
Chelation Combination—A Strategy to Mitigate the Neurotoxicity of Manganese, Iron, and Copper?

Abstract
:1. Introduction
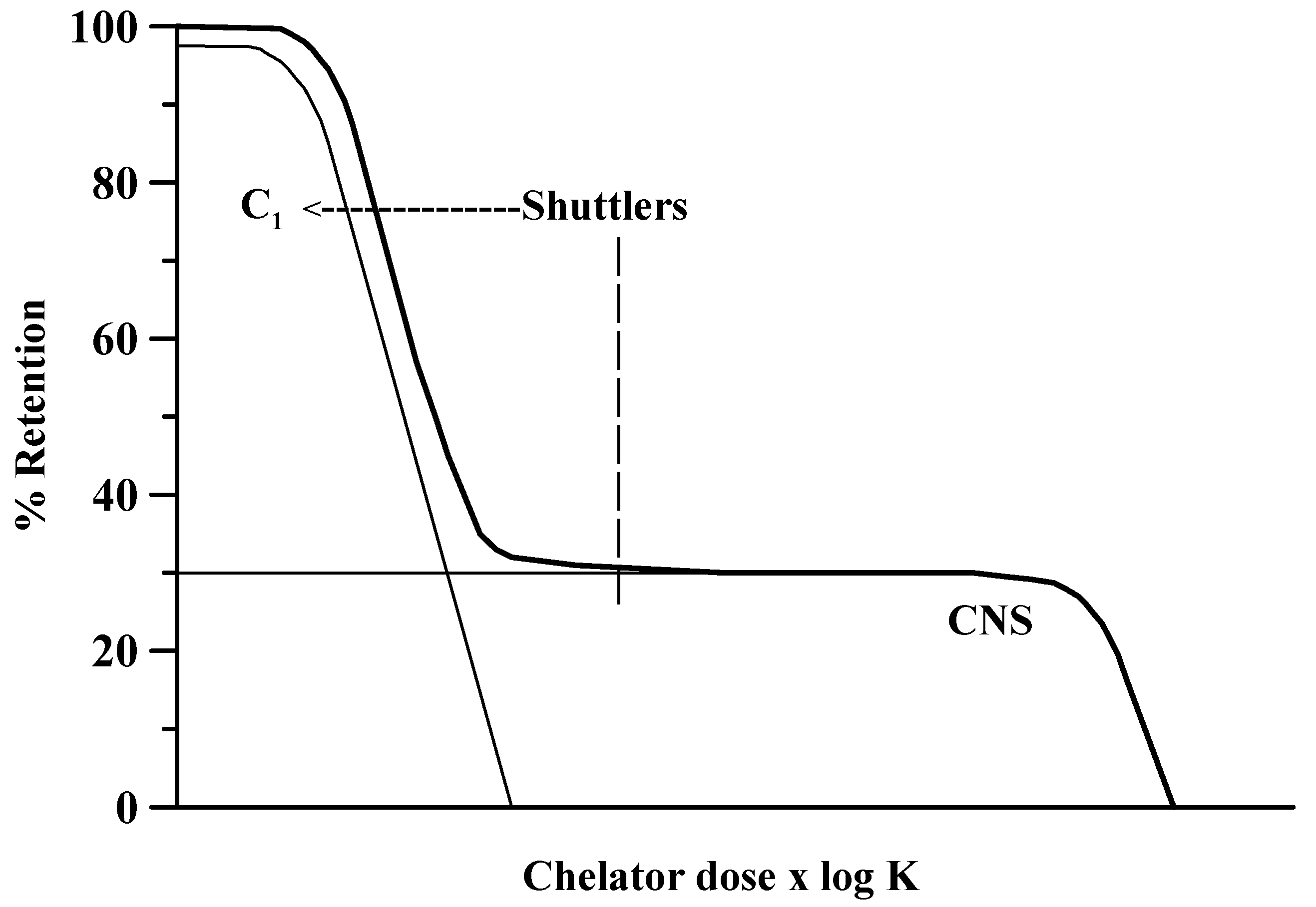
2. Principles of Metal Chelation
3. Chelator Combination versus Monotherapy as Therapeutic Strategy
3.1. Manganese Neurotoxicity and Chelation
3.2. Iron Neurotoxicity and Chelation
3.3. Copper Neurotoxicity and Chelation
4. Summary and Perspectives
- (1)
- Symptoms on neurotoxicity should ideally be identified early and chelation therapy should ideally be started before substantial irreversible brain damage has occurred. However, although symptoms on neurodegenerative diseases cannot be reversed, it will be of great interest if the disease progression can be prevented.
