
A Focus on Astrocyte Contribution to Parkinson’s Disease Etiology

{kind=link}
{kind=link}
Abstract
:1. An Introduction to Parkinson’s Disease
2. PD as a Non-Neuronal Cell Autonomous Disease
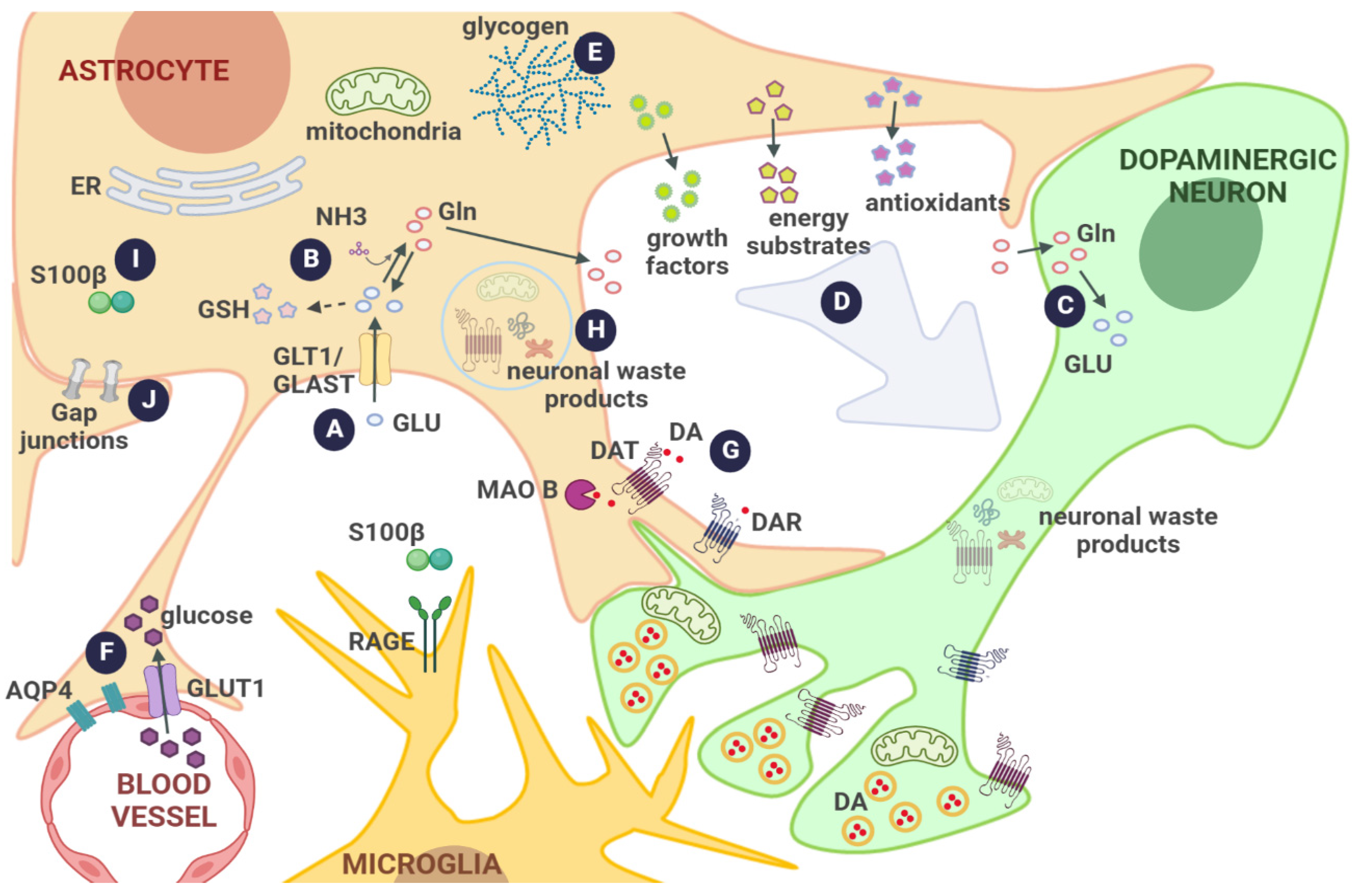
3. Interplay between Astrocytes and Dopaminergic Neurons under Physiological Conditions
4. Evidence for Astrocyte Roles in PD as a Non-Neuronal Cell Autonomous Disease
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharm. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Olanow, C.W.; Brundin, P. Parkinson’s disease and alpha synuclein: Is Parkinson’s disease a prion-like disorder? Mov. Disord. 2013, 28, 31–40. [Google Scholar] [CrossRef]
- Lunati, A.; Lesage, S.; Brice, A. The genetic landscape of Parkinson’s disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Bové, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, J.; Pötschke, C.; Liss, B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. S1), 156–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J. Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 2018, 285, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Jana, S.; Sinha, M.; Chanda, D.; Roy, T.; Banerjee, K.; Munshi, S.; Patro, B.S.; Chakrabarti, S. Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochim. Biophys. Acta 2011, 1812, 663–673. [Google Scholar] [CrossRef] [Green Version]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Kannarkat, G.T.; Boss, J.M.; Tansey, M.G. The role of innate and adaptive immunity in Parkinson’s disease. J. Park. Dis. 2013, 3, 493–514. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debré, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Venkateshappa, C.; Harish, G.; Mythri, R.B.; Mahadevan, A.; Bharath, M.M.; Shankar, S.K. Increased oxidative damage and decreased antioxidant function in aging human substantia nigra compared to striatum: Implications for Parkinson’s disease. Neurochem. Res. 2012, 37, 358–369. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Zhang, P.; Agid, Y.; Javoy-Agid, F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience 1993, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Nedergaard, M.; Verkhratsky, A. Artifact versus reality—How astrocytes contribute to synaptic events. Glia 2012, 60, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Piperi, C. Emerging role of S100B protein implication in Parkinson’s disease pathogenesis. Cell. Mol. Life Sci. 2021, 78, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef]
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem. Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Teismann, P.; Tieu, K.; Cohen, O.; Choi, D.K.; Wu, D.C.; Marks, D.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Pathogenic role of glial cells in Parkinson’s disease. Mov. Disord. 2003, 18, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Lindqvist, D.; Kaufman, E.; Brundin, L.; Hall, S.; Surova, Y.; Hansson, O. Non-motor symptoms in patients with Parkinson’s disease—Correlations with inflammatory cytokines in serum. PLoS ONE 2012, 7, e47387. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Xue, G.; Cheng, H.; Meng, P.; Lian, X.; Holscher, C.; Li, D. The GLP-1/GIP dual-receptor agonist DA5-CH inhibits the NF-kappaB inflammatory pathway in the MPTP mouse model of Parkinson’s disease more effectively than the GLP-1 single-receptor agonist NLY01. Brain Behav. 2021, 11, e2231. [Google Scholar] [CrossRef] [PubMed]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-Neuron Metabolic Crosstalk in Neurodegeneration: A Mitochondrial Perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J. Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 2006, 209, 2304–2311. [Google Scholar] [CrossRef] [Green Version]
