

The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis
Abstract
:1. Introduction
2. Ferroptosis
2.1. Definition of Ferroptosis
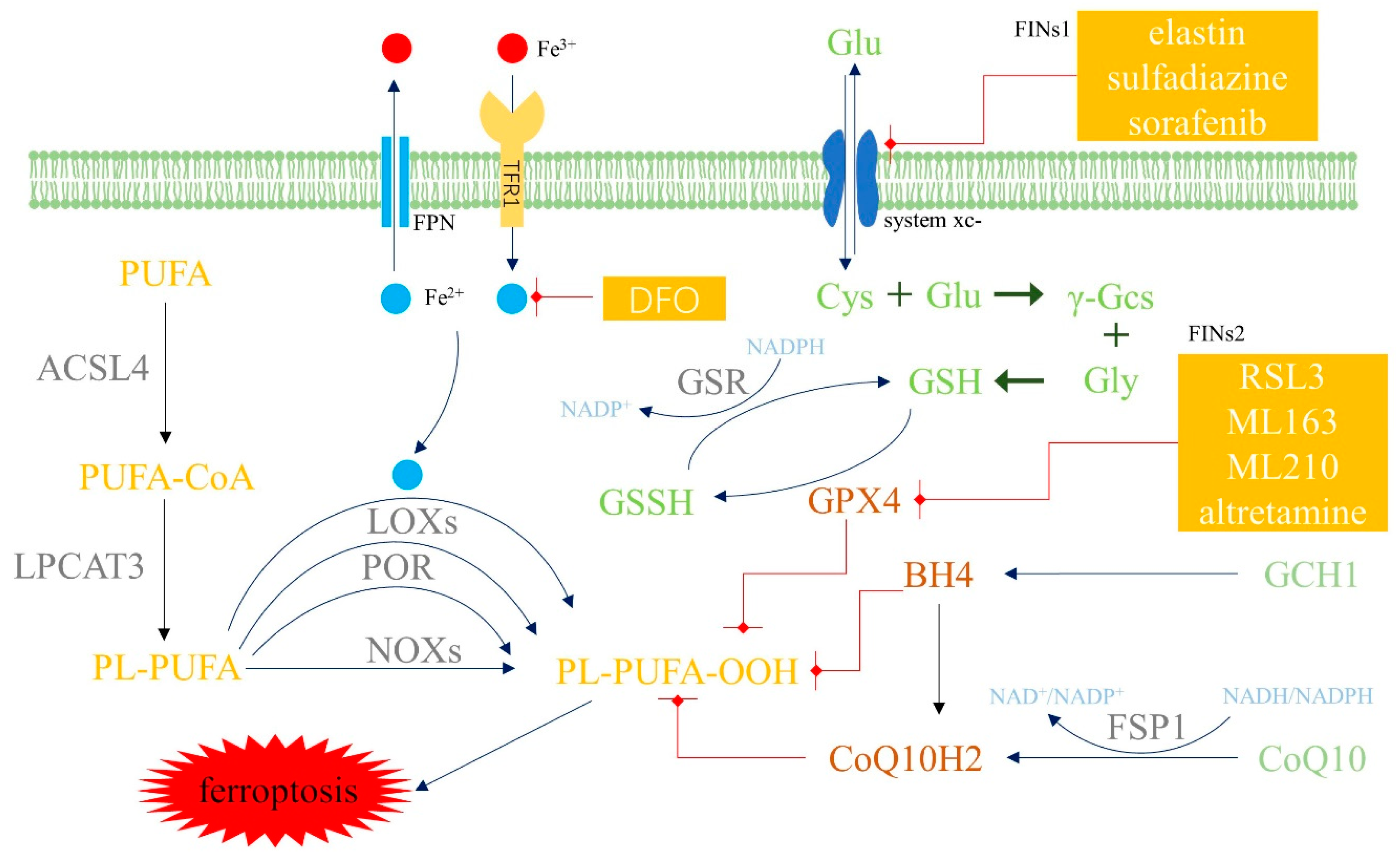
2.2. Mechanism and Regulatory Factors of Ferroptosis
2.3. Anti-Ferroptosis Systems in Cells
2.4. Atherosclerosis and Ferroptosis
3. Macrophages and Iron Homeostasis
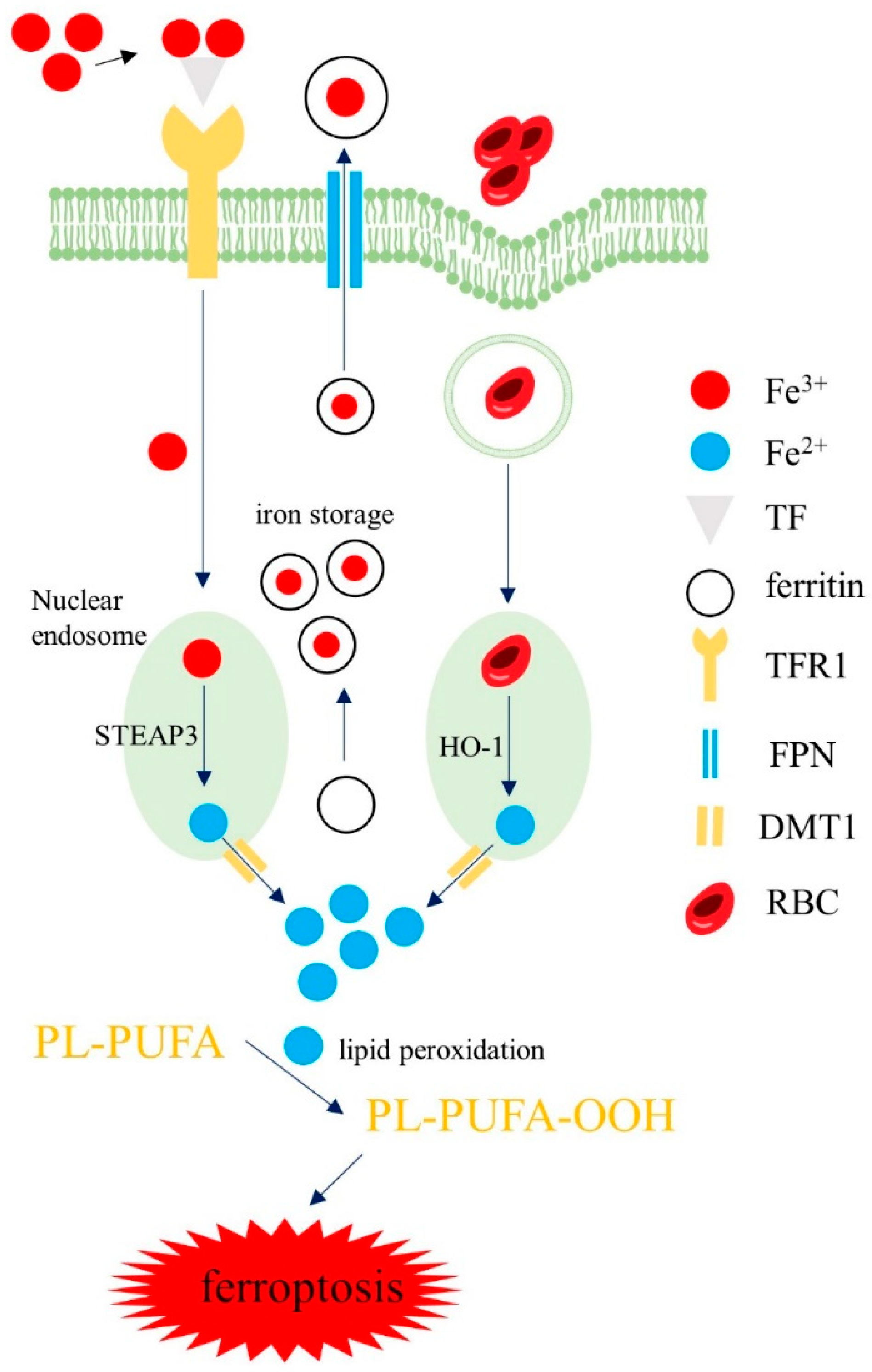
3.1. Iron Homeostasis in Macrophages
3.2. Iron Regulation in Macrophages
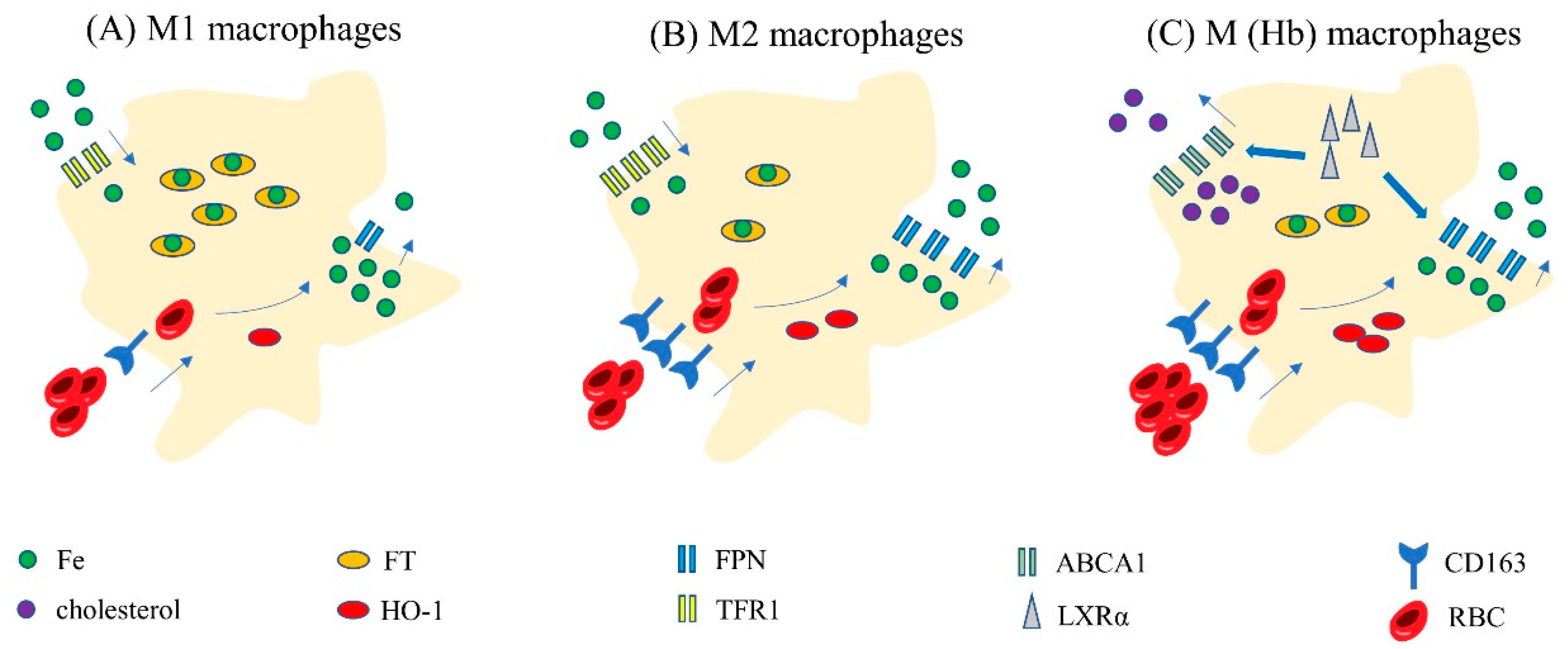
3.3. Iron Homeostasis in Different Subtypes of Macrophages
3.4. Iron Homeostasis in Special Macrophages: Hemoglobin-Stimulated Macrophages
4. Iron Overload and Ferroptosis in Macrophages
4.1. Iron Overload and Ferroptosis in Macrophages Promote the Progression of AS
4.2. Iron Overload, Ferroptosis and Polarization in Macrophages
4.3. Iron Metabolism and Ferroptosis in M (Hb) Type Macrophages
5. Prospect
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CVDs | cardiovascular diseases |
| ASCVD | atherosclerotic cardiovascular disease |
| ESC | European Society of Cardiology |
| AS | atherosclerosis |
| LPS | lipopolysaccharide |
| LPO | lipid peroxide |
| ROS | reactive oxygen species |
| Fe2+ | ferrous iron |
| PUFAs | polyunsaturated fatty acids |
| TFR1 | transferrin receptor 1 |
| FT | ferritin |
| LIP | labile iron pool |
| FPN | ferroportin |
| LOXs | lipoxygenases |
| POR | cytochrome P450 oxidoreductase |
| NOXs | reduced nicotinamide adenine dinucleotide phosphate oxidase |
| NCOA4 | nuclear receptor coactivator 4 |
| Fer-1 | ferrostatin-1 |
| DFO | deferoxamine |
| GPX4 | glutathione peroxidase 4 |
| GSH | glutathione |
| SLC7A11 | solute carrier family 7 member 11 |
| SLC3A2 | solute carrier family 3 member 2 |
| FINs1 | Ferroptosis-inducing compounds 1 |
| RSL3 | Ras Selective Lethal 3 |
| NADPH | reduced form of nicotinamide-adenine dinucleotide phosphate |
| FSP1 | ferroptosis suppressor protein 1 |
| AIFM2 | apoptosis-inducing factor mitochondrial 2 |
| CoQ10H2 | ubiquinol |
| CoQ10 | ubiquinone |
| DHODH | dihydroorotate dehydrogenase |
| GCH1 | GTP-cyclohydrolase1 |
| BH4 | tetrahydrobiopterin |
| PTGS2 | prostaglandin endoperoxide synthase 2 |