- (2)
- Doses and circulating chelator levels must be controlled to enhance intended metal removal.
- (3)
- The levels of essential metals must remain sufficient within cells for metalloenzyme biosynthesis.
- (4)
- Regular control of blood and tissue metal pools is needed.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Aaseth, J.; Skaug, M.A.; Cao, Y.; Andersen, O. Chelation in metal intoxication-principles and paradigms. J. Trace Elem. Med. Biol. 2015, 31, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Chartrand, M.S.; Aaseth, J. Manganese exposure and neurotoxic effects in children. Environ. Res. 2017, 155, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hulsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Choo, X.Y.; Alukaidey, L.; White, A.R.; Grubman, A. Neuroinflammation and copper in Alzheimer’s disease. Int. J. Alzheimers Dis. 2013, 2013, 145345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Skaug, M.A.; Andersen, O.; Aaseth, J. Chelation therapy in intoxications with mercury, lead and copper. J. Trace Elem. Med. Biol. 2015, 31, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A. Manganism in the 21st century: The Hanninen lecture. Neurotoxicology 2014, 45, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Harrison, F.E.; Aschner, M.; Bowman, A.B. Exposing the role of metals in neurological disorders: A focus on manganese. Trends Mol. Med. 2022, 28, 555–568. [Google Scholar] [CrossRef]
- Herrero Hernandez, E.; Discalzi, G.; Valentini, C.; Venturi, F.; Chio, A.; Carmellino, C.; Rossi, L.; Sacchetti, A.; Pira, E. Follow-up of patients affected by manganese-induced Parkinsonism after treatment with CaNa2EDTA. Neurotoxicology 2006, 27, 333–339. [Google Scholar] [CrossRef]
- Dusek, P.; Roos, P.M.; Litwin, T.; Schneider, S.A.; Flaten, T.P.; Aaseth, J. The neurotoxicity of iron, copper and manganese in Parkinson’s and Wilson’s diseases. J. Trace Elem. Med. Biol. 2015, 31, 193–203. [Google Scholar] [CrossRef]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. Biometals 2018, 31, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusek, P.; Aaseth, J. Chelation Therapy in the Treatment of Metal Storage Diseases; Aaseth, J., Crisponi, G., Anderson, O., Eds.; Academic Press: London, UK, 2016; pp. 285–311. [Google Scholar]
- Bjørklund, G.; Tinkov, A.A.; Hosnedlová, B.; Kizek, R.; Ajsuvakova, O.P.; Chirumbolo, S.; Skalny, A.V. The role of glutathione redox imbalance in autism spectrum disorder: A review. Free Radic. Biol. Med. 2020, 160, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, D.; Møller, L.B.; Aaseth, J. Handbook on the Toxicology of Metals; Nordberg, G., Fowler, B.A., Nordberg, M., Eds.; Academic Press: London, UK, 2014; pp. 765–797. [Google Scholar]
- Cox, D.W.; Moore, S.D. Copper transporting P-type ATPases and human disease. J. Bioenerg. Biomembr. 2002, 34, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Zhang, L.; Meng, R.N.; Gao, J.; Jin, M.; Wang, X.P. Effect of metal ions on Alzheimer’s disease. Brain Behav. 2022, 12, e2527. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Tonali, N.; Shepard, W.; Nencetti, S.; Orlandini, E. Physiological Metals Can Induce Conformational Changes in Transthyretin Structure: Neuroprotection or Misfolding Induction? Crystals 2021, 11, 354. [Google Scholar] [CrossRef]
- Ciccone, L.; Fruchart-Gaillard, C.; Mourier, G.; Savko, M.; Nencetti, S.; Orlandini, E.; Shepard, W. Copper mediated amyloid-β binding to Transthyretin. Sci. Rep. 2018, 8, 13744. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Siotto, M.; Ventriglia, M.; Benussi, L.; Paterlini, A.; Magri, M.; Binetti, G.; Cassetta, E.; Caprara, D.; et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann. Neurol. 2014, 75, 574–580. [Google Scholar] [CrossRef]
