metabolism
5.1. PTG and Fatty Acid Metabolism
5.2. PTG and Fat Synthesis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adeva-Andany, M.M.; Pérez-Felpete, N.; Fernández-Fernández, C.; Donapetry-García, C.; Pazos-García, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36, e00416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Su, X.; Rohatgi, N.; Zhang, Y.; Brestoff, J.R.; Shoghi, K.I.; Xu, Y.; Semenkovich, C.F.; Harris, C.A.; Peterson, L.L.; et al. Hepatic lipids promote liver metastasis. JCI Insight 2020, 5, e136215. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Saltiel, A.R. Insulin signaling in health and disease. J. Clin. Investig. 2021, 131, e142241. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. CB 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, S.R.; Paul-Heng, M.; Krycer, J.R.; Fazakerley, D.J.; Sharland, A.F.; Hoy, A.J. Lipid and glucose metabolism in hepatocyte cell lines and primary mouse hepatocytes: A comprehensive resource for in vitro studies of hepatic metabolism. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E578–E589. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017, 13, 572–587. [Google Scholar] [CrossRef] [Green Version]
- López-Soldado, I.; Bertini, A.; Adrover, A.; Duran, J.; Guinovart, J.J. Maintenance of liver glycogen during long-term fasting preserves energy state in mice. FEBS Lett. 2020, 594, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, S.; Chen, J.; Su, Z. Unraveling the Regulation of Hepatic Gluconeogenesis. Front. Endocrinol. 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, Q.; Wang, M.; Du, Y.; Tan, X.; Xu, B.; Cheung, U.; Li, E.; Gilbert, R.G.; Tang, D. Effects of fasting on liver glycogen structure in rats with type 2 diabetes. Carbohydr. Polym. 2020, 237, 116144. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzym. Res. 2011, 2011, 329098. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Kurochkina, N.; Bhaskar, M.; Yadav, S.P.; Pant, H.C. Phosphorylation, Dephosphorylation, and Multiprotein Assemblies Regulate Dynamic Behavior of Neuronal Cytoskeleton: A Mini-Review. Front. Mol. Neurosci. 2018, 11, 373. [Google Scholar] [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. TEM 2015, 26, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Shaytan, A.; Panchenko, A.R. Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 2014, 5, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, E.K.; Sosale, N.G.; Lazzara, M.J. Cell signaling regulation by protein phosphorylation: A multivariate, heterogeneous, and context-dependent process. Curr. Opin. Biotechnol. 2016, 40, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blachnio-Zabielska, A.U.; Hady, H.R.; Markowski, A.R.; Kurianiuk, A.; Karwowska, A.; Górski, J.; Zabielski, P. Inhibition of Ceramide De Novo Synthesis Affects Adipocytokine Secretion and Improves Systemic and Adipose Tissue Insulin Sensitivity. Int. J. Mol. Sci. 2018, 19, 3995. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.H.; Yang, X.; Osipovich, A.B.; Cabrera, O.; Hayashi, M.L.; Magnuson, M.A.; Gu, G.; Kaverina, I. Glucose Regulates Microtubule Disassembly and the Dose of Insulin Secretion via Tau Phosphorylation. Diabetes 2020, 69, 1936–1947. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, J.; Li, P.; Liu, B.; Wen, X.; Yang, J. Ilexgenin A restrains CRTC2 in the cytoplasm to prevent SREBP1 maturation via AMP kinase activation in the liver. Br. J. Pharm. 2021, 179, 958–978. [Google Scholar] [CrossRef]
- Kim, H.K.; Jeong, J.; Kang, E.Y.; Go, G.W. Red Pepper (Capsicum annuum L.) Seed Extract Improves Glycemic Control by Inhibiting Hepatic Gluconeogenesis via Phosphorylation of FOXO1 and AMPK in Obese Diabetic db/db Mice. Nutrients 2020, 12, 2546. [Google Scholar] [CrossRef]
- Brady, M.J.; Printen, J.A.; Mastick, C.C.; Saltiel, A.R. Role of protein targeting to glycogen (PTG) in the regulation of protein phosphatase-1 activity. J. Biol. Chem. 1997, 272, 20198–20204. [Google Scholar] [CrossRef] [Green Version]
