
Drug Repositioning Based on the Enhanced Message Passing and Hypergraph Convolutional Networks

Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
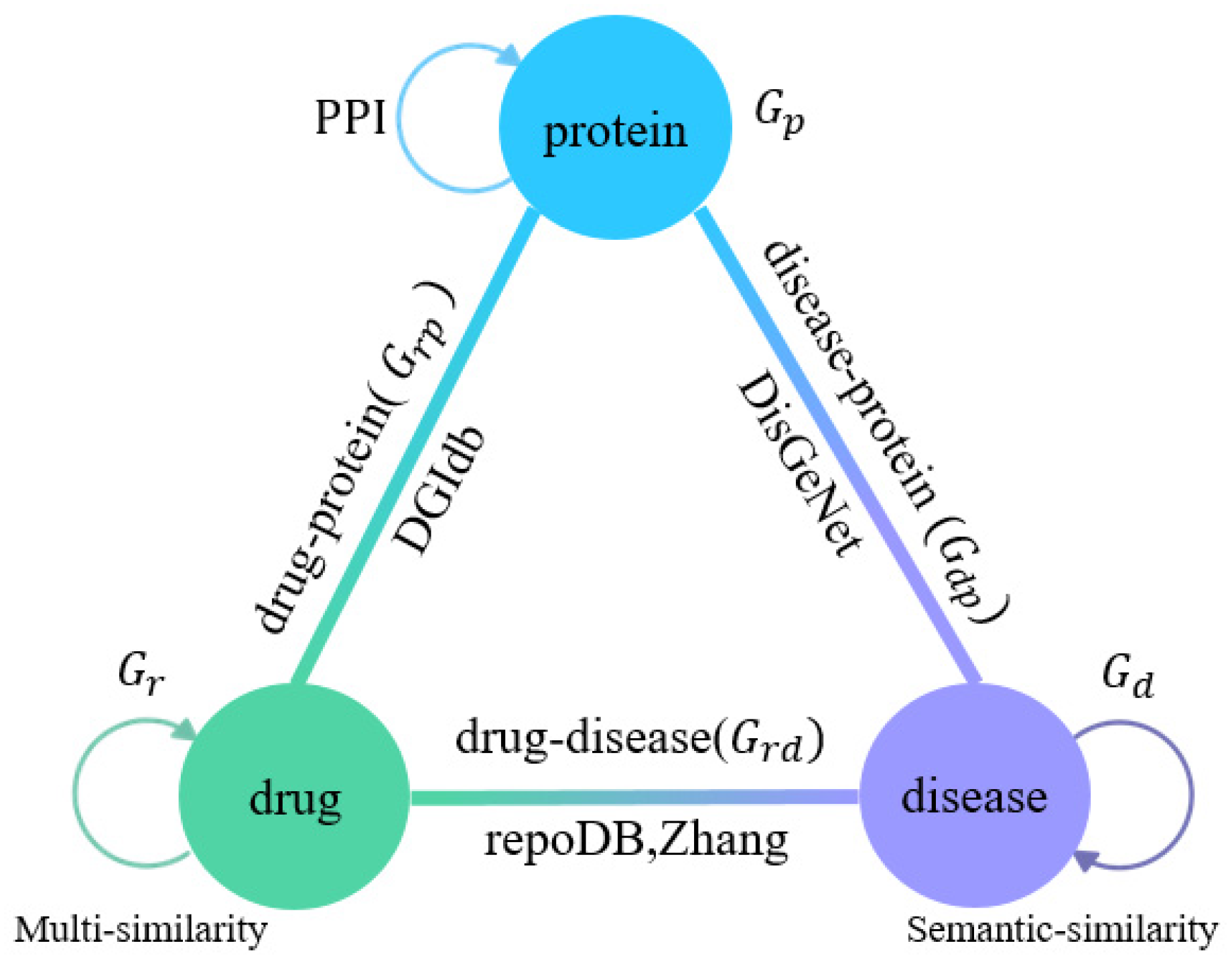
2.2. Multi-Association Network Construction
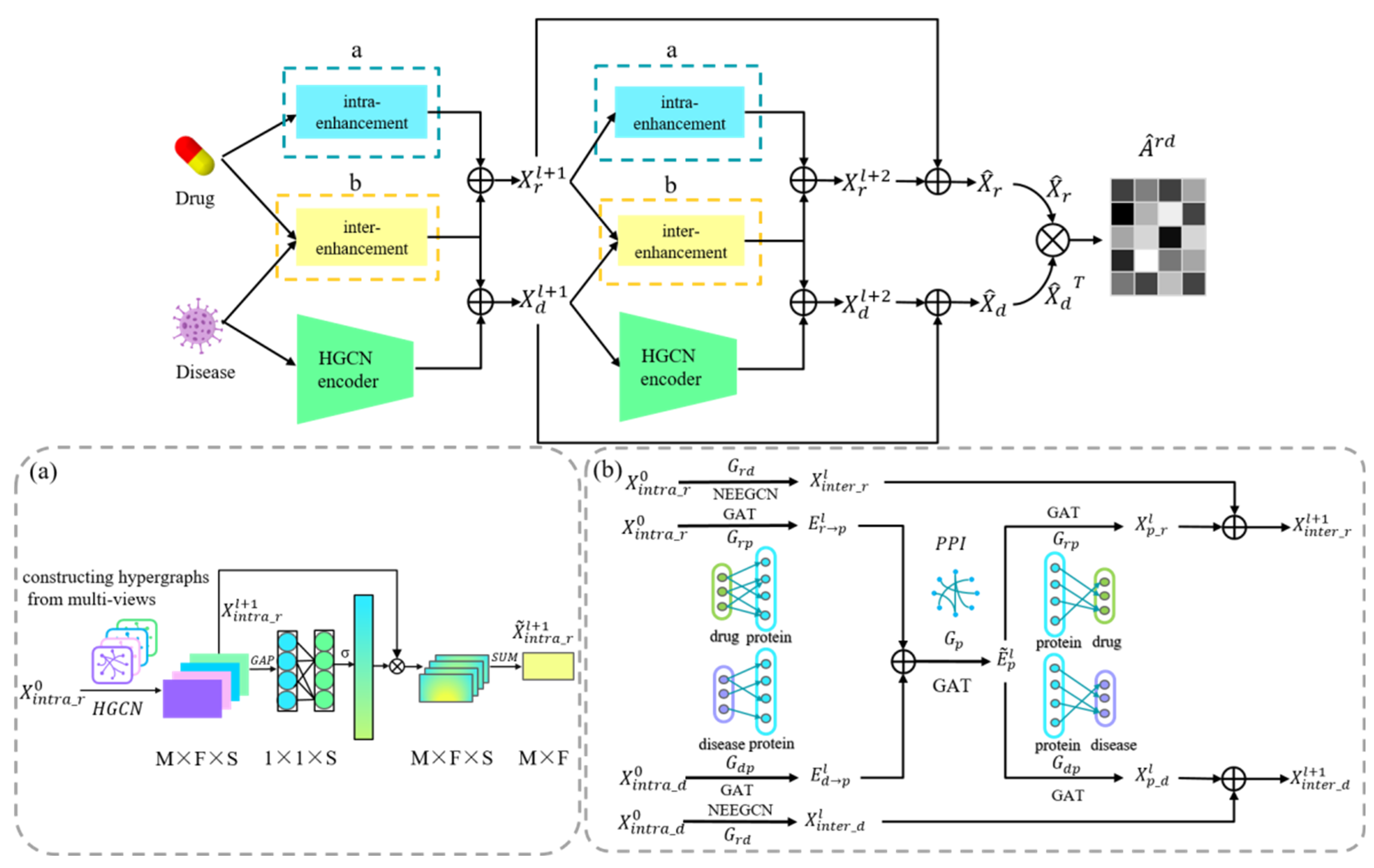
2.3. Model Framework
2.4. Drug and Disease Intra-Domain Message Passing
2.5. Drug and Disease Inter-Domain Message Passing
2.6. Known Drug-Disease Inter-Domain Message Passing
2.7. Protein-Related Inter-Domain Message Passing
2.8. Association Prediction for Drugs and Diseases
2.9. Optimization and Parameter Setting
