
Essential Components of Synthetic Infectious Prion Formation De Novo

Abstract
:1. Introduction
2. Defining a Prion
2.1. PK Resistance
2.2. Toxicity
2.3. Infectivity
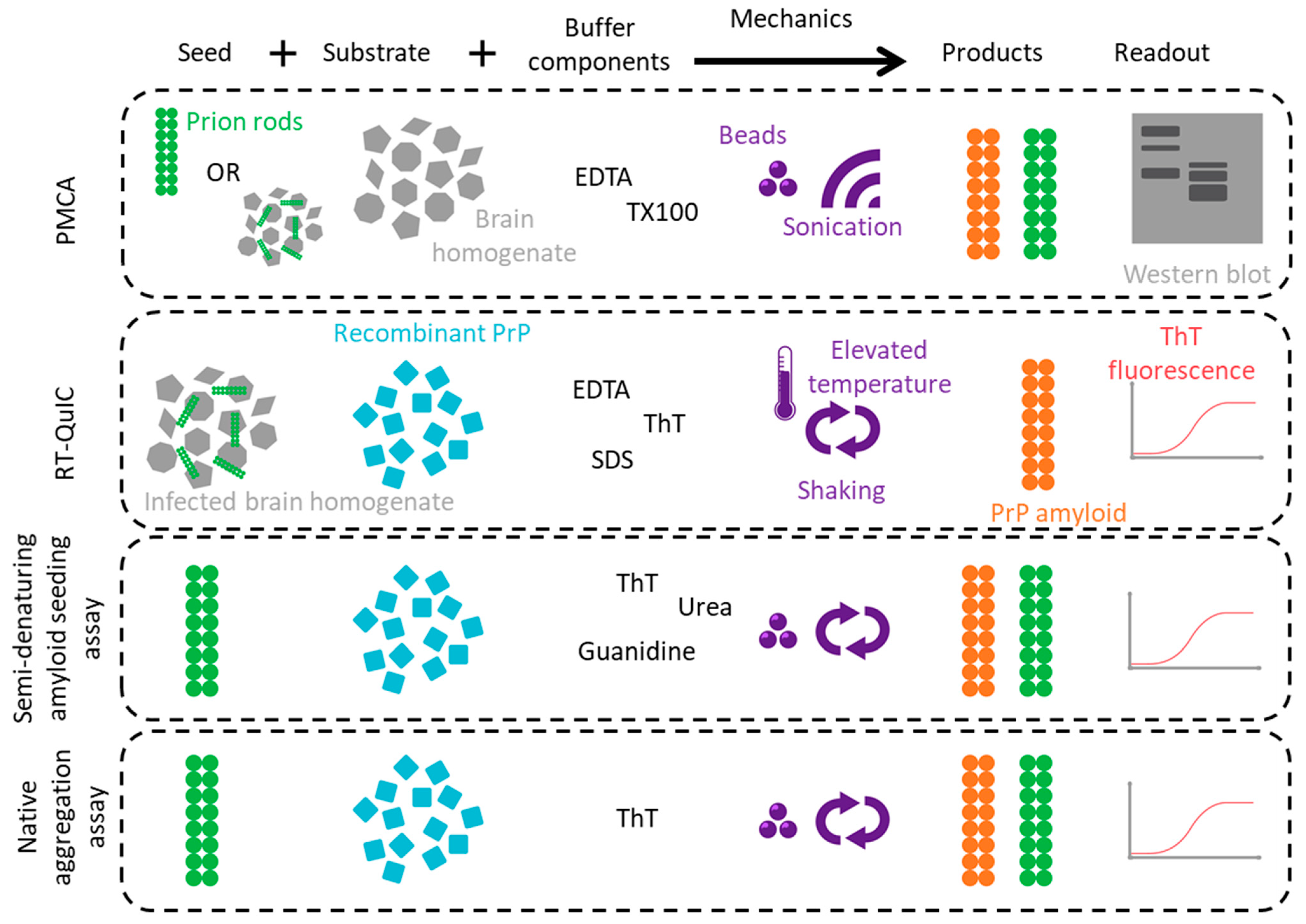
3. Synthetic Prion Generation
3.1. PMCA
3.2. Amyloid Seeding Assay/RT-QuIC
3.3. Semi-Denaturing Amyloid Seeding Assays
3.4. Native Aggregation Assays
4. Non-Protein Requirements
4.1. Post-Translational Modifications
4.2. Lipids and RNA
5. Current State of the Field
Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Liberski, P.P. Historical overview of prion diseases: A view from afar. Folia Neuropathol. 2012, 50, 1–12. [Google Scholar]
- Sternbach, G.; Dibble, C.L.; Varon, J. From Creutzfeldt-Jakob disease to the mad cow epidemic. J. Emerg. Med. 1997, 15, 701–705. [Google Scholar] [CrossRef]
- Gajdusek, D.C.; Gibbs, C.J. Attempts to demonstrate a transmissible agent in Kuru, amylotrophic lateral sclerosis, and other sub-acute and chornic nervous system degenerations of man. Nature 1964, 204, 257–259. [Google Scholar] [CrossRef]
- Hadlow, W.J. Scrapie and Kuru. Lancet 1959, 274, 289–290. [Google Scholar] [CrossRef]
- Brown, P.; Bradley, R. 1755 and all that: A historical primer of transmissible spongiform encephalopathy. BMJ 1998, 317, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Manka, S.W.; Zhang, W.; Wenborn, A.; Betts, J.; Joiner, S.; Saibil, H.R.; Collinge, J.; Wadsworth, J.D.F. 2.7 Å cryo-EM structure of ex vivo RML prion fibrils. Nat. Commun. 2022, 13, 4004. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar]
- Weissmann, C. The state of the prion. Nat. Rev. Microbiol. 2004, 2, 861–871. [Google Scholar] [CrossRef]
- van Rheede, T.; Smolenaars, M.M.; Madsen, O.; de Jong, W.W. Molecular evolution of the mammalian prion protein. Mol. Biol. Evol. 2003, 20, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Dong, X.P. Epidemiological characteristics of human prion diseases. Infect. Dis. Poverty 2016, 5, 47. [Google Scholar] [CrossRef] [Green Version]
- Nathanson, N.; Wilesmith, J.; Griot, C. Bovine spongiform encephalopathy (BSE): Causes and consequences of a common source epidemic. Am. J. Epidemiol. 1997, 145, 959–969. [Google Scholar] [CrossRef]
- Saunders, S.E.; Bartelt-Hunt, S.L.; Bartz, J.C. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerg. Infect. Dis. 2012, 18, 369–376. [Google Scholar] [CrossRef]
- Aranda-Anzaldo, A. Possible cell-free prion replication. Med. Hypotheses 1992, 38, 249–251. [Google Scholar] [CrossRef]
- Yakupova, E.I.; Bobyleva, L.G.; Vikhlyantsev, I.M.; Bobylev, A.G. Congo Red and amyloids: History and relationship. Biosci. Rep. 2019, 39, BSR20181415. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-resolution structure and strain comparison of infectious mammalian prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Wenborn, A.; Terry, C.; Gros, N.; Joiner, S.; D’Castro, L.; Panico, S.; Sells, J.; Cronier, S.; Linehan, J.M.; Brandner, S.; et al. A novel and rapid method for obtaining high titre intact prion strains from mammalian brain. Sci. Rep. 2015, 5, 10062. [Google Scholar] [CrossRef] [Green Version]