| ACSL4 | acyl-CoA synthetase long-chain family member 4 |
| ABCB6 | ATP-binding cassette transporter B6 |
| RBC | red blood cells |
| HO-1 | heme oxygenase-1 |
| Fe3+ | ferric iron |
| TF | transferrin |
| STEAP3 | six-segment transmembrane epithelial antigen of prostate 3 |
| DMT1 | divalent metal transporter 1 |
| FTH | ferritin heavy chain |
| FTL | ferritin light chain |
| ox-LDL | oxidized low-density lipoprotein |
| LXRα | liver X receptor alpha |
| ABCA1 | ATP-binding cassette transporterA1 |
| BACH1 | BTB domain and CNC homolog 1 |
| NEDD4L | NEDD4-like E3 ubiquitin protein ligase |
| Hb | hemoglobin |
| Hp | haptoglobin |
| HH | hemoglobin-haptoglobin complex |
| M (Hb) | hemoglobin-stimulated macrophages |
| CD206 | mannose receptor |
| MMP | matrix metalloproteinases |
| NOD1 | binding oligomerization domain 1 |
| SPION | superparamagnetic iron oxide nanoparticles |
| IFN-Y | interferon-Y |
| RRM2 | ribonucleotide reductase small subunit M2 |
| ECAR | extracellular acidification rate |
| PHDs | prolyl hydroxylase protein |
| HIF-1α | hypoxia-inducible factor 1α |
| VEGF | vascular endothelial growth factor |
References
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. J. Prev. Cardiol. 2022, 29, 5–115. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Jia, Y.; Yang, Y.; Wen, W.; Huang, M.; Liu, S.; Yang, Y.; Dong, Y.; Zhang, M. Thyroid hormones inhibit apoptosis of macrophage induced by oxidized low-density lipoprotein. Biofactors 2022, 48, 86–99. [Google Scholar] [CrossRef]
- Mao, C.; Li, D.; Zhou, E.; Zhang, J.; Wang, C.; Xue, C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging 2021, 13, 7627–7643. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yu, F.; Zhang, Y.; Li, M.; Di, M.; Chen, W.; Liu, X.; Zhang, Y.; Zhang, M. Traditional Chinese Medication Tongxinluo Attenuates Lipidosis in Ox-LDL-Stimulated Macrophages by Enhancing Beclin-1-Induced Autophagy. Front. Pharmacol. 2021, 12, 673366. [Google Scholar] [CrossRef]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hu, H.; Chen, Y.; Jing, L.; Zhai, C.; Shen, L. The Link Between Ferroptosis and Cardiovascular Diseases: A Novel Target for Treatment. Front. Cardiovasc. Med. 2021, 8, 710963. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Yang, W.; Wang, S.; Geng, C.; Guan, X. SIRT1-autophagy axis inhibits excess iron-induced ferroptosis of foam cells and subsequently increases IL-1Β and IL-18. Biochem. Biophys. Res. Commun. 2021, 561, 33–39. [Google Scholar] [CrossRef]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Chang, L.C.; Chiang, S.K.; Chen, S.E.; Yu, Y.L.; Chou, R.H.; Chang, W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayır, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Henson, E.S.; Chen, Y.; Gibson, S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Rong, L. Cascade reaction-mediated efficient ferroptosis synergizes with immunomodulation for high-performance cancer therapy. Biomater. Sci. 2020, 8, 6272–6285. [Google Scholar] [CrossRef]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef]
- Kose, T.; Vera-Aviles, M.; Sharp, P.A.; Latunde-Dada, G.O. Curcumin and (-)- Epigallocatechin-3-Gallate Protect Murine MIN6 Pancreatic Beta-Cells Against Iron Toxicity and Erastin-Induced Ferroptosis. Pharmaceuticals 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Zheng, C.; Zhang, Y.; Chang, Y.Z.; Qian, Z.M.; Shen, X. Heat shock protein 27 downregulates the transferrin receptor 1-mediated iron uptake. Int. J. protection against Biochem. Cell Biol. 2006, 38, 1402–1416. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Garcia-Bermudez, J.; Birsoy, K. A mitochondrial gatekeeper that helps cells escape death by ferroptosis. Nature 2021, 593, 514–515. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Liu, X.; Zhang, Y.; Lei, G.; Yan, Y.; Lee, H.; Koppula, P.; Wu, S.; Zhuang, L.; Fang, B.; et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 2021, 593, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K.; Stockwell, B.R. SnapShot: Ferroptosis. Cell 2020, 181, 1188–1188.e1. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, H.; Hua, L.; Hou, C.; Jia, Q.; Chen, J.; Zhang, S.; Wang, Y.; He, S.; Jia, E. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic. Biol. Med. 2021, 171, 55–68. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Li, C.; Chen, J.W.; Liu, Z.H.; Shen, Y.; Ding, F.H.; Gu, G.; Liu, J.; Qiu, J.P.; Gao, J.; Zhang, R.Y.; et al. CTRP5 promotes transcytosis and oxidative modification of low-density lipoprotein and the development of atherosclerosis. Atherosclerosis 2018, 278, 197–209. [Google Scholar] [CrossRef]