- Squitti, R.; Ghidoni, R.; Simonelli, I.; Ivanova, I.D.; Colabufo, N.A.; Zuin, M.; Benussi, L.; Binetti, G.; Cassetta, E.; Rongioletti, M.; et al. Copper dyshomeostasis in Wilson disease and Alzheimer’s disease as shown by serum and urine copper indicators. J. Trace Elem. Med. Biol. 2018, 45, 181–188. [Google Scholar] [CrossRef]
- Bourassa, M.W.; Leskovjan, A.C.; Tappero, R.V.; Farquhar, E.R.; Colton, C.A.; Van Nostrand, W.E.; Miller, L.M. Elevated copper in the amyloid plaques and iron in the cortex are observed in mouse models of Alzheimer’s disease that exhibit neurodegeneration. Biomed. Spectrosc. Imaging 2013, 2, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, B.; Sass-Kortsak, A.; Clarke, R.; Laurie, S.H.; Wei, P. A comparative study of in vitro and in vivo interaction of D-penicillamine and triethylenetetramine with copper. Proc. R. Soc. Med. 1977, 70 (Suppl. S3), 13–18. [Google Scholar] [CrossRef]
- Horn, N.; Moller, L.B.; Nurchi, V.M.; Aaseth, J. Chelating principles in Menkes and Wilson diseases: Choosing the right compounds in the right combinations at the right time. J. Inorg. Biochem. 2019, 190, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Schneider, S.A.; Aaseth, J. Iron chelation in the treatment of neurodegenerative diseases. J. Trace Elem. Med. Biol. 2016, 38, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O. Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev. Med. Chem. 2004, 4, 11–21. [Google Scholar] [PubMed]
- Peana, M.; Pelucelli, A.; Medici, S.; Cappai, R.; Nurchi, V.M.; Zoroddu, M.A. Metal Toxicity and Speciation: A Review. Curr. Med. Chem. 2021, 28, 7190–7208. [Google Scholar] [CrossRef]
- Nurchi, V.M.; Cappai, R.; Chand, K.; Chaves, S.; Gano, L.; Crisponi, G.; Peana, M.; Zoroddu, M.A.; Santos, M.A. New strong extrafunctionalizable tris(3,4-HP) and bis(3,4-HP) metal sequestering agents: Synthesis, solution and in vivo metal chelation. Dalton Trans. 2019, 48, 16167–16183. [Google Scholar] [CrossRef]
- Aaseth, J.; Crisponi, G.; Anderson, O. Chelation Therapy in the Treatment of Metal Intoxication; Academic Press: London, UK, 2016. [Google Scholar]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood–brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef]
- Aaseth, J.; Ajsuvakova, O.P.; Skalny, A.V.; Skalnaya, M.G.; Tinkov, A.A. Chelator combination as therapeutic strategy in mercury and lead poisonings. Coord. Chem. Rev. 2018, 358, 1–12. [Google Scholar]
- Aaseth, J. Recent advance in the therapy of metal poisonings with chelating agents. Hum. Toxicol. 1983, 2, 257–272. [Google Scholar] [CrossRef]
- Catsch, A.; Harmuth-Hoene, A.E. Pharmacology and therapeutic applications of agents used in heavy metal poisoning. In Pharmacology & Therapeutics. Part A: Chemotherapy, Toxicology and Metabolic Inhibitors; Elsevier: Amsterdam, The Netherlands, 1976; Volume 1, pp. 1–118. [Google Scholar]
- Ringbom, A.; Harju, L. Determination of stability constants of chelate complexes: Part II. Applications. Anal. Chim. Acta 1972, 59, 49–58. [Google Scholar] [CrossRef]
- Catsch, A.; Harmuth-Hoene, A.E. Commentary. New developments in metal antidotal properties of chelating agents. Biochem. Pharm. 1975, 24, 1557–1562. [Google Scholar] [CrossRef]
- Ringbom, A.; Still, E. The calculation and use of a coefficients. Anal. Chim. Acta 1972, 59, 143–146. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part 1: Fundamental principles. J. Chem. Educ. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Ohrvik, H.; Aaseth, J.; Horn, N. Orchestration of dynamic copper navigation—New and missing pieces. Metallomics 2017, 9, 1204–1229. [Google Scholar] [CrossRef]