- Printen, J.A.; Brady, M.J.; Saltiel, A.R. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science 1997, 275, 1475–1478. [Google Scholar] [CrossRef]
- Mullard, A. Phosphatases start shedding their stigma of undruggability. Nat. Rev. Drug Discov. 2018, 17, 847–849. [Google Scholar] [CrossRef]
- Krzyzosiak, A.; Sigurdardottir, A.; Luh, L.; Carrara, M.; Das, I.; Schneider, K.; Bertolotti, A. Target-Based Discovery of an Inhibitor of the Regulatory Phosphatase PPP1R15B. Cell 2018, 174, 1216–1228.e1219. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriou, P.; Geronikaki, A.; Petrou, A. PTP1b Inhibition, A Promising Approach for the Treatment of Diabetes Type II. Curr. Top. Med. Chem. 2019, 19, 246–263. [Google Scholar] [CrossRef]
- Cohen, P.T. Protein phosphatase 1--targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Heroes, E.; Lesage, B.; Görnemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Korrodi-Gregório, L.; Esteves, S.L.; Fardilha, M. Protein phosphatase 1 catalytic isoforms: Specificity toward interacting proteins. Transl. Res. J. Lab. Clin. Med. 2014, 164, 366–391. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, A. The split protein phosphatase system. Biochem. J. 2018, 475, 3707–3723. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, D.; Huang, H.; Chen, S.; Wang, L.; Zhu, L.; Jiang, X.; Ruan, X.; Luo, X.; Cao, P.; et al. Regulation of glucose homeostasis and lipid metabolism by PPP1R3G-mediated hepatic glycogenesis. Mol. Endocrinol. 2014, 28, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhao, Q.; Zhang, J.; Zhou, L.; Zhang, W.; Chua, B.; Chen, Y.; Xu, L.; Li, P. The Protein Phosphatase 1 Complex Is a Direct Target of AKT that Links Insulin Signaling to Hepatic Glycogen Deposition. Cell Rep. 2019, 28, 3406–3422. [Google Scholar] [CrossRef] [Green Version]
- Doherty, M.J.; Young, P.R.; Cohen, P.T. Amino acid sequence of a novel protein phosphatase 1 binding protein (R5) which is related to the liver- and muscle-specific glycogen binding subunits of protein phosphatase 1. FEBS Lett. 1996, 399, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.K.; O’Doherty, R.M.; Anderson, P.; Newgard, C.B. Overexpression of protein targeting to glycogen (PTG) in rat hepatocytes causes profound activation of glycogen synthesis independent of normal hormone- and substrate-mediated regulatory mechanisms. J. Biol. Chem. 1998, 273, 26421–26425. [Google Scholar] [CrossRef]
- Cheng, A.; Zhang, M.; Crosson, S.M.; Bao, Z.Q.; Saltiel, A.R. Regulation of the mouse protein targeting to glycogen (PTG) promoter by the FoxA2 forkhead protein and by 3',5'-cyclic adenosine 5'-monophosphate in H4IIE hepatoma cells. Endocrinology 2006, 147, 3606–3612. [Google Scholar] [CrossRef] [PubMed]
- Takane, K.; Midorikawa, Y.; Yagi, K.; Sakai, A.; Aburatani, H.; Takayama, T.; Kaneda, A. Aberrant promoter methylation of PPP1R3C and EFHD1 in plasma of colorectal cancer patients. Cancer Med. 2014, 3, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ye, G.; Wang, Y.; Luo, D. MiR-4461 Inhibits Tumorigenesis of Renal Cell Carcinoma by Targeting PPP1R3C. Cancer Biother. Radiopharm. 2022, 37, 503–514. [Google Scholar] [CrossRef]
- Crosson, S.M.; Khan, A.; Printen, J.; Pessin, J.E.; Saltiel, A.R. PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance. J. Clin. Investig. 2003, 111, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Bridges, D.; Yang, Y.; Fisher, K.; Cheng, A.; Chang, L.; Meng, Z.X.; Lin, J.D.; Downes, M.; Yu, R.T.; et al. Metabolic crosstalk: Molecular links between glycogen and lipid metabolism in obesity. Diabetes 2014, 63, 2935–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, S.; Ceulemans, H.; Bollen, M.; Diplexcito, J.; Cohen, P.T. A novel glycogen-targeting subunit of protein phosphatase 1 that is regulated by insulin and shows differential tissue distribution in humans and rodents. FEBS J. 2005, 272, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Pellerin, L.; Magistretti, P.J. Protein targeting to glycogen mRNA expression is stimulated by noradrenaline in mouse cortical astrocytes. Glia 2000, 30, 382–391. [Google Scholar] [CrossRef]