3. Results
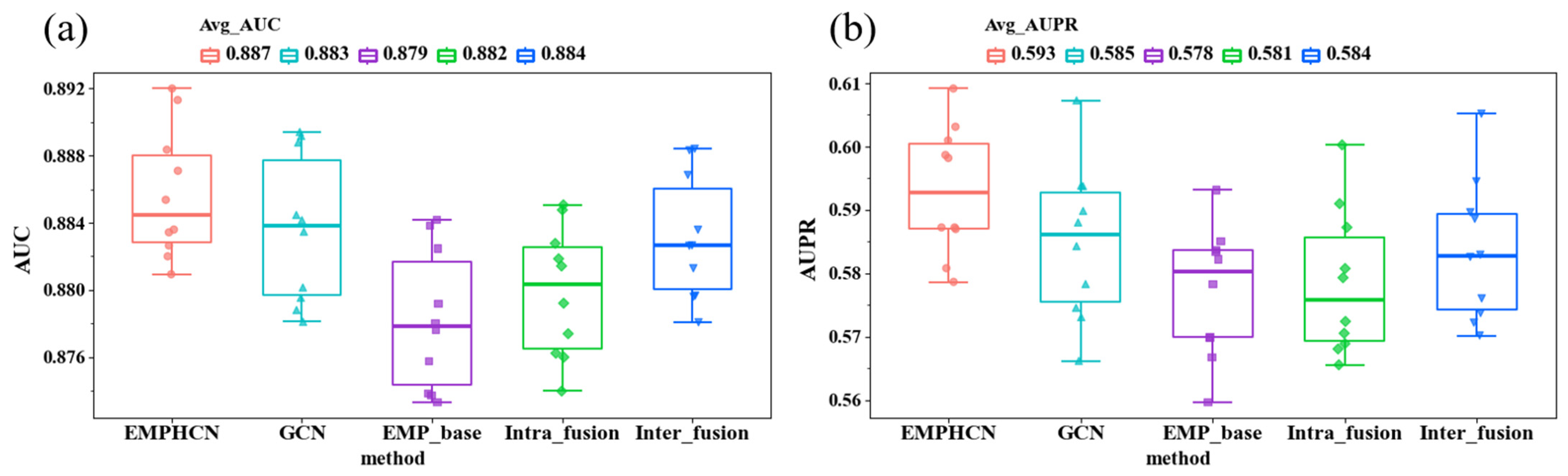
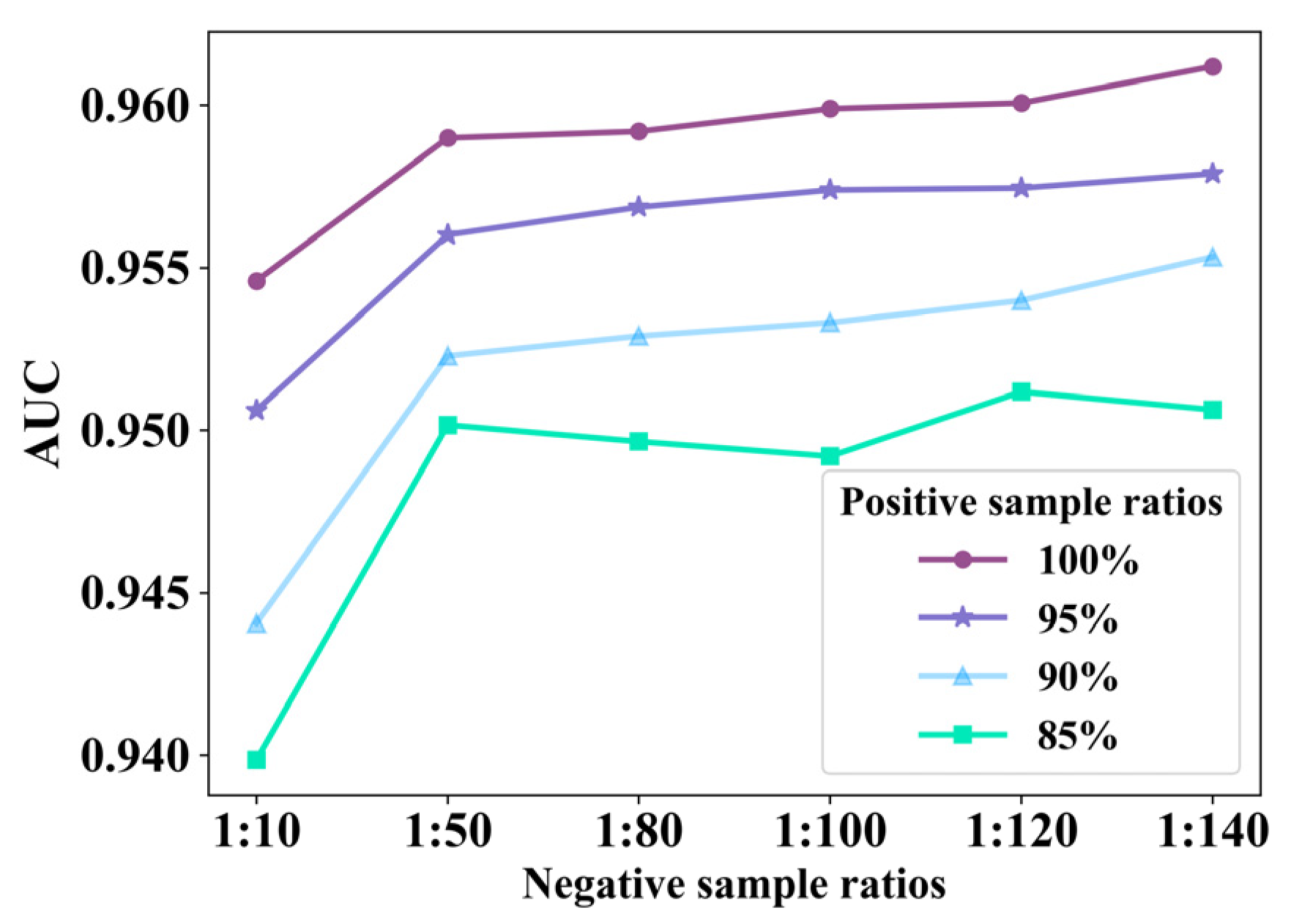
3.1. Ablation Experiment of EMPHCN Model
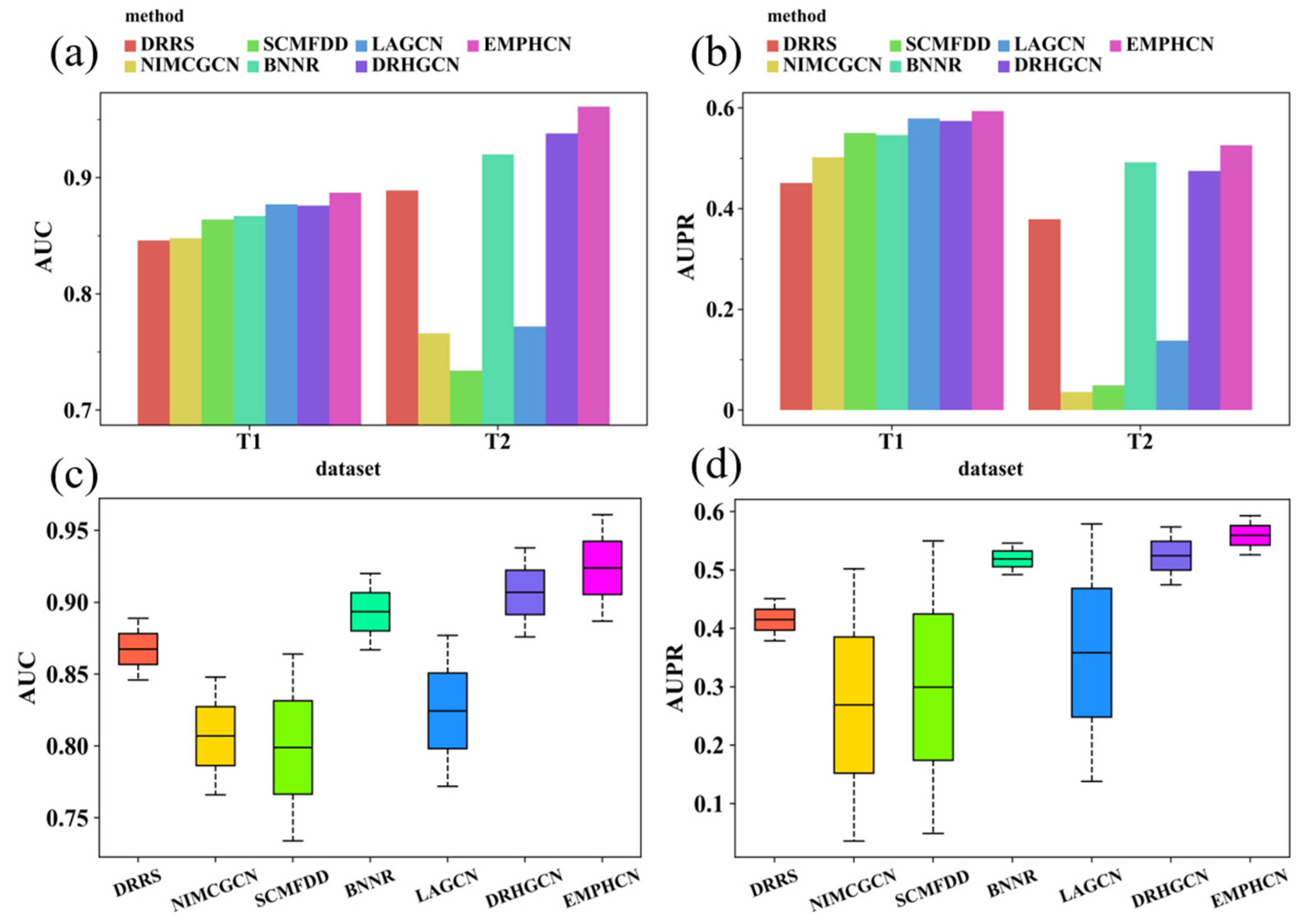
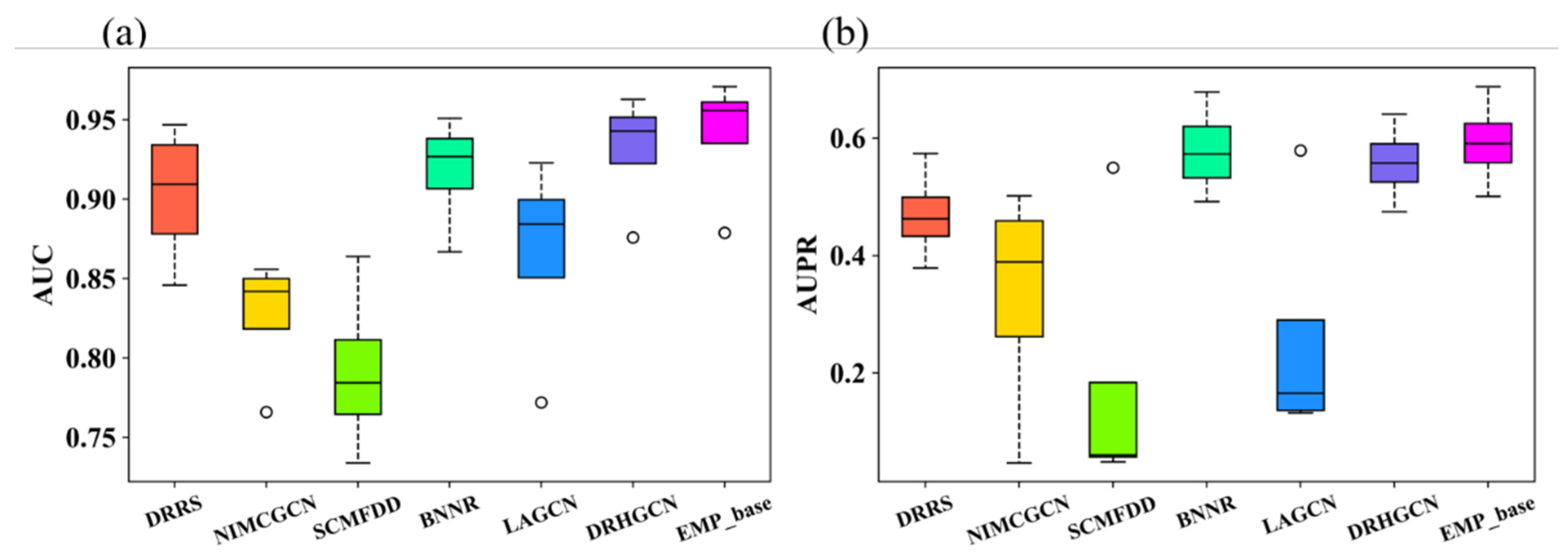
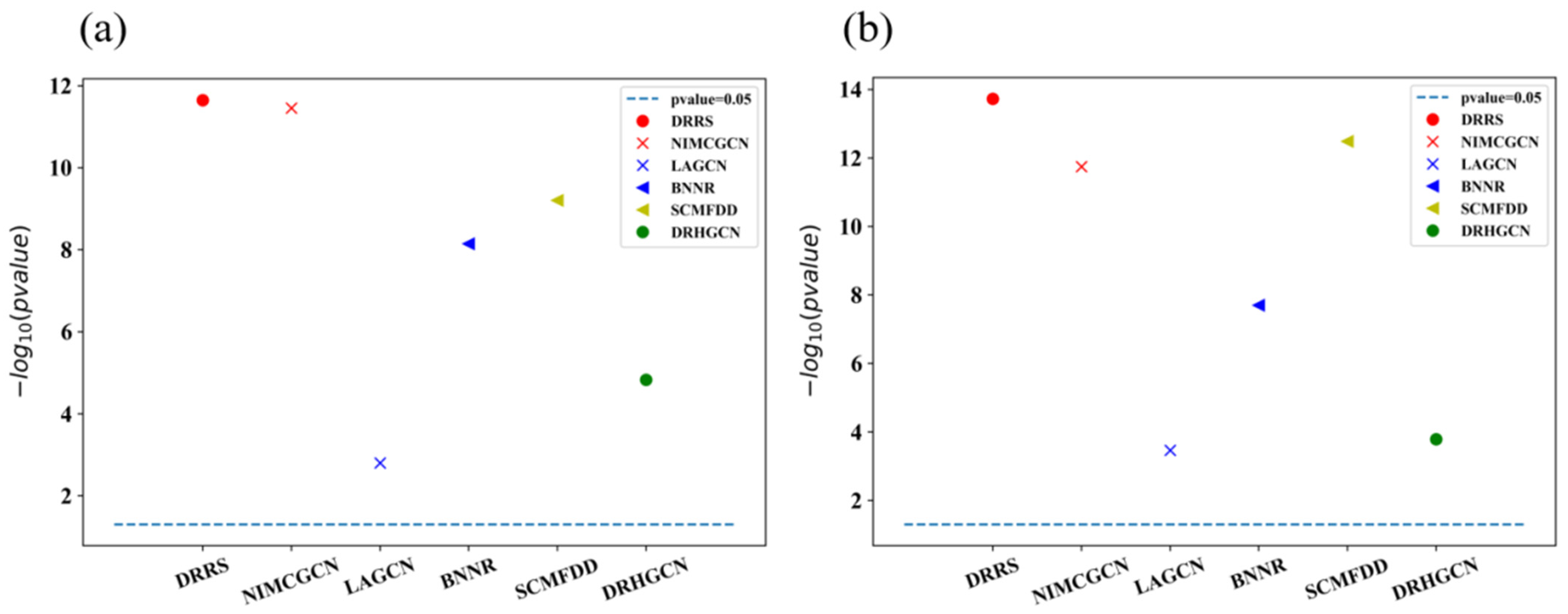
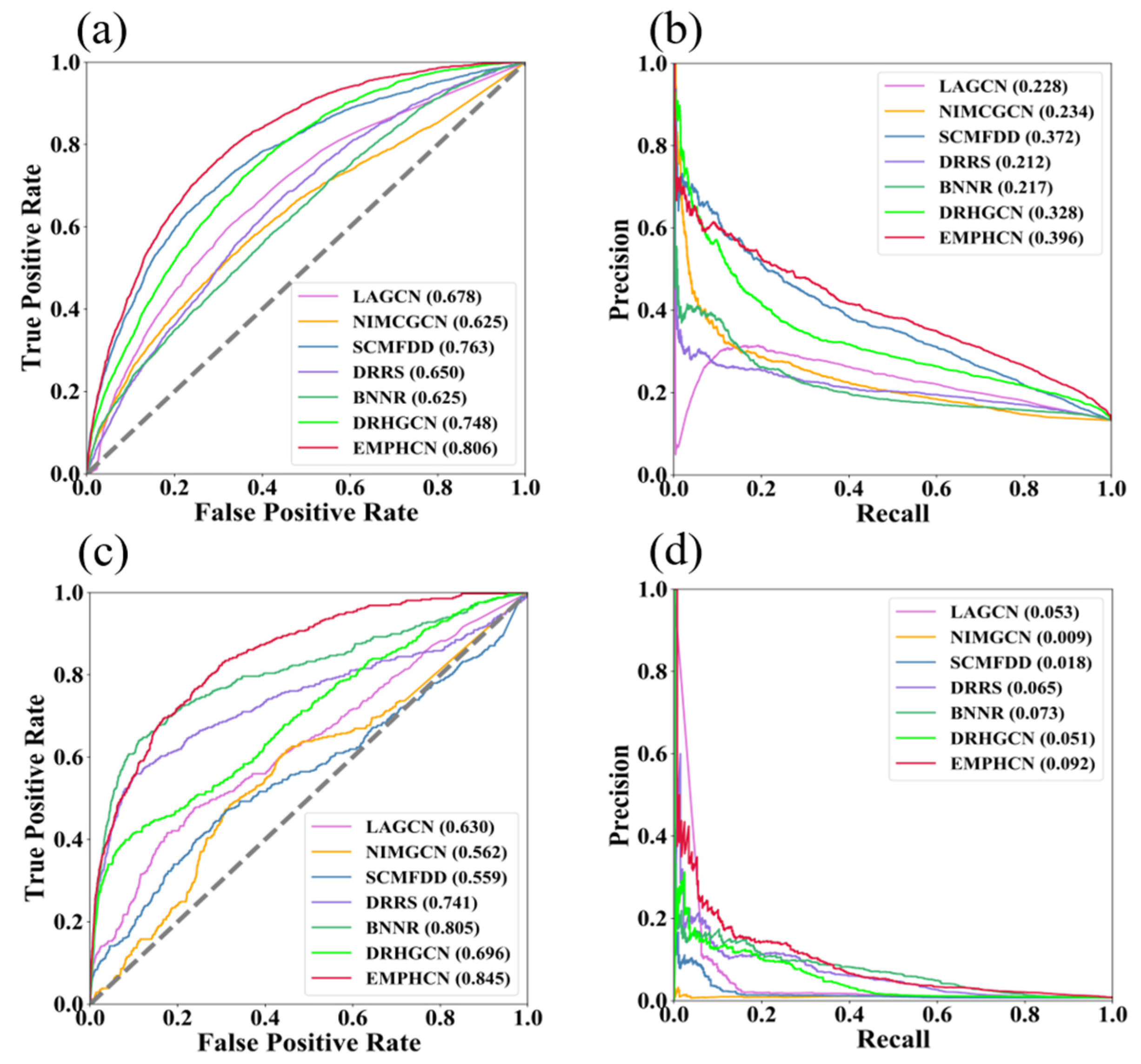
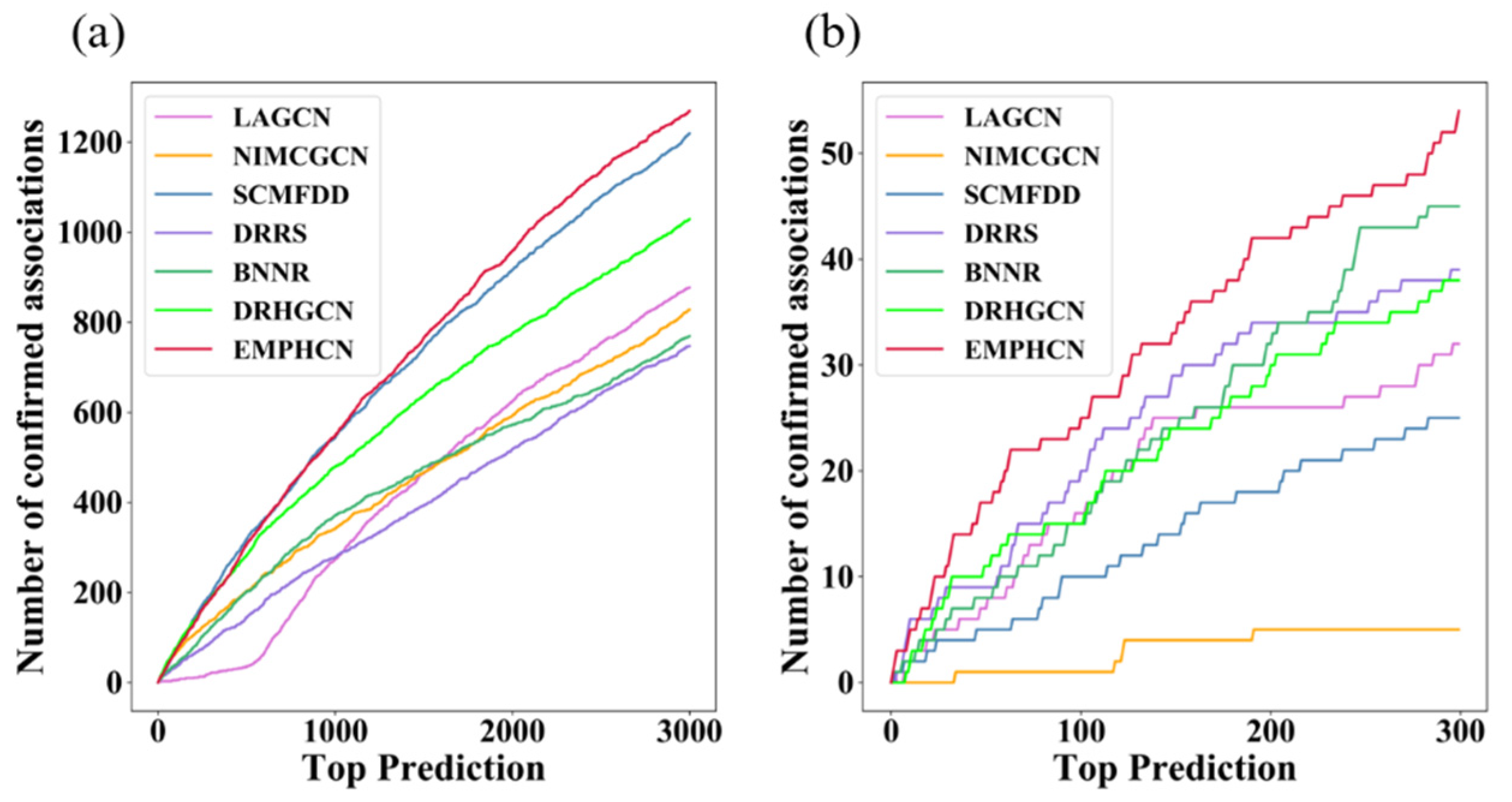
3.2. Comparison between EMPHCN and Other Methods
3.3. Investigation of Novel Predictions
3.4. Case Study
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Esoctt, J.K.