- Andersen, O.; Aaseth, J. A review of pitfalls and progress in chelation treatment of metal poisonings. J. Trace Elem. Med. Biol. 2016, 38, 74–80. [Google Scholar] [CrossRef]
- Ghassaban, K.; He, N.; Sethi, S.K.; Huang, P.; Chen, S.; Yan, F.; Haacke, E.M. Regional high iron in the substantia nigra differentiates Parkinson’s disease patients from healthy controls. Front. Aging Neurosci. 2019, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Mertz, W. Risk assessment of essential trace elements: New approaches to setting recommended dietary allowances and safety limits. Nutr. Rev. 1995, 53, 179–185. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30, 475–483. [Google Scholar] [CrossRef]
- Tuschl, K.; Mills, P.B.; Clayton, P.T. Manganese and the brain. Int. Rev. Neurobiol. 2013, 110, 277–312. [Google Scholar]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.-A.; Di Toro Mammarella, L.; Mignarri, A.; Monti, L.; et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Olanow, C.W. Manganese-induced parkinsonism and Parkinson’s disease. Ann. N. Y. Acad. Sci. 2004, 1012, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P.; Olanow, C.W. The neuropathology of manganese-induced Parkinsonism. J. Neuropathol. Exp. Neurol. 2007, 66, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Aschner, M.; Erikson, K.M.; Herrero Hernandez, E.; Tjalkens, R. Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromol. Med. 2009, 11, 252–266. [Google Scholar] [CrossRef]
- Soares, A.T.G.; Silva, A.C.; Tinkov, A.A.; Khan, H.; Santamaria, A.; Skalnaya, M.G.; Skalny, A.V.; Tsatsakis, A.; Bowman, A.B.; Aschner, M.; et al. The impact of manganese on neurotransmitter systems. J. Trace Elem. Med. Biol. 2020, 61, 126554. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Aschner, M. Role of astrocytes in manganese mediated neurotoxicity. BMC Pharmacol. Toxicol. 2013, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossgrove, J.; Zheng, W. Manganese toxicity upon overexposure. NMR Biomed. 2004, 17, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Kaviani, S.; Shahab, S.; Sheikhi, M.; Khaleghian, M.; Al Saud, S. Characterization of the binding affinity between some anti-Parkinson agents and Mn2+, Fe3+ and Zn2+ metal ions: A DFT insight. Inorg. Chem. Commun. 2021, 128, 108582. [Google Scholar] [CrossRef]
- Jiang, Y.M.; Mo, X.A.; Du, F.Q.; Fu, X.; Zhu, X.Y.; Gao, H.Y.; Xie, J.L.; Liao, F.L.; Pira, E.; Zheng, W. Effective treatment of manganese-induced occupational Parkinsonism with p-aminosalicylic acid: A case of 17-year follow-up study. J. Occup. Environ. Med. 2006, 48, 644–649. [Google Scholar] [CrossRef]
- Lachowicz, J.I.; Nurchi, V.M.; Crisponi, G.; Cappai, I.; Cappai, R.; Busato, M.; Aaseth, J. para-Aminosalicylic acid in the treatment of manganese toxicity. Complexation of Mn2+ with 4-amino-2-hydroxybenzoic acid and its N-acetylated metabolite. New J. Chem. 2018, 42, 8035–8049. [Google Scholar] [CrossRef] [Green Version]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- Bjørklund, G.; Hofer, T.; Nurchi, V.M.; Aaseth, J. Iron and other metals in the pathogenesis of Parkinson’s disease: Toxic effects and possible detoxification. J. Inorg. Biochem. 2019, 199, 110717. [Google Scholar] [CrossRef] [PubMed]
- Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; Medici, S.; Peana, M.; Zoroddu, M.A. Chemical features of in use and in progress chelators for iron overload. J. Trace Elem. Med. Biol. 2016, 38, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Gong, N.J.; Dibb, R.; Bulk, M.; van der Weerd, L.; Liu, C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer’s disease using quantitative susceptibility mapping MRI. Neuroimage 2019, 191, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Halliday, G.M.; Fedorow, H.; Rickert, C.H.; Gerlach, M.; Riederer, P.; Double, K.L. Evidence for specific phases in the development of human neuromelanin. J. Neural Transm. 2006, 113, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Tampellini, D.; Gatti, A.; Crippa, R.; Eisner, M.; Sulzer, D.; Ito, S.; Fariello, R.; Gallorini, M. The neuromelanin of human substantia nigra and its interaction with metals. J. Neural Transm. 2002, 109, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Nurchi, V.M.; Cappai, R.; Spano, N.; Sanna, G. A friendly complexing agent for spectrophotometric determination of total iron. Molecules 2021, 26, 3071. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Kleanthous, M.; Kontoghiorghe, C.N. The History of Deferiprone (L1) and the Paradigm of the Complete Treatment of Iron Overload in Thalassaemia. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020011. [Google Scholar] [CrossRef]
- Vlachodimitropoulou Koumoutsea, E.; Garbowski, M.; Porter, J. Synergistic intracellular iron chelation combinations: Mechanisms and conditions for optimizing iron mobilization. Br. J. Haematol. 2015, 170, 874–883. [Google Scholar] [CrossRef]
- Piga, A.; Gaglioti, C.; Fogliacco, E.; Tricta, F. Comparative effects of deferiprone and deferoxamine on survival and cardiac disease in patients with thalassemia major: A retrospective analysis. Haematologica 2003, 88, 489–496. [Google Scholar]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaten, T.P.; Aaseth, J.; Andersen, O.; Kontoghiorghes, G.J. Iron mobilization using chelation and phlebotomy. J. Trace Elem. Med. Biol. 2012, 26, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, G.; Nurchi, V.M.; Lachowicz, J.I. Iron Chelation for Iron Overload in Thalassemia. In Metal Ions in Life Sciences; Walter de Gruyter GmbH: Berlin, Germany, 2019; Volume 19. [Google Scholar]
- Mourad, F.H.; Hoffbrand, A.V.; Sheikh-Taha, M.; Koussa, S.; Khoriaty, A.I.; Taher, A. Comparison between desferrioxamine and combined therapy with desferrioxamine and deferiprone in iron overloaded thalassaemia patients. Br. J. Haematol. 2003, 121, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.C.; Kang, B.P.; Thompson, A.; Giardina, P.; Harmatz, P.; Glynos, T.; Paley, C.; Coates, T.D. The effect of deferasirox on cardiac iron in thalassemia major: Impact of total body iron stores. Blood 2010, 116, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, H.; Qin, Y.; Chen, G.; Zhao, Y. Effectiveness and Safety of Deferasirox in Thalassemia with Iron Overload: A Meta-Analysis. Acta Haematol. 2019, 141, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Totadri, S.; Bansal, D.; Bhatia, P.; Attri, S.V.; Trehan, A.; Marwaha, R.K. The deferiprone and deferasirox combination is efficacious in iron overloaded patients with beta-thalassemia major: A prospective, single center, open-label study. Pediatr. Blood Cancer 2015, 62, 1592–1596. [Google Scholar] [CrossRef]
- Aydinok, Y.; Kattamis, A.; Cappellini, M.D.; El-Beshlawy, A.; Origa, R.; Elalfy, M.; Kilinc, Y.; Perrotta, S.; Karakas, Z.; Viprakasit, V.; et al. Effects of deferasirox-deferoxamine on myocardial and liver iron in patients with severe transfusional iron overload. Blood 2015, 125, 3868–3877. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural Transm. 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Lamichhane, J.R.; Osdaghi, E.; Behlau, F.; Köhl, J.; Jones, J.B.; Aubertot, J.N. Thirteen decades of antimicrobial copper compounds applied in agriculture. A review. Agron. Sustain. Dev. 2018, 38, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Georgopoulos, P.G.; Roy, A.; Yonone-Lioy, M.J.; Opiekun, R.E.; Lioy, P.J. Environmental copper: Its dynamics and human exposure issues. J. Toxicol. Environ. Health B Crit. Rev. 2001, 4, 341–394. [Google Scholar]