- Vernia, S.; Solaz-Fuster, M.C.; Gimeno-Alcañiz, J.V.; Rubio, T.; García-Haro, L.; Foretz, M.; de Córdoba, S.R.; Sanz, P. AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J. Biol. Chem. 2009, 284, 8247–8255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, Y.; Petersen, M.C.; Zhang, D.; Vatner, D.F.; Perry, R.J.; Abulizi, A.; Haedersdal, S.; Zhang, X.M.; Butrico, G.M.; Samuel, V.T.; et al. Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 8166–8176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.Q.; Zhao, T.T.; Gui, D.K.; Gao, C.L.; Gu, J.L.; Gan, W.J.; Huang, W.; Xu, Y.; Zhou, H.; Chen, W.N.; et al. Sodium Butyrate Improves Liver Glycogen Metabolism in Type 2 Diabetes Mellitus. J. Agric. Food Chem. 2019, 67, 7694–7705. [Google Scholar] [CrossRef] [PubMed]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Zhang, D.; Guerra, M.T.; Brill, A.L.; Goedeke, L.; Nasiri, A.R.; Rabin-Court, A.; Wang, Y.; Peng, L.; Dufour, S.; et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 2020, 579, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Cherrington, A.D.; Moore, M.C.; Sindelar, D.K.; Edgerton, D.S. Insulin action on the liver in vivo. Biochem. Soc. Trans. 2007, 35, 1171–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceperuelo-Mallafré, V.; Ejarque, M.; Serena, C.; Duran, X.; Montori-Grau, M.; Rodríguez, M.A.; Yanes, O.; Núñez-Roa, C.; Roche, K.; Puthanveetil, P.; et al. Adipose tissue glycogen accumulation is associated with obesity-linked inflammation in humans. Mol. Metab. 2016, 5, 5–18. [Google Scholar] [CrossRef]
- O'Doherty, R.M.; Jensen, P.B.; Anderson, P.; Jones, J.G.; Berman, H.K.; Kearney, D.; Newgard, C.B. Activation of direct and indirect pathways of glycogen synthesis by hepatic overexpression of protein targeting to glycogen. J. Clin. Investig. 2000, 105, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, C.C.; Danos, A.M.; Brady, M.J. Central role for protein targeting to glycogen in the maintenance of cellular glycogen stores in 3T3-L1 adipocytes. Mol. Cell. Biol. 2006, 26, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, C.C.; Meredith, K.N.; Yan, L.; Brady, M.J. Protein targeting to glycogen overexpression results in the specific enhancement of glycogen storage in 3T3-L1 adipocytes. J. Biol. Chem. 2003, 278, 30835–30842. [Google Scholar] [CrossRef] [Green Version]
- López-Soldado, I.; Guinovart, J.J.; Duran, J. Increasing hepatic glycogen moderates the diabetic phenotype in insulin-deficient Akita mice. J. Biol. Chem. 2021, 296, 100498. [Google Scholar] [CrossRef]
- Ruchti, E.; Roach, P.J.; DePaoli-Roach, A.A.; Magistretti, P.J.; Allaman, I. Protein targeting to glycogen is a master regulator of glycogen synthesis in astrocytes. IBRO Rep. 2016, 1, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Petit, J.M.; Tobler, I.; Allaman, I.; Borbély, A.A.; Magistretti, P.J. Sleep deprivation modulates brain mRNAs encoding genes of glycogen metabolism. Eur. J. Neurosci. 2002, 16, 1163–1167. [Google Scholar] [CrossRef]
- Petit, J.M.; Tobler, I.; Kopp, C.; Morgenthaler, F.; Borbély, A.A.; Magistretti, P.J. Metabolic response of the cerebral cortex following gentle sleep deprivation and modafinil administration. Sleep 2010, 33, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Tadi, M.; Allaman, I.; Lengacher, S.; Grenningloh, G.; Magistretti, P.J. Learning-Induced Gene Expression in the Hippocampus Reveals a Role of Neuron -Astrocyte Metabolic Coupling in Long Term Memory. PLoS ONE 2015, 10, e0141568. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Yao, T.; Wang, R.; Guo, S.; Wang, X.; Zhou, Z.; Zhang, Y.; Zhuo, X.; Wang, R.; Li, J.Z.; et al. IRF4 in Skeletal Muscle Regulates Exercise Capacity via PTG/Glycogen Pathway. Adv. Sci. 2020, 7, 2001502. [Google Scholar] [CrossRef] [PubMed]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, S.; Qiu, X.; Li, J.; Liang, J.; Li, W.; Zhang, C.; Zhang, Z.N.; Luan, B. Glucagon-induced extracellular cAMP regulates hepatic lipid metabolism. J. Endocrinol. 2017, 234, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.A.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156. [Google Scholar] [CrossRef]