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. NeuroSci. 2010, 11, 155–159. [Google Scholar] [CrossRef] [Green Version]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Purro, S.A.; Farrow, M.A.; Linehan, J.; Nazari, T.; Thomas, D.X.; Chen, Z.; Mengel, D.; Saito, T.; Saido, T.; Rudge, P.; et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018, 564, 415–419. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Sajnani, G.; Requena, J.R. Prions, proteinase K and infectivity. Prion 2012, 6, 430–432. [Google Scholar] [CrossRef] [Green Version]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Betts, J.; Joiner, S.; Saibil, H.R.; Collinge, J.; Wadsworth, J.D.F. A structural basis for prion strain diversity. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar] [CrossRef]
- Li, Q.; Wang, F.; Xiao, X.; Kim, C.; Bohon, J.; Kiselar, J.; Safar, J.G.; Ma, J.; Surewicz, W.K. Structural attributes of mammalian prion infectivity: Insights from studies with synthetic prions. J. Biol. Chem. 2018, 293, 18494–18503. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Reilly, M.; Terry, C.; Wenborn, A.; Schmidt, C.; Marinho, A.T.; Risse, E.; Al-Doujaily, H.; Wiggins De Oliveira, M.; Sandberg, M.K.; et al. Highly infectious prions are not directly neurotoxic. Proc. Natl. Acad. Sci. USA 2020, 117, 23815. [Google Scholar] [CrossRef]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat. Commun. 2014, 5, 4347. [Google Scholar] [CrossRef] [Green Version]
- van der Merwe, J.; Aiken, J.; Westaway, D.; McKenzie, D. The standard scrapie cell assay: Development, utility and prospects. Viruses 2015, 7, 180–198. [Google Scholar] [CrossRef]
- Klöhn, P.C.; Stoltze, L.; Flechsig, E.; Enari, M.; Weissmann, C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc. Natl. Acad. Sci. USA 2003, 100, 11666–11671. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef]
- Ayers, J.I.; Lee, J.; Monteiro, O.; Woerman, A.L.; Lazar, A.A.; Condello, C.; Paras, N.A.; Prusiner, S.B. Different α-synuclein prion strains cause dementia with Lewy bodies and multiple system atrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2113489119. [Google Scholar] [CrossRef]
- Kim, C.; Xiao, X.; Chen, S.; Haldiman, T.; Smirnovas, V.; Kofskey, D.; Warren, M.; Surewicz, K.; Maurer, N.R.; Kong, Q.; et al. Artificial strain of human prions created in vitro. Nat. Commun. 2018, 9, 2166. [Google Scholar] [CrossRef] [Green Version]
- Post, K.; Pitschke, M.; Schäfer, O.; Wille, H.; Appel, T.R.; Kirsch, D.; Mehlhorn, I.; Serban, H.; Prusiner, S.B.; Riesner, D. Rapid acquisition of beta-sheet structure in the prion protein prior to multimer formation. Biol. Chem. 1998, 379, 1307–1317. [Google Scholar] [CrossRef]
- Saborio, G.P.; Permanne, B.; Soto, C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001, 411, 810–813. [Google Scholar] [CrossRef]