- Thomassen, Y.; Nieboer, E.; Romanova, N.; Nikanov, A.; Hetland, S.; VanSpronsen, E.P.; Odland, J.O.; Chashchin, V. Multi-component assessment of worker exposures in a copper refinery. Part 1. Environmental monitoring. J. Environ. Monit. 2004, 6, 985–991. [Google Scholar] [CrossRef]
- Squitti, R.; Lupoi, D.; Pasqualetti, P.; Dal Forno, G.; Vernieri, F.; Chiovenda, P.; Rossi, L.; Cortesi, M.; Cassetta, E.; Rossini, P.M. Elevation of serum copper levels in Alzheimer’s disease. Neurology 2002, 59, 1153–1161. [Google Scholar] [CrossRef]
- Singh, S.K.; Sinha, P.; Mishra, L.; Srikrishna, S. Neuroprotective Role of a Novel Copper Chelator against Abeta 42 Induced Neurotoxicity. Int. J. Alzheimers Dis. 2013, 2013, 567128. [Google Scholar] [PubMed] [Green Version]
- Squitti, R.; Salustri, C.; Rongioletti, M.; Siotto, M. Commentary: The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front. Neurol. 2017, 8, 503. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, F.; Vecchio, F.; Ventriglia, M.; Porcaro, C.; Miraglia, F.; Siotto, M. Non-ceruloplasmin copper distinguishes a distinct subtype of Alzheimer’s disease: A study of EEG-derived brain activity. Curr. Alzheimer Res. 2016, 13, 1374–1384. [Google Scholar] [CrossRef]
- Kaur, D.; Behl, T.; Sehgal, A.; Singh, S.; Sharma, N.; Chigurupati, S.; Bungau, S. Decrypting the potential role of α-lipoic acid in Alzheimer’s disease. Life Sci. 2021, 284, 119899. [Google Scholar] [CrossRef]
- Aaseth, J.; Skalny, A.V.; Roos, P.M.; Alexander, J.; Aschner, M.; Tinkov, A.A.; Rossini, P.M.; Rongioletti, M.; Squitti, R. Copper, Iron, Selenium and lipo-glycemic dysmetabolism in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 9461. [Google Scholar] [CrossRef] [PubMed]
- Møller, L.B.; Aaseth, J. Copper. In Handbook on the Toxicology of Metals; Nordberg, G., Costa, M., Eds.; Academic Press: London, UK, 2022; Volume 2, pp. 243–266. [Google Scholar]
- Tanzi, R.E.; Petrukhin, K.; Chernov, I.; Pellequer, J.L.; Wasco, W.; Ross, B.; Romano, D.M.; Parano, E.; Pavone, L.; Brzustowicz, L.M.; et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat. Genet. 1993, 5, 344–350. [Google Scholar] [CrossRef]
- Telianidis, J.; Hui Hung, Y.; Materia, S. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.M.; Davies, K.M.; Hare, D.J.; Cottam, V.; Chen, N.; Hilgers, L.; Halliday, G.; Mercer, J.F.B.; Double, K.L. Localization of copper and copper transporters in the human brain. Metallomics 2013, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Walshe, J.M. Penicillamine: The treatment of first choice for patients with Wilson’s disease. Mov. Disord. 1999, 14, 545–550. [Google Scholar] [CrossRef]
- Netter, P.; Bannwarth, B.; Pere, P.; Nicolas, A. Clinical pharmacokinetics of D-penicillamine. Clin. Pharmacokinet. 1987, 13, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Singh, A.P.; Wheeler, N.; Galindo, C.L.; Kim, J.J. Safety profile of D-penicillamine: A comprehensive pharmacovigilance analysis by FDA adverse event reporting system. Expert Opin. Drug Saf. 2021, 20, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Patra, B.R.; Irtaza, M.; Rao, P.K.; Giri, S.; Darak, H.; Gopan, A.; Kale, A.; Shukla, A. Adverse Events with d-penicillamine Therapy in Hepatic Wilson’s Disease: A Single-Center Retrospective Audit. Clin. Drug Investig. 2022, 42, 177–184. [Google Scholar] [CrossRef]