- Veloz Castillo, M.F.; Magistretti, P.J.; Cali, C. l-Lactate: Food for Thoughts, Memory and Behavior. Metabolites 2021, 11, 548. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte glutamine synthetase: Pivotal in health and disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Hertz, E.; Juurlink, B.H.; Yu, P.H. Development of monoamine oxidase activity and monoamine effects on glutamate release in cerebellar neurons and astrocytes. Neurochem. Res. 1989, 14, 1039–1046. [Google Scholar] [CrossRef]
- Saura, J.; Kettler, R.; Da Prada, M.; Richards, J.G. Quantitative enzyme radioautography with 3H-Ro 41-1049 and 3H-Ro 19-6327 in vitro: Localization and abundance of MAO-A and MAO-B in rat CNS, peripheral organs, and human brain. J. Neurosci. 1992, 12, 1977–1999. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.; Scheffler, P.; Lohr, C. Dopamine-induced calcium signaling in olfactory bulb astrocytes. Sci. Rep. 2020, 10, 631. [Google Scholar] [CrossRef] [Green Version]
- Vaarmann, A.; Gandhi, S.; Abramov, A.Y. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J. Biol. Chem. 2010, 285, 25018–25023. [Google Scholar] [CrossRef] [Green Version]
- Xin, W.; Schuebel, K.E.; Jair, K.W.; Cimbro, R.; De Biase, L.M.; Goldman, D.; Bonci, A. Ventral midbrain astrocytes display unique physiological features and sensitivity to dopamine D2 receptor signaling. Neuropsychopharmacology 2019, 44, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Requardt, R.P.; Wilhelm, F.; Rillich, J.; Winkler, U.; Hirrlinger, J. The biphasic NAD(P)H fluorescence response of astrocytes to dopamine reflects the metabolic actions of oxidative phosphorylation and glycolysis. J. Neurochem. 2010, 115, 483–492. [Google Scholar] [CrossRef]
- Kinor, N.; Geffen, R.; Golomb, E.; Zinman, T.; Yadid, G. Dopamine increases glial cell line-derived neurotrophic factor in human fetal astrocytes. Glia 2001, 33, 143–150. [Google Scholar] [CrossRef]
- Li, A.; Guo, H.; Luo, X.; Sheng, J.; Yang, S.; Yin, Y.; Zhou, J.; Zhou, J. Apomorphine-induced activation of dopamine receptors modulates FGF-2 expression in astrocytic cultures and promotes survival of dopaminergic neurons. Faseb J. 2006, 20, 1263–1265. [Google Scholar] [CrossRef]
- Ohta, K.; Kuno, S.; Inoue, S.; Ikeda, E.; Fujinami, A.; Ohta, M. The effect of dopamine agonists: The expression of GDNF, NGF, and BDNF in cultured mouse astrocytes. J. Neurol. Sci. 2010, 291, 12–16. [Google Scholar] [CrossRef]
- Lin, L.F.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, E.L.; Fletcher, E.J.R.; Duty, S. Fibroblast growth factor 20 is protective towards dopaminergic neurons in vivo in a paracrine manner. Neuropharmacology 2018, 137, 156–163. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Makar, T.K.; Nedergaard, M.; Preuss, A.; Gelbard, A.S.; Perumal, A.S.; Cooper, A.J. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: Evidence that astrocytes play an important role in antioxidative processes in the brain. J. Neurochem. 1994, 62, 45–53. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M.; Kikkawa, Y.; Takeshima, M.; Murakami, S.; Miyoshi, K.; Sogawa, N.; Kita, T. Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 2011, 59, 435–451. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Tyacke, R.J.; Polychronis, S.; Myers, J.; Gunn, R.N.; Rabiner, E.A.; Nutt, D.; Politis, M. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: An in vivo 11C-BU99008 PET study. Brain 2019, 142, 3116–3128. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lim, T.M. Glutathione conjugates with dopamine-derived quinones to form reactive or non-reactive glutathione-conjugates. Neurochem. Res. 2010, 35, 1805–1818. [Google Scholar] [CrossRef]