- Vilchez, D.; Ros, S.; Cifuentes, D.; Pujadas, L.; Vallès, J.; García-Fojeda, B.; Criado-García, O.; Fernández-Sánchez, E.; Medraño-Fernández, I.; Domínguez, J.; et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 2007, 10, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, P.; Bao, Z.; Wang, T.; Liu, S.; Huang, F. PGC-1α Promotes Ureagenesis in Mouse Periportal Hepatocytes through SIRT3 and SIRT5 in Response to Glucagon. Sci. Rep. 2016, 6, 24156. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Pan, Q.; Yan, H.; Zhang, K.; Guo, X.; Xu, Z.; Yang, W.; Qi, Y.; Guo, C.A.; Hornsby, C.; et al. Novel Mechanism of Foxo1 Phosphorylation in Glucagon Signaling in Control of Glucose Homeostasis. Diabetes 2018, 67, 2167–2182. [Google Scholar] [CrossRef]
- Uebi, T.; Tamura, M.; Horike, N.; Hashimoto, Y.K.; Takemori, H. Phosphorylation of the CREB-specific coactivator TORC2 at Ser(307) regulates its intracellular localization in COS-7 cells and in the mouse liver. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E413–E425. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Guo, Y.; Song, Y.; Yu, C.; Zhao, L.; Fang, L.; Kong, D.; Zhao, J.; Gao, L. Follicle-stimulating hormone enhances hepatic gluconeogenesis by GRK2-mediated AMPK hyperphosphorylation at Ser485 in mice. Diabetologia 2018, 61, 1180–1192. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Wang, S.; Tang, H.; Zhang, Y.; Zhou, F.; Zhang, L.; Zhu, Q.; Zhu, K.; Liu, Q.; Liu, Y.; et al. PPP1R3C mediates metformin-inhibited hepatic gluconeogenesis. Metabolism 2019, 98, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Gluconeogenesis and glycogenolysis in health and diabetes. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2004, 52, 375–378. [Google Scholar] [CrossRef]
- Nuttall, F.Q.; Gilboe, D.P.; Gannon, M.C.; Niewoehner, C.B.; Tan, A.W. Regulation of glycogen synthesis in the liver. Am. J. Med. 1988, 85, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef]
- Watt, M.J.; Miotto, P.M.; De Nardo, W.; Montgomery, M.K. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr. Rev. 2019, 40, 1367–1393. [Google Scholar] [CrossRef]
- Staehr, P.; Hother-Nielsen, O.; Beck-Nielsen, H. The role of the liver in type 2 diabetes. Rev. Endocr. Metab. Disord. 2004, 5, 105–110. [Google Scholar] [CrossRef]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [Green Version]
- Vasconcellos, R.; Alvarenga, É.C.; Parreira, R.C.; Lima, S.S.; Resende, R.R. Exploring the cell signalling in hepatocyte differentiation. Cell. Signal. 2016, 28, 1773–1788. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Du, W.; Liu, Y.; Cheng, M.; Wang, X.; Zhang, C.; Lv, X.; Li, F.; Zhao, S.; Hao, J. Prolonged high-glucose exposure decreased SREBP-1/FASN/ACC in Schwann cells of diabetic mice via blocking PI3K/Akt pathway. J. Cell Biochem. 2019, 120, 5777–5789. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Shimano, H. CREBH Regulates Systemic Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Li, C.; Zhang, H.; Liu, X.; Zhou, G. Functional study of PPP1R3C gene during differentiation of 3T3-L1 preadipocytes. J. Liaocheng Univ. (Nat. Sci. Ed.) 2021, 34, 95–102. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, X.; Wang, C.; Xia, Y.; Yuan, G. Protein Targeting to Glycogen (PTG): A Promising Player in Glucose and Lipid Metabolism. Biomolecules 2022, 12, 1755. https://doi.org/10.3390/biom12121755
Deng X, Wang C, Xia Y, Yuan G. Protein Targeting to Glycogen (PTG): A Promising Player in Glucose and Lipid Metabolism. Biomolecules. 2022; 12(12):1755. https://doi.org/10.3390/biom12121755
Chicago/Turabian StyleDeng, Xia, Chenxi Wang, Yue Xia, and Guoyue Yuan. 2022. "Protein Targeting to Glycogen (PTG): A Promising Player in Glucose and Lipid Metabolism" Biomolecules 12, no. 12: 1755. https://doi.org/10.3390/biom12121755