- Chiu, M.H.; Heydari, B.; Batulan, Z.; Maarouf, N.; Subramanya, V.; Schenck-Gustafsson, K.; O’Brien, E.R. Coronary artery disease in post-menopausal women: Are there appropriate means of assessment? Clin. Sci. 2018, 132, 1937–1952. [Google Scholar] [CrossRef]
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. [Google Scholar] [CrossRef] [Green Version]
- Carpi-Santos, R.; Calaza, K.C. Alterations in System x(c)(-) Expression in the Retina of Type 1 Diabetic Rats and the Role of Nrf2. Mol. NeuroBiol. 2018, 55, 7941–7948. [Google Scholar] [CrossRef]
- Yang, K.; Song, H.; Yin, D. PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 2021, 77, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted inhibition of STAT3 as a potential treatment strategy for atherosclerosis. Theranostics 2019, 9, 6424–6442. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019, 47, 598–606. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Luo, G.; Guo, X.; Jiang, C.; Zeng, H.; Zhou, F.; Li, Y.; Yu, J.; Yao, P. Macrophage iron retention aggravates atherosclerosis: Evidence for the role of autocrine formation of hepcidin in plaque macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158531. [Google Scholar] [CrossRef]
- Malhotra, R.; Wunderer, F.; Barnes, H.J.; Bagchi, A.; Buswell, M.D.; O’Rourke, C.D.; Slocum, C.L.; Ledsky, C.D.; Peneyra, K.M.; Sigurslid, H.; et al. Hepcidin Deficiency Protects Against Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 178–187. [Google Scholar] [CrossRef]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem 2020, 295, 69–82. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Liu, J.; Kang, R.; Tang, D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem. Biophys. Res. Commun. 2020, 531, 581–587. [Google Scholar] [CrossRef]