- Castilla, J.; Saá, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Bieschke, J.; Weber, P.; Sarafoff, N.; Beekes, M.; Giese, A.; Kretzschmar, H. Autocatalytic self-propagation of misfolded prion protein. Proc. Natl. Acad. Sci. USA 2004, 101, 12207–12211. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Harris, B.T.; Rees, J.R.; Supattapone, S. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. USA 2007, 104, 9741–9746. [Google Scholar] [CrossRef] [Green Version]
- Moudjou, M.; Chapuis, J.; Mekrouti, M.; Reine, F.; Herzog, L.; Sibille, P.; Laude, H.; Vilette, D.; Andréoletti, O.; Rezaei, H.; et al. Glycoform-independent prion conversion by highly efficient, cell-based, protein misfolding cyclic amplification. Sci. Rep. 2016, 6, 29116. [Google Scholar] [CrossRef] [Green Version]
- Baskakov, I.V.; Legname, G.; Baldwin, M.A.; Prusiner, S.B.; Cohen, F.E. Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 2002, 277, 21140–21148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.S.; Hosszu, L.L.; Power, A.; Hill, A.F.; Kenney, J.; Saibil, H.; Craven, C.J.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999, 283, 1935–1937. [Google Scholar] [CrossRef] [Green Version]
- Colby, D.W.; Zhang, Q.; Wang, S.; Groth, D.; Legname, G.; Riesner, D.; Prusiner, S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA 2007, 104, 20914–20919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieschke, J.; Giese, A.; Schulz-Schaeffer, W.; Zerr, I.; Poser, S.; Eigen, M.; Kretzschmar, H. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc. Natl. Acad. Sci. USA 2000, 97, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieschke, J.; Schwille, P. Aggregation of prion protein investigated by dual-color fluorescence cross-correlation spectroscopy. In Fluorescence Microscopy and Fluorescent Probes; Plenum Press: New York, NY, USA, 1998; pp. 81–86. [Google Scholar]
- do Carmo Ferreira, N.; Caughey, B. Cell-free prion protein conversion assays in screening for anti-prion drug candidates. Curr. Opin. Pharm. 2019, 44, 1–7. [Google Scholar] [CrossRef]
- Pitschke, M.; Prior, R.; Haupt, M.; Riesner, D. Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat. Med. 1998, 4, 832–834. [Google Scholar] [CrossRef]
- Atarashi, R.; Wilham, J.M.; Christensen, L.; Hughson, A.G.; Moore, R.A.; Johnson, L.M.; Onwubiko, H.A.; Priola, S.A.; Caughey, B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat. Methods 2008, 5, 211–212. [Google Scholar] [CrossRef]
- Orru, C.D.; Wilham, J.M.; Vascellari, S.; Hughson, A.G.; Caughey, B. New generation QuIC assays for prion seeding activity. Prion 2012, 6, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Orru, C.D.; Groveman, B.R.; Hughson, A.G.; Manca, M.; Raymond, L.D.; Raymond, G.J.; Campbell, K.J.; Anson, K.J.; Kraus, A.; Caughey, B. RT-QuIC Assays for Prion Disease Detection and Diagnostics. Methods Mol. Biol. 2017, 1658, 185–203. [Google Scholar] [CrossRef]