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Reichert, J. A guide to drug discovery: Trends in development and approval times for new therapeutics in the United States. Nat. Rev. Drug Discov. 2003, 2, 695–702. [Google Scholar] [CrossRef]
- Jarada, T.N.; Rokne, J.G.; Alhajj, R. A review of computational drug repositioning: Strategies, approaches, opportunities, challenges, and directions. J. Cheminform. 2020, 12, 46. [Google Scholar] [CrossRef]
- Martinez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug–disease prioritization by integrating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous net-work model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and bi-random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Subramanian, A.; Reich, M.; Brunet, J.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Chen, X.; Liao, M.; Li, W.; Lian, B.; Wang, L.; Lian, B.; Meng, F.; Liu, X.; Chen, X.; et al. Identification of links between small molecules and miRNAs in human cancers based on transcriptional responses. Sci. Rep. 2012, 2, 282. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, M.; Yang, M.; Wu, F.; Li, Y.; Wang, J. Biomedical data and computational models for drug repositioning: A comprehensive review. Brief Bioinform. 2021, 22, 1604–1619. [Google Scholar] [CrossRef]
- Gottlieb, A.; Stein, G.; Ruppin, E.; Sharan, R. PREDICT: A method for inferring novel drug indications with application to personalized medicine. Mol. Syst. Biol. 2011, 7, 496. [Google Scholar] [CrossRef]
- Yang, L.; Agarwal, P. Systematic drug repositioning based on clinical side-effects. PLoS ONE 2011, 6, e28025. [Google Scholar] [CrossRef]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC Bioinform. 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Luo, H.; Li, Y.; Wang, J. Drug repositioning based on bounded nuclear norm regularization. Bioinformatics 2019, 35, i455–i463. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Qiu, Y.; Zeng, J.; Lei, X.; Zhang, P. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat. Mach. Intell. 2021, 3, 247–257. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. In Proceedings of the International Conference on Learning Representations (ICLR), Toulon, France, 24–26 April 2017; Available online: https://iclr.cc/archive/www/2017.html (accessed on 1 January 2022).