- Ji, J.; Zhou, Y.; Hao, S.; Wang, Q.; Li, K.; Qiao, T. Low expression of ferroxidases is implicated in the iron retention in human atherosclerotic plaques. Biochem. Biophys. Res. Commun. 2015, 464, 1134–1138. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Recalcati, S.; Locati, M.; Gammella, E.; Invernizzi, P.; Cairo, G. Iron levels in polarized macrophages: Regulation of immunity and autoimmunity. Autoimmun. Rev. 2012, 11, 883–889. [Google Scholar] [CrossRef] [PubMed]
- de Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1- and M2-Type Macrophage Responses Are Predictive of Adverse Outcomes in Human Atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- Nairz, M.; Schroll, A.; Demetz, E.; Tancevski, I.; Theurl, I.; Weiss, G. ‘Ride on the ferrous wheel’--the cycle of iron in macrophages in health and disease. Immunobiology 2015, 220, 280–294. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Knutson, M.D.; Vafa, M.R.; Haile, D.J.; Wessling-Resnick, M. Iron loading and erythrophagocytosis increase ferroportin 1 (FPN1) expression in J774 macrophages. Blood 2003, 102, 4191–4197. [Google Scholar] [CrossRef]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Habib, A.; Finn, A.V. The role of iron metabolism as a mediator of macrophage inflammation and lipid handling in atherosclerosis. Front. Pharmacol. 2014, 5, 195. [Google Scholar] [CrossRef]
- Chawla, A.; Boisvert, W.A.; Lee, C.H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Haschka, D.; Hoffmann, A.; Weiss, G. Iron in immune cell function and host defense. Semin. Cell Dev. Biol. 2021, 115, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, M.; Liu, Y.; Li, H.; Shang, L.; Xu, T.; Chen, Z.; Wang, F.; Qiao, T.; Li, K. Iron accumulation in macrophages promotes the formation of foam cells and development of atherosclerosis. Cell BioSci. 2020, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Marcil, V.; Lavoie, J.C.; Emonnot, L.; Seidman, E.; Levy, E. Analysis of the effects of iron and vitamin C co-supplementation on oxidative damage, antioxidant response and inflammation in THP-1 macrophages. Clin. Biochem. 2011, 44, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Dufrusine, B.; Di Francesco, A.; Oddi, S.; Scipioni, L.; Angelucci, C.B.; D’Addario, C.; Serafini, M.; Häfner, A.K.; Steinhilber, D.; Maccarrone, M.; et al. Iron-Dependent Trafficking of 5-Lipoxygenase and Impact on Human Macrophage Activation. Front. Immunol. 