- Mok, T.H.; Nihat, A.; Luk, C.; Sequeira, D.; Batchelor, M.; Mead, S.; Collinge, J.; Jackson, G.S. Bank vole prion protein extends the use of RT-QuIC assays to detect prions in a range of inherited prion diseases. Sci. Rep. 2021, 11, 5231. [Google Scholar] [CrossRef] [PubMed]
- Orru, C.D.; Groveman, B.R.; Hughson, A.G.; Zanusso, G.; Coulthart, M.B.; Caughey, B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. mBio 2015, 6, e02451-14. [Google Scholar] [CrossRef]
- Cramm, M.; Schmitz, M.; Karch, A.; Mitrova, E.; Kuhn, F.; Schroeder, B.; Raeber, A.; Varges, D.; Kim, Y.S.; Satoh, K.; et al. Stability and Reproducibility Underscore Utility of RT-QuIC for Diagnosis of Creutzfeldt-Jakob Disease. Mol. Neurobiol. 2016, 53, 1896–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabareesan, A.T.; Udgaonkar, J.B. The G126V Mutation in the Mouse Prion Protein Hinders Nucleation-Dependent Fibril Formation by Slowing Initial Fibril Growth and by Increasing the Critical Concentration. Biochemistry 2017, 56, 5931–5942. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, L.; Lowe, J.; Self, T.; Kenward, N.; Landon, M.; McBride, T.; Farquhar, C.; McConnell, I.; Brown, J.; Hope, J. Lysosomes as key organelles in the pathogenesis of prion encephalopathies. J. Pathol. 1992, 166, 333–341. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Daggett, V. Influence of pH on the human prion protein: Insights into the early steps of misfolding. Biophys. J. 2010, 99, 2289–2298. [Google Scholar] [CrossRef] [Green Version]
- Sangar, D.; Jack, K.; Batchelor, M.; Mistry, B.; Bieschke, J. Syntaxin 6 delays prion protein fibril formation and prolongs presence of toxic aggregation intermediates. bioRxiv 2022. [Google Scholar] [CrossRef]
- Appel, T.R.; Dumpitak, C.; Matthiesen, U.; Riesner, D. Prion rods contain an inert polysaccharide scaffold. Biol. Chem. 1999, 380, 1295–1306. [Google Scholar] [CrossRef]
- Burke, C.M.; Walsh, D.J.; Steele, A.D.; Agrimi, U.; Di Bari, M.A.; Watts, J.C.; Supattapone, S. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog. 2019, 15, e1007662. [Google Scholar] [CrossRef]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [Green Version]
- Imamura, M.; Kato, N.; Yoshioka, M.; Okada, H.; Iwamaru, Y.; Shimizu, Y.; Mohri, S.; Yokoyama, T.; Murayama, Y. Glycosylphosphatidylinositol anchor-dependent stimulation pathway required for generation of baculovirus-derived recombinant scrapie prion protein. J. Virol. 2011, 85, 2582–2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Timmes, A.G.; Moore, R.A.; Fischer, E.R.; Priola, S.A. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS ONE 2013, 8, e71081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Wang, F. Prion disease and the ‘protein-only hypothesis’. Essays Biochem. 