- Li, J.; Zhang, S.; Liu, T.; Ning, C.; Zhang, Z.; Zhou, W. Neural inductive matrix completion with graph convolutional networks for miRNA-disease association prediction. Bioinformatics 2020, 36, 2538–2546. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, K.; Qin, S.; Zhong, Y.; Elofsson, A. GCSENet: A GCN, CNN and SENet ensemble model for microRNA-disease association prediction. PLoS Comput. Biol. 2021, 17, e1009048. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, M.; Arnold, C. Toward heterogeneous information fusion: Bipartite graph convolutional networks for in silico drug repurposing. Bioinformatics 2020, 36 (Suppl. 1), i525–i533. [Google Scholar] [CrossRef]
- Yu, Z.; Huang, F.; Zhao, X.; Xiao, W.; Zhang, W. Predicting drug–disease associations through layer attention graph convolutional network. Brief Bioinform. 2021, 22, bbaa243. [Google Scholar] [CrossRef]
- Cai, L.; Lu, C.; Xu, J.; Meng, Y.; Wang, P.; Fu, X.; Zeng, X.; Su, Y. Drug repositioning based on the heterogeneous information fusion graph convolutional network. Brief Bioinform. 2021, 22, bbab319. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; You, H.; Zhang, Z.; Ji, R.; Gao, Y. Hypergraph neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 3558–3565. [Google Scholar]
- Davis, A.; Grondin, C.; Johnson, R.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.; Mattingly, C. The comparative toxicogenomics database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Patel, C. A standard database for drug repositioning. Sci. Data 2017, 14, 170029. [Google Scholar] [CrossRef] [Green Version]
- Tanimoto, T. An Elementary Mathematical Theory of Classification and Prediction; International Business Machines Corporation: New York, NY, USA, 1958. [Google Scholar]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org/ (accessed on 1 January 2022).
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Cotto, K.; Wagner, A.; Feng, Y.; Kiwala, S.; Coffman, A.; Spies, C.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M.; et al. DGIdb 3.0: A redesign and expansion of the drug–gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Hewett, M.; Oliver, D.E.; Rubin, D.L.; Easton, K.L.; Stuart, J.M.; Altman, R.B.; Klein, T.E. PharmGKB: The pharmacogenetics knowledge base. Nucleic Acids Res. 2002, 30, 163–165. [Google Scholar] [CrossRef]
- Anna, G.; Bellis, L.J.; Patricia, B.A.; Jon, C.; Mark, D.; Anne, H.; Yvonne, L.; Shaun, M.; David, M.; Bissan, A.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar]
- Chen, X.; Ji, Z.L.; Chen, Y.Z. TTD: Therapeutic target database. Nucleic Acids Res. 2002, 30, 412–415. [Google Scholar] [CrossRef] [Green Version]
- Piero, J.; Bravo, L.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar]
- Lipscomb, C.E. Medical subject headings (MeSH). Bull. Med. Libr. Assoc. 2000, 88, 265–266. [Google Scholar]
- Bodenreider, O. The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32 (Suppl. 1), D267–D270. [Google Scholar] [CrossRef] [Green Version]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Wu, B.; Zhu, P.; Li, P.; Zuo, W.; Hu, Q. ECA-Net: Efficient Channel Attention for Deep Convolutional Neural Networks. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), Seattle, WA, USA, 13–19 June 2022. [Google Scholar]
- He, X.; Deng, K.; Wang, X.; Li, Y.; Zhang, Y.; Wang, M. LightGCN: Simplifying and powering graph convolution network for recommendation. In Proceedings of the SIGIR 2020, Xi’an, China, 25–30 July 2020; Available online: https://sigir.org/sigir2020/accepted-papers/ (accessed on 1 January 2022).
- Chen, C.; Ma, W.; Zhang, M.; Wang, Z.; He, X.; Wang, C.; Liu, Y.; Ma, S. Graph heterogeneous multi-relational recommendation. In Proceedings of the AAAI Conference on Artificial Intelligence, Virtual, 2–9 February 2021; Volume 35, pp. 3958–3966. [Google Scholar]
- Velickovic, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph attention networks. In Proceedings of the International Conference on Learning Representations (ICLR), Vancouver, BC, Canada, 30 April–3 May 2018. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. In Proceedings of the International Conference on Learning Representations (ICLR), San Diego, CA, USA, 7–9 May 2015; Available online: https://iclr.cc/archive/www/2015.html (accessed on 1 January 2022).