- Shih, A.Y.; Erb, H.; Sun, X.; Toda, S.; Kalivas, P.W.; Murphy, T.H. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006, 26, 10514–10523. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, J.A. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef]
- Mullett, S.J.; Di Maio, R.; Greenamyre, J.T.; Hinkle, D.A. DJ-1 expression modulates astrocyte-mediated protection against neuronal oxidative stress. J. Mol. Neurosci. 2013, 49, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Mullett, S.J.; Hinkle, D.A. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J. Neurochem. 2011, 117, 375–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura-Aguilar, J.; Mannervik, B.; Inzunza, J.; Varshney, M.; Nalvarte, I.; Munoz, P. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural. Regen. Res. 2022, 17, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcon, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine receptor D3 signalling in astrocytes promotes neuroinflammation. J. Neuroinflammation 2019, 16, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.; Zhang, S.Z.; Tang, M.; Zhang, X.H.; Zhou, Z.; Yin, Y.Q.; Zhou, Q.B.; Huang, Y.Y.; Liu, Y.J.; Wawrousek, E.; et al. Suppression of neuroinflammation by astrocytic dopamine D2 receptors via alphaB-crystallin. Nature 2013, 494, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Hu, Z.; Han, X.; Wang, D.; Jiang, Q.; Ding, J.; Xiao, M.; Wang, C.; Lu, M.; Hu, G. Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of beta-arrestin2 and NLRP3. Cell Death Differ. 2018, 25, 2037–2049. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Gai, W.P.; Xu, Y.H.; Sachdev, P.; Guillemin, G.J.; Jiang, X.M.; Ballard, J.W.; Horan, M.P.; Fang, Z.M.; Chong, B.H.; et al. Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl. Neurodegener. 2013, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Sanchez, A.; Puertas-Avendaño, R.; Rodriguez-Sabate, C.; Perez-Barreto, A.; Rodriguez, M. Neuroglial transmitophagy and Parkinson’s disease. Glia 2020, 68, 2277–2299. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Aoki-Yoshino, K.; Uchihara, T.; Duyckaerts, C.; Nakamura, A.; Hauw, J.J.; Wakayama, Y. Enhanced expression of aquaporin 4 in human brain with inflammatory diseases. Acta Neuropathol. 2005, 110, 281–288. [Google Scholar] [CrossRef]
- Neal, M.L.; Boyle, A.M.; Budge, K.M.; Safadi, F.F.; Richardson, J.R. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation 2018, 15, 73. [Google Scholar] [CrossRef] [Green Version]
- Knott, C.; Wilkin, G.P.; Stern, G. Astrocytes and microglia in the substantia nigra and caudate-putamen in Parkinson’s disease. Park. Relat. Disord. 1999, 5, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Mirza, B.; Hadberg, H.; Thomsen, P.; Moos, T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 2000, 95, 425–432. [Google Scholar] [CrossRef]
- Song, Y.J.; Halliday, G.M.; Holton, J.L.; Lashley, T.; O’Sullivan, S.S.; McCann, H.; Lees, A.J.; Ozawa, T.; Williams, D.R.; Lockhart, P.J.; et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J. Neuropathol. Exp. Neurol. 2009, 68, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Ang, L.C.; Williams, B.; Furukawa, Y.; Fitzmaurice, P.; Guttman, M.; Boileau, I.; Hornykiewicz, O.; Kish, S.J. Low levels of astroglial markers in Parkinson’s disease: Relationship to alpha-synuclein accumulation. Neurobiol. Dis. 2015, 82, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorshkov, K.; Aguisanda, F.; Thorne, N.; Zheng, W. Astrocytes as targets for drug discovery. Drug. Discov. Today 2018, 23, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ganesan, P.; Rengarajan, T.; Choi, D.K.; Arulselvan, P. Cellular phenotypes as inflammatory mediators in Parkinson’s disease: Interventional targets and role of natural products. Biomed. Pharm. 2018, 106, 1052–1062. [Google Scholar] [CrossRef]