2014, 56, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Orru, C.D.; Groveman, B.R.; Surewicz, K.; Abskharon, R.; Imamura, M.; Yokoyama, T.; Kim, Y.S.; Vander Stel, K.J.; et al. Self-propagating, protease-resistant, recombinant prion protein conformers with or without in vivo pathogenicity. PLoS Pathog. 2017, 13, e1006491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Zhang, Z.; Wang, X.; Li, J.; Zha, L.; Yuan, C.G.; Weissmann, C.; Ma, J. Genetic informational RNA is not required for recombinant prion infectivity. J. Virol. 2012, 86, 1874–1876. [Google Scholar] [CrossRef] [Green Version]
- Deleault, N.R.; Walsh, D.J.; Piro, J.R.; Wang, F.; Wang, X.; Ma, J.; Rees, J.R.; Supattapone, S. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. USA 2012, 109, E1938–E1946. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Fizet, J.; Properzi, F.; Batchelor, M.; Sandberg, M.K.; Edgeworth, J.A.; Afran, L.; Ho, S.; Badhan, A.; Klier, S.; et al. A systematic investigation of production of synthetic prions from recombinant prion protein. Open Biol. 2015, 5, 150165. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, K.; Ball, H.L.; Wille, H.; Zhang, H.; Groth, D.; Torchia, M.; Tremblay, P.; Safar, J.; Prusiner, S.B.; DeArmond, S.J.; et al. A synthetic peptide initiates Gerstmann-Sträussler-Scheinker (GSS) disease in transgenic mice. J. Mol. Biol. 2000, 295, 997–1007. [Google Scholar] [CrossRef]
- Barria, M.A.; Mukherjee, A.; Gonzalez-Romero, D.; Morales, R.; Soto, C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 2009, 5, e1000421. [Google Scholar] [CrossRef] [Green Version]
- Colby, D.W.; Giles, K.; Legname, G.; Wille, H.; Baskakov, I.V.; DeArmond, S.J.; Prusiner, S.B. Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. USA 2009, 106, 20417–20422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarava, N.; Kovacs, G.G.; Bocharova, O.; Savtchenko, R.; Alexeeva, I.; Budka, H.; Rohwer, R.G.; Baskakov, I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010, 119, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Wain, R.; Baskakov, I.V.; Legname, G.; Palmer, C.G.; Nguyen, H.O.; Lemus, A.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Protease-sensitive synthetic prions. PLoS Pathog. 2010, 6, e1000736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhang, Y.; Wang, F.; Wang, X.; Xu, Y.; Yang, H.; Yu, G.; Yuan, C.; Ma, J. De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J. 2013, 27, 4768–4775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moda, F.; Le, T.N.; Aulic, S.; Bistaffa, E.; Campagnani, I.; Virgilio, T.; Indaco, A.; Palamara, L.; Andreoletti, O.; Tagliavini, F.; et al. Synthetic prions with novel strain-specified properties. PLoS Pathog. 2015, 11, e1005354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koloteva-Levine, N.; Aubrey, L.D.; Marchante, R.; Purton, T.J.; Hiscock, J.R.; Tuite, M.F.; Xue, W.F. Amyloid particles facilitate surface-catalyzed cross-seeding by acting as promiscuous nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2104148118. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, C.; Li, J.; Mahal, S.P.; Browning, S. Prions on the move. EMBO Rep. 2011, 12, 1109–1117. [Google Scholar] [CrossRef]