2019, 10, 1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Cai, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. Iron-load exacerbates the severity of atherosclerosis via inducing inflammation and enhancing the glycolysis in macrophages. J. Cell Physiol. 2019, 234, 18792–18800. [Google Scholar] [CrossRef]
- Fernández-García, V.; González-Ramos, S.; Avendaño-Ortiz, J.; Martín-Sanz, P.; Delgado, C.; Castrillo, A.; Boscá, L. NOD1 splenic activation confers ferroptosis protection and reduces macrophage recruitment under pro-atherogenic conditions. BioMed. Pharmacother. 2022, 148, 112769. [Google Scholar] [CrossRef]
- Yu, W.; Liu, W.; Xie, D.; Wang, Q.; Xu, C.; Zhao, H.; Lv, J.; He, F.; Chen, B.; Yamamoto, T.; et al. High Level of Uric Acid Promotes Atherosclerosis by Targeting NRF2-Mediated Autophagy Dysfunction and Ferroptosis. Oxid. Med. Cell Longev. 2022, 2022, 9304383. [Google Scholar] [CrossRef]
- Zhou, Y.; Que, K.T.; Zhang, Z.; Yi, Z.J.; Zhao, P.X.; You, Y.; Gong, J.P.; Liu, Z.J. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018, 7, 4012–4022. [Google Scholar] [CrossRef] [Green Version]
- Laskar, A.; Eilertsen, J.; Li, W.; Yuan, X.M. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochem. Biophys. Res. Commun. 2013, 441, 737–742. [Google Scholar] [CrossRef]
- Agoro, R.; Taleb, M.; Quesniaux, V.F.J.; Mura, C. Cell iron status influences macrophage polarization. PLoS ONE 2018, 13, e0196921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J. Clin. Invest 2011, 121, 985–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, L.; Negre-Salvayre, A.; Costa, L.; Canonne-Hergaux, F. Iron gene expression profile in atherogenic Mox macrophages. Biochim. Biophys. Acta 2016, 1862, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.W.; Wen, Y.H.; Ma, R.Q.; Chen, L.; Zeng, X.L.; Wen, W.P.; Sun, W. Ferroptosis Driver SOCS1 and Suppressor FTH1 Independently Correlate With M1 and M2 Macrophage Infiltration in Head and Neck Squamous Cell Carcinoma. Front. Cell Dev. Biol. 2021, 9, 727762. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xu, W.; Wang, Y.; Zhu, J.; Wang, H.; Tu, J.; Weng, Q.; Kong, C.; Yang, Y.; Qiu, R.; et al. Identification of critical ferroptosis regulators in lung adenocarcinoma that RRM2 facilitates tumor immune infiltration by inhibiting ferroptotic death. Clin. Immunol. 2021, 232, 108872. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Gu, Z.; Liu, T.; Liu, C.; Yang, Y.; Tang, J.; Song, H.; Wang, Y.; Yang, Y.; Yu, C. Ferroptosis-Strengthened Metabolic and Inflammatory Regulation of Tumor-Associated Macrophages Provokes Potent Tumoricidal Activities. Nano Lett. 2021, 21, 6471–6479. [Google Scholar] [CrossRef]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Invest. 2018, 128, 1106–1124. [Google Scholar] [CrossRef]
- Guo, L.; Sakamoto, A.; Cornelissen, A.; Hong, C.C.; Finn, A.V. Ironing-Out the Role of Hepcidin in Atherosclerosis. Arter. Thromb. Vasc. Biol. 2019, 39, 303–305. [Google Scholar] [CrossRef]
- Youssef, L.A.; Rebbaa, A.; Pampou, S.; Weisberg, S.P.; Stockwell, B.R.; Hod, E.A.; Spitalnik, S.L. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood 2018, 131, 2581–2593. [Google Scholar] [CrossRef]
- Liu, W.; Östberg, N.; Yalcinkaya, M.; Dou, H.; Endo-Umeda, K.; Tang, Y.; Hou, X.; Xiao, T.; Fidler, T.P.; Abramowicz, S.; et al. Erythroid lineage Jak2V617F expression promotes atherosclerosis through erythrophagocytosis and macrophage ferroptosis. J. Clin. Investig. 2022, 132, 13. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, Y.; Ruan, J.; Chen, Y.; Wang, X.; Zhou, F.; Meng, F. Decreased Iron Ion Concentrations in the Peripheral Blood Correlate with Coronary Atherosclerosis. Nutrients 2022, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.L.; Imran Ul-Haq, M.; Abbina, S.; Kalathottukaren, M.T.; Lai, B.F.; Hatef, A.; Unniappan, S.; Kizhakkedathu, J.N. In vivo efficacy, toxicity and biodistribution of ultra-long circulating desferrioxamine based polymeric iron chelator. Biomaterials 2016, 102, 58–71. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Forms of Death | Influence | Reference |
|---|---|---|
| Apoptosis | Protective in early stage and harmful in late stage | [6] |