- Rydbirk, R.; Elfving, B.; Andersen, M.D.; Langbøl, M.A.; Folke, J.; Winge, K.; Pakkenberg, B.; Brudek, T.; Aznar, S. Cytokine profiling in the prefrontal cortex of Parkinson’s Disease and Multiple System Atrophy patients. Neurobiol. Dis. 2017, 106, 269–278. [Google Scholar] [CrossRef]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2022, 145, 964–978. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Sonninen, T.M.; Hamalainen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.P.; Salin, P.; Kerkerian-Le Goff, L. Glutamate transporter 1-expressing glia in the rat substantia nigra-Morphometric analysis and relationships to synapses. Glia 2020, 68, 2028–2039. [Google Scholar] [CrossRef]
- Avola, R.; Graziano, A.C.E.; Pannuzzo, G.; Albouchi, F.; Cardile, V. New insights on Parkinson’s disease from differentiation of SH-SY5Y into dopaminergic neurons: An involvement of aquaporin4 and 9. Mol. Cell Neurosci. 2018, 88, 212–221. [Google Scholar] [CrossRef]
- Benga, O.; Huber, V.J. Brain water channel proteins in health and disease. Mol. Asp. Med. 2012, 33, 562–578. [Google Scholar] [CrossRef]
- Küppers, E.; Gleiser, C.; Brito, V.; Wachter, B.; Pauly, T.; Hirt, B.; Grissmer, S. AQP4 expression in striatal primary cultures is regulated by dopamine--implications for proliferation of astrocytes. Eur. J. Neurosci. 2008, 28, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Thenral, S.T.; Vanisree, A.J. Peripheral assessment of the genes AQP4, PBP and TH in patients with Parkinson’s disease. Neurochem. Res. 2012, 37, 512–515. [Google Scholar] [CrossRef]
- Fan, Y.; Kong, H.; Shi, X.; Sun, X.; Ding, J.; Wu, J.; Hu, G. Hypersensitivity of aquaporin 4-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine and astrocytic modulation. Neurobiol. Aging 2008, 29, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Behnam, M.; Pourattar, M.A.; Jafarpour, H.; Asemi, Z. Aquaporin 4: A key player in Parkinson’s disease. J. Cell Physiol. 2019, 234, 21471–21478. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 mediates communication between astrocyte and microglia: Implications of neuroinflammation in experimental Parkinson’s disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, B.; Sun, H.; Zhou, Y.; Liu, M.; Ding, J.; Fang, F.; Fan, Y.; Hu, G. Aquaporin-4 deficiency diminishes the differential degeneration of midbrain dopaminergic neurons in experimental Parkinson’s disease. Neurosci. Lett. 2016, 614, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, E.A.; Srinivasan, R. Emerging Roles for Aberrant Astrocytic Calcium Signals in Parkinson’s Disease. Front. Physiol. 2021, 12, 812212. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [Green Version]
- Ofori, E.; Pasternak, O.; Planetta, P.J.; Li, H.; Burciu, R.G.; Snyder, A.F.; Lai, S.; Okun, M.S.; Vaillancourt, D.E. Longitudinal changes in free-water within the substantia nigra of Parkinson’s disease. Brain 2015, 138, 2322–2331. [Google Scholar] [CrossRef]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef] [Green Version]
- Olivera, S.; Fernandez, A.; Latini, A.; Rosillo, J.C.; Casanova, G.; Wajner, M.; Cassina, P.; Barbeito, L. Astrocytic proliferation and mitochondrial dysfunction induced by accumulated glutaric acidemia I (GAI) metabolites: Possible implications for GAI pathogenesis. Neurobiol. Dis. 2008, 32, 528–534. [Google Scholar] [CrossRef]