- Eigen, M.; Fau-Schuster, P.; Schuster, P. Part A: Emergence of the hypercycle. In The Hypercycle: A Principle of Natural Self-Organization; Springer: Berlin/Heidelberg, Germany, 1977; Volume 64, pp. 541–565. [Google Scholar] [CrossRef]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [Green Version]
- Piro, J.R.; Wang, F.; Walsh, D.J.; Rees, J.R.; Ma, J.; Supattapone, S. Seeding specificity and ultrastructural characteristics of infectious recombinant prions. Biochemistry 2011, 50, 7111–7116. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Year | Name of Strain | Seed | Substrate Source | Co-Factors | Aggregation Mechanism | Seeding Ability | Animal Strain | Specific Infectivity |
|---|---|---|---|---|---|---|---|---|
| 2000 [70] | None | Artificially synthesized PrP (89–143, Pro101Leu) peptide | None | 3-week acetonitrile incubation at 4°, followed by 3 lyophilisation and washing steps | N/A | Mice expressing low levels of a transgene coding for PrP (Pro101Leu) | Not mentioned | |
| 2004 [42] | MoSP1 | None | Recombinant, bacterial, Mouse PrP 89–230 | None | Denaturing aggregation assay | Serial transmission to FVB mice and Tg4053 mice gave mean incubation time of 154 and 90 days, respectively | Mice overexpressing PrP (89–231) (known as Tg9949 mice) | Not mentioned |
| 2007 [39] | Sc237 PrP 27–30 | Purified hamster PrP | Co-purified lipid, poly(A) RNA | PMCA | N/A | Golden Syrian hamsters | ~5 × 104 LD(50) per mL | |
| 139H | Purified hamster PrP | Co-purified lipid, poly(A) RNA | PMCA | N/A | Golden Syrian hamsters | Not mentioned | ||
| None | Purified hamster PrP | Co-purified lipid, poly(A) RNA | PMCA | N/A | Golden Syrian hamsters | ~5 × 103 LD(50) per mL | ||
| 2009 [71] | None | Sonicated brain homogenate from Syrian hamsters | Sonicated brain homogenate from Syrian hamsters | PMCA | Serial passage being carried out at time of publication | Syrian hamsters | Not mentioned | |
| 2009 [72] | “MoSP5”, “MoSP6”, and “MoSP7” | None | Recombinant, bacterial, Mouse PrP 89–230 (MoSP6 and MoSP6) or Mouse PrP 23–230 (MoSP5) | None | Denaturing aggregation assay | Brain homogenates containing MoSP5, MoSP6, and MoSP7 transmitted disease to healthy Tg4053 mice ([72], Figure 3A and Table S5); MoSP6 and MoSP7 also transmitted disease to wild-type FVB mice | Mice overexpressing full-length, wild-type PrP (known as Tg4053 mice) | Not mentioned |
| 2010 [63] | rPrP-res(RNA)/OSU strain | None | Recombinant, bacterial, Mouse PrP 23–230 | RNA (mouse liver), POPG | PMCA | Able to propagate with normal mouse brain homogenate PMCA | CD-1 mice | Not mentioned |
| 2010 [73] | SSLOW | None | Recombinant, bacterial, Golden Syrian Hamster PrP 23–231 | Fibrils annealed with normal brain homogenate (with sonication) | Denaturing aggregation assay | Carried out serial passage, with some controls also producing PK resistant material | Golden Syrian hamsters | Not mentioned |