| Pyroptosis | Enlarges necrotic core and destroys plaque stability | [5] |
| Necroptosis | Enlarges necrotic core and induces inflammation | [7] |
| Autophagy | Reduces ROS and plays a protective role in AS | [8] |
| Regulators of Ferroptosis | Effects | Subjects | Reference | |
|---|---|---|---|---|
| Inducers | Ammonium ferric citrate | Increases intracellular iron load | Foam cells | [14] |
| NCOA4 | Increases intracellular iron storage and degrades ferritin | Macrophages | [15] | |
| BAY 11-7085 | Up-regulates HO-1 through Nrf2-SLC7A11-HO-1 pathway and increases free iron | Cancer cells | [16] | |
| HO-1 and Hb | Increases free iron by decomposing Hb | Cancer cells | [16] | |
| WA | Decreases the activity of GPX4; activates HO-1 by KEAP1 | Neuroblastoma cells | [17] | |
| Siramesine and lapatinib | Increase transferrin and iron entering cells; decrease FPN and iron leaving cells | Breast cancer cells | [18] | |
| CaFe@DMSN/C NPs | Increases intracellular iron and ROS | 4T1 and Raw264.7 cells | [19] | |
| Inhibitors | Fer-1 | Decreases intracellular iron and ROS | MAECs | [20] |
| Curcumin and EGCG | Chelate iron and limit the accumulation of iron in cells | Mouse MIN6 pancreatic-cells | [21] | |
| Heat shock protein 27 | Restricts LIP; Reduces iron intake through TfR1 | Chinese hamster lung fibroblast cells | [22] | |
| CISD1,2 | Decreases iron and ROS in mitochondria | Human HCC cell lines/ HNC and SNU cell lines | [23,24] | |
| DFO | Chelates iron and limits iron accumulation in cells | HT-1080 | [10] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Zhang, H.; Chen, Y.; Liu, X.; Tian, J.; Shen, W. The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis. Biomolecules 2022, 12, 1702. https://doi.org/10.3390/biom12111702
Ma J, Zhang H, Chen Y, Liu X, Tian J, Shen W. The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis. Biomolecules. 2022; 12(11):1702. https://doi.org/10.3390/biom12111702
Chicago/Turabian StyleMa, Jiedong, Hongqi Zhang, Yufei Chen, Xiaojin Liu, Jiamin Tian, and Wei Shen. 2022. "The Role of Macrophage Iron Overload and Ferroptosis in Atherosclerosis" Biomolecules 12, no. 11: 1702. https://doi.org/10.3390/biom12111702