- Sathe, K.; Maetzler, W.; Lang, J.D.; Mounsey, R.B.; Fleckenstein, C.; Martin, H.L.; Schulte, C.; Mustafa, S.; Synofzik, M.; Vukovic, Z.; et al. S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain 2012, 135, 3336–3347. [Google Scholar] [CrossRef] [Green Version]
- Morales, I.; Sanchez, A.; Rodriguez-Sabate, C.; Rodriguez, M. The astrocytic response to the dopaminergic denervation of the striatum. J. Neurochem. 2016, 139, 81–95. [Google Scholar] [CrossRef]
- Batassini, C.; Broetto, N.; Tortorelli, L.S.; Borsoi, M.; Zanotto, C.; Galland, F.; Souza, T.M.; Leite, M.C.; Goncalves, C.A. Striatal Injury with 6-OHDA Transiently Increases Cerebrospinal GFAP and S100B. Neural. Plast. 2015, 2015, 387028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iuvone, T.; Esposito, G.; De Filippis, D.; Bisogno, T.; Petrosino, S.; Scuderi, C.; Di Marzo, V.; Steardo, L. Cannabinoid CB1 receptor stimulation affords neuroprotection in MPTP-induced neurotoxicity by attenuating S100B up-regulation in vitro. J. Mol. Med. 2007, 85, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, H.; Zhang, L.; Xu, Y.; Deng, W.; Zhu, H.; Qin, C. S100B transgenic mice develop features of Parkinson’s disease. Arch. Med. Res. 2011, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tadayon, R.; Shaw, G.S. Monitoring Interactions Between S100B and the Dopamine D2 Receptor Using NMR Spectroscopy. Methods Mol. Biol. 2019, 1929, 311–324. [Google Scholar] [CrossRef]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef]
- Tremblay, M.E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol. Brain 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. Faseb J. 2006, 20, 2000–2008. [Google Scholar] [CrossRef] [PubMed]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell Neurosci. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, E.H.; Weber, S.S.; Kahle, P.J. Exogenous α-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 2015, 16, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef]
- Drouin-Ouellet, J.; St-Amour, I.; Saint-Pierre, M.; Lamontagne-Proulx, J.; Kriz, J.; Barker, R.A.; Cicchetti, F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014, 18, pyu103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botia, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [Green Version]
- Piccinini, A.M.; Midwood, K.S. DAMPening inflammation by modulating TLR signalling. Mediat. Inflamm. 2010, 2010, 672395. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Lee, S.; Cho, I.H.; Lee, H.K.; Kim, D.; Choi, S.Y.; Oh, S.B.; Park, K.; Kim, J.S.; Lee, S.J. TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: Differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia 2006, 53, 248–256. [Google Scholar] [CrossRef]
- Diaz, J.; Lévesque, D.; Griffon, N.; Lammers, C.H.; Martres, M.P.; Sokoloff, P.; Schwartz, J.C. Opposing roles for dopamine D2 and D3 receptors on neurotensin mRNA expression in nucleus accumbens. Eur. J. Neurosci. 1994, 6, 1384–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, F.; Xu, P.; Qu, S. Recent Advance in the Relationship between Excitatory Amino Acid Transporters and Parkinson’s Disease. Neural. Plast. 2016, 2016, 8941327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrand, A.Q.; Gregory, R.A.; Backman, C.M.; Helke, K.L.; Boger, H.A. Altered glutamate release in the dorsal striatum of the MitoPark mouse model of Parkinson’s disease. Brain Res. 2016, 1651, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhou, S.; Sun, J. Exercise increases striatal Glu reuptake and improves motor dysfunction in 6-OHDA-induced Parkinson’s disease rats. Exp. Brain Res. 2021, 239, 3277–3287. [Google Scholar] [CrossRef]