- Weiss, K.H.; Thurik, F.; Gotthardt, D.N.; Schafer, M.; Teufel, U.; Wiegand, F.; Merle, U.; Ferenci-Foerster, D.; Maieron, A.; Stauber, R.; et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin. Gastroenterol. Hepatol. 2013, 11, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Plitz, T.; Boyling, L. Metabolic disposition of WTX101 (bis-choline tetrathiomolybdate) in a rat model of Wilson disease. Xenobiotica 2019, 49, 332–338. [Google Scholar] [CrossRef]
- Brewer, G.J.; Hedera, P.; Kluin, K.J.; Carlson, M.; Askari, F.; Dick, R.B.; Sitterly, J.; Fink, J.K. Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch. Neurol. 2003, 60, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Ren, M.S.; Yang, R.M.; Zhang, B.; Xu, S.H. Comparison of therapeutic effects between unithiol, succimer and penicillamine on hepatolenticular degeneration. Chin. J. New Drugs Clin. Remedies 1998, 17, 23–25. [Google Scholar]
- Zhang, J.; Xiao, L.; Yang, W. Combined sodium Dimercapto propanesulfonate and zinc versus D-penicillamine as first-line therapy for neurological Wilson’s disease. BMC Neurol. 2020, 20, 255. [Google Scholar] [CrossRef] [Green Version]
- Li, K. Iron Pathophysiology in Friedreich’s Ataxia. Adv. Exp. Med. Biol. 2019, 1173, 125–143. [Google Scholar]
- Zhang, S.L.; Yue, Z.; Arnold, D.M.; Artiushin, G.; Sehgal, A. A circadian clock in the blood-brain barrier regulates xenobiotic efflux. Cell 2018, 173, 130–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, J.; Kabin, E.; Järving, I.; Bragina, O.; Tõugu, V.; Plitz, T.; Palumaa, P. Copper(I)-binding properties of de-coppering drugs for the treatment of Wilson disease. α-Lipoic acid as a potential anti-copper agent. Sci. Rep. 2018, 8, 1463. [Google Scholar] [CrossRef] [PubMed]
- Kuria, A.; Fang, X.; Li, M.; Han, H.; He, J.; Aaseth, J.; Cao, Y. Does dietary intake of selenium protect against cancer? A systematic review and meta-analysis of population-based prospective studies. Crit. Rev. Food Sci. Nutr. 2020, 60, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Ramchandani, D.; Vahdat, L. Copper depletion as a therapeutic strategy in cancer. Met. Ions Life Sci. 2019, 19, 303–330. [Google Scholar]



{kind=link}
{kind=link}
| Disease | Metal Deposition |
|---|---|
| Manganism | Long-term manganese exposure [2] |
| Parkinson’s disease | Cerebral iron deposition in substantia nigra [3] |
| Aceruloplasminemia | Cerebral iron deposition [4] |
| Neurologic Wilson’s disease | Cerebral copper deposition [5] |
| Alzheimer’s disease | Associated with copper deposits in brain [6] |
| Metal Ions | Coordinating Groups | ||||
|---|---|---|---|---|---|
| Hard (Oxygen-Seekers) | Intermediate | Soft | Hard | Intermediate | Soft |
| Mg2+, Ca2+, Sr2+, Mn2+, Cr3+, Fe3+ | Pb2+, Fe2+, Cu2+ | Hg2+, CH3Hg+, Cu+ | OH−, RCOO−, RO− | RNH2 | RSH, RS−, R-Se− |
| Disease | Chelator Combination |
|---|---|
| Manganism | CaEDTA + PAS |
| Cerebral iron deposition | DFOA + deferiprone, or DFOA + deferasirox |
| Neurologic Wilson’s disease | DMSA + TTM, or Trien + TTM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aaseth, J.O.; Nurchi, V.M. Chelation Combination—A Strategy to Mitigate the Neurotoxicity of Manganese, Iron, and Copper? Biomolecules 2022, 12, 1713. https://doi.org/10.3390/biom12111713
Aaseth JO, Nurchi VM. Chelation Combination—A Strategy to Mitigate the Neurotoxicity of Manganese, Iron, and Copper? Biomolecules. 2022; 12(11):1713. https://doi.org/10.3390/biom12111713
Chicago/Turabian StyleAaseth, Jan O., and Valeria M. Nurchi. 2022. "Chelation Combination—A Strategy to Mitigate the Neurotoxicity of Manganese, Iron, and Copper?" Biomolecules 12, no. 11: 1713. https://doi.org/10.3390/biom12111713