| 2010 [74] | MoSP1 | Recombinant, bacterial, Mouse PrP 89–230 | Co-purified from PTA prion precipitation (seeds) | Denaturing aggregation assay | Serial passage carried out caused disease | Tg9949 mice | Not mentioned | |
| 2012 [61] | rPrP-res(RNA) | Recombinant, bacterial, Mouse PrP 23–231 | Synthetic PE | PMCA | Propagation in many rounds of sPMCA | C57BL/6 mice | Not mentioned | |
| 2012 [67] | rPrP-res(RNA) | Recombinant, bacterial, Mouse PrP 23–230 | poly(rA) RNA, POPG | PMCA | Able to infect neuronal CAD5 cells | CD-1 mice | Not mentioned | |
| 2012 [68] | OSU co-factor PrP | rPrP-res(RNA) | Recombinant, bacterial, Mouse PrP 23–230 | Purified PE (mouse brain) | PMCA | Propagation of an ~18kD conformer maintained indefinitely | C57BL/6 mice | ∼2.2 × 106 LD50 units/μg PrP * |
| OSU protein-only PrP | OSU co-factor PrP | Recombinant, bacterial, Mouse PrP 23–230 | None | PMCA | 40%: no propagation, 60%: adaption to ~16kD band (which can be propagated indefinitely with rPrP), no propagation with normal BH | C57BL/6 mice | N/A | |
| 2013 [64] | rPrP-res(NIH) | None | Recombinant, bacterial, Mouse PrP 23–230 | RNA (mouse liver), POPG | PMCA | No prion formation in scrapie susceptible cell lines (SN56 or CF10) | C57BL/10 mice | N/A |
| 2013 [75] | Same as 2010, but in prion-free lab | None | Recombinant, bacterial, Mouse PrP 23–230 | RNA (mouse liver), POPG | PMCA | Able to propagate with normal mouse brain homogenate PMCA | CD-1 mice | Not mentioned |
| 2015 [76] | None | Recombinant, bacterial, Mouse PrP 23–231 | None | Denaturing aggregation assay | Able to seed mouse hypothalamic GT1 cells and mouse neuroblastoma N2a cells | CD-1 mice | N/A | |
| 2017 [66] | rPrP-res(RNA-low) | rPrP-res (RNA) | Recombinant, bacterial, Mouse PrP 23–230 | RNA (mouse liver), POPG | PMCA | No prion formation in CAD5 cells, but able to seed RT-QuIC reaction | C57BL/10 mice or Tga20 mice (which overexpress PrP 23–231) | N/A |
| 2019 [60] | OSU co = factor PrP | Bank vole brain homogenate | Bank vole BH | PMCA | PMCA product propagates at 27–30 kD | M109 bank voles | Not mentioned | |
| OSU protein-only PrP | Bank vole brain homogenate | Bank vole BH | PMCA | PMCA product propagates at 27–30 kD | M109 bank voles | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jack, K.; Jackson, G.S.; Bieschke, J. Essential Components of Synthetic Infectious Prion Formation De Novo. Biomolecules 2022, 12, 1694. https://doi.org/10.3390/biom12111694
Jack K, Jackson GS, Bieschke J. Essential Components of Synthetic Infectious Prion Formation De Novo. Biomolecules. 2022; 12(11):1694. https://doi.org/10.3390/biom12111694
Chicago/Turabian StyleJack, Kezia, Graham S. Jackson, and Jan Bieschke. 2022. "Essential Components of Synthetic Infectious Prion Formation De Novo" Biomolecules 12, no. 11: 1694. https://doi.org/10.3390/biom12111694