- Chassain, C.; Cladiere, A.; Tsoutsos, C.; Pereira, B.; Boumezbeur, F.; Debilly, B.; Marques, A.R.; Thobois, S.; Durif, F. Glutamate cycle changes in the putamen of patients with de novo Parkinson’s disease using (1)H MRS. Park. Relat. Disord. 2022, 99, 65–72. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Shang, P.; Baker, M.; Banks, S.; Hong, S.I.; Choi, D.S. Emerging Nondopaminergic Medications for Parkinson’s Disease: Focusing on A2A Receptor Antagonists and GLP1 Receptor Agonists. J. Mov. Disord. 2021, 14, 193–203. [Google Scholar] [CrossRef]
- An, H.; Heo, J.Y.; Lee, C.J.; Nam, M.H. The Pathological Role of Astrocytic MAOB in Parkinsonism Revealed by Genetic Ablation and Over-expression of MAOB. Exp. Neurobiol. 2021, 30, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291 e279. [Google Scholar] [CrossRef]
- Mallajosyula, J.K.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Nicholls, D.G.; Di Monte, D.A.; Macarthur, H.; Andersen, J.K. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS ONE 2008, 3, e1616. [Google Scholar] [CrossRef]
- Nam, M.H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease. Int. J. Mol. Sci 2022, 23, 4453. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Jenner, P.; Chen, S.D. Monoamine Oxidase-B Inhibitors for the Treatment of Parkinson’s Disease: Past, Present, and Future. J. Park. Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef]
- Huntington, T.E.; Srinivasan, R. Adeno-Associated Virus Expression of alpha-Synuclein as a Tool to Model Parkinson’s Disease: Current Understanding and Knowledge Gaps. Aging Dis. 2021, 12, 1120–1137. [Google Scholar] [CrossRef]
- Okubo, Y.; Kanemaru, K.; Suzuki, J.; Kobayashi, K.; Hirose, K.; Iino, M. Inositol 1,4,5-trisphosphate receptor type 2-independent Ca(2+) release from the endoplasmic reticulum in astrocytes. Glia 2019, 67, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Mashima, K. Neuroprotection and Disease Modification by Astrocytes and Microglia in Parkinson Disease. Antioxidants 2022, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Maatouk, L.; Yi, C.; Carrillo-de Sauvage, M.A.; Compagnion, A.C.; Hunot, S.; Ezan, P.; Hirsch, E.C.; Koulakoff, A.; Pfrieger, F.W.; Tronche, F.; et al. Glucocorticoid receptor in astrocytes regulates midbrain dopamine neurodegeneration through connexin hemichannel activity. Cell Death Differ. 2019, 26, 580–596. [Google Scholar] [CrossRef]
- Kawasaki, A.; Hayashi, T.; Nakachi, K.; Trosko, J.E.; Sugihara, K.; Kotake, Y.; Ohta, S. Modulation of connexin 43 in rotenone-induced model of Parkinson’s disease. Neuroscience 2009, 160, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.F.; Labra, V.C.; Alvear, T.F.; Mellado, L.A.; Inostroza, C.A.; Oyarzun, J.E.; Salgado, N.; Quintanilla, R.A.; Orellana, J.A. Connexin 43 hemichannels and pannexin-1 channels contribute to the alpha-synuclein-induced dysfunction and death of astrocytes. Glia 2019, 67, 1598–1619. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prunell, G.; Olivera-Bravo, S. A Focus on Astrocyte Contribution to Parkinson’s Disease Etiology. Biomolecules 2022, 12, 1745. https://doi.org/10.3390/biom12121745
Prunell G, Olivera-Bravo S. A Focus on Astrocyte Contribution to Parkinson’s Disease Etiology. Biomolecules. 2022; 12(12):1745. https://doi.org/10.3390/biom12121745
Chicago/Turabian StylePrunell, Giselle, and Silvia Olivera-Bravo. 2022. "A Focus on Astrocyte Contribution to Parkinson’s Disease Etiology" Biomolecules 12, no. 12: 1745. https://doi.org/10.3390/biom12121745