- Oberhoff, C.; Kieback, D.; Wurstlein, R.; Deertz, H.; Sehouli, J.; Soest, C.; Hilfrich, J.; Mesrogli, M.; Staab, H.; Schindler, A.; et al. Topotecan chemotherapy in patients with breast cancer and brain metastases: Results of a pilot study. Onkologie 2001, 24, 256–260. [Google Scholar] [CrossRef]
- Martin, M.; Diaz-Rubio, E.; Casado, A.; Santabárbara, P.; Adrover, E.; Lenaz, L. Carboplatin: An active drug in metastatic breast cancer. J. Clin. Oncol. 1992, 10, 433–437. [Google Scholar] [CrossRef]
- Campana, L.; Galuppo, S.; Valpione, S.; Brunello, A.; Ghiotto, C.; Ongaro, A.; Rossi, C. Bleomycin electrochemotherapy in elderly metastatic breast cancer patients: Clinical outcome and management considerations. J. Cancer Res. Clin. 2014, 140, 1557–1565. [Google Scholar] [CrossRef]
- Heinemann, V. Role of gemcitabine in the treatment of advanced and metastatic breast cancer. Oncology 2003, 64, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.; Richardson, A.; Eklund, A.; Wang, Z.; Szallasi, Z.; Li, Q.; Calogrias, D.; Leong, C.; Juul, N.; Fatima, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Alavi, S.; Esfahani, M.; Alavi, F.; Movahedi, F.; Akbarzadeh, A. Drug delivery of hydroxyurea to breast cancer using liposomes. Indian, J. Clin. Biochem. 2013, 28, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Jolivet, J.; Cowan, K.; Curt, G.; Neil, J.; Bruce, A. The pharmacology and clinical use of methotrexate. N. Engl. J. Med. 1983, 309, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Samuels, B.; Bitran, J. High-dose intravenous melphalan: A review. J. Clin. Oncol. 1995, 13, 1786–1799. [Google Scholar] [CrossRef] [PubMed]
- Comte, A.; Jdid, W.; Guilhaume, M.; Kriegel, I.; Piperno, S.; Dieras, V.; Dorval, T.; Pierga, J.; Cottu, P.; Mignot, L.; et al. Survival of breast cancer patients with meningeal carcinomatosis treated by intrathecal thiotepa. J. Neuro-Oncol. 2013, 115, 445–452. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Zambelli, A. Trabectedin for the treatment of breast cancer. Expert. Opin. Investig. Drug 2016, 25, 105–115. [Google Scholar] [CrossRef]
- Mizuno, Y.; Kondo, T.; Narabayashi, H. Pergolide in the treatment of Parkinson’s disease. Neurology 1995, 45, S13–S21. [Google Scholar] [CrossRef]
- Aarsland, D.; Hutchinson, M.; Larsen, J. Cognitive, psychiatric and motor response to galantamine in Parkinson’s disease with dementia. Int. J. Geriatr. Psychiatry 2003, 18, 937–941. [Google Scholar] [CrossRef]
- Priori, A.; Foffani, G.; Pesenti, A.; Tamma, F.; Bianchi, A.; Pellegrini, M.; Locatelli, M.; Villani, R. Rhythm-specific pharmacological modulation of subthalamic activity in Parkinson’s disease. Exp. Neurol. 2004, 189, 369–379. [Google Scholar] [CrossRef]
- Aarsland, D.; Laake, K.; Larsen, J.; Janvin, C. Donepezil for cognitive impairment in Parkinson’s disease: A randomised controlled study. J. Neurol. Neuprsurg. Psychatry 2002, 72, 708–712. [Google Scholar] [CrossRef] [Green Version]
- Rinne, U.; Bracco, F.; Chouza, C.; Dupont, E.; Gershanik, O.; Marti Masso, J.L.; Montastruc, C. Marsden Early treatment of Parkinson’s disease with cabergoline delays the onset of motor complications. Drugs 1998, 55, 23–30. [Google Scholar] [CrossRef]
- Tourtellotte, W.; Potvin, A.; Costanza, A.; Hirsch, B.; Ko, S. Cyclobenzaprine: A new type of anti-parkinsonian drug. Prog. Neuro-Psychopharmacol. 1978, 2, 553–578. [Google Scholar] [CrossRef]
- Olson, W.; Gruenthal, M.; Mueller, M.; Olson, W.H. Gabapentin for Parkinsonism: A double-blind, placebo-controlled, crossover trial. Am. J. Med. 1997, 102, 60–66. [Google Scholar] [CrossRef]
- Lorimer, A.R.; MacFarlane, P.; Pringle, S.; Barbour, M.P.; Fox, Y.; Lawrie, T. The effects of felodipine in angina pectoris. Eur. J. Clin. Pharmacol. 1990, 38, 415–419. [Google Scholar] [CrossRef]
- Messerli, F.H. Vasodilatory edema: A common side effect of antihypertensive therapy. Curr. Cardiol. Rep. 2002, 4, 479–482. [Google Scholar] [CrossRef]
- Faulds, D.; Sorkin, E.M. Felodipine. Drug Aging 1992, 2, 374–388. [Google Scholar] [CrossRef]
- Dunselman, P.; Kuntze, C.E.E.; Van Bruggen, A. Efficacy of felodipine in congestive heart failure. Eur. Heart J. 1989, 10, 354–364. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | P | Drug | Disease | Protein | Domain | Interaction | Sparsity |
|---|---|---|---|---|---|---|---|
| T1 | 4 | 263 | 480 | 6059 | drug-disease | 15,630 | 0.1238 |
| drug-protein | 5620 | ||||||
| disease-protein | 20,019 | ||||||
| protein-protein | 49,406 | ||||||
| T2 | 3 | 850 | 339 | 5105 | drug-disease | 1921 | 0.0067 |
| drug-protein | 10,872 | ||||||
| disease-protein | 12,625 | ||||||
| protein-protein | 41,903 | ||||||
| Cdataset | 1 | 663 | 409 | ---------- | drug-disease | 2532 | 0.0093 |
| Fdataset | 1 | 593 | 313 | ---------- | drug-disease | 1933 | 0.0104 |
| Dataset | DRRS | NIMCGCN | SCMFDD | BNNR | LAGCN | DRHGCN | EMP_base | EMPHCN |
|---|---|---|---|---|---|---|---|---|
| AUROC | ||||||||
| T1 | 0.846 ± 0.002 | 0.848 ± 0.004 | 0.864 ± 0.002 | 0.867 ± 0.001 | 0.877 ± 0.002 | 0.876 ± 0.001 | 0.879 ± 0.001 | 0.887 ± 0.002 |
| T2 | 0.889 ± 0.002 | 0.766 ± 0.002 | 0.734 ± 0.004 | 0.920 ± 0.001 | 0.772 ± 0.003 | 0.938 ± 0.002 | 0.958 ± 0.003 | 0.961 ± 0.003 |
| Cdataset | 0.947 ± 0.002 | 0.856 ± 0.004 | 0.794 ± 0.001 | 0.951 ± 0.001 | 0.923 ± 0.004 | 0.963 ± 0.001 | 0.971 ± 0.001 | --------- |
| Fdataset | 0.930 ± 0.002 | 0.836 ± 0.004 | 0.775 ± 0.001 | 0.934 ± 0.001 | 0.892 ± 0.003 | 0.948 ± 0.002 | 0.954 ± 0.002 | --------- |
| Avg | 0.903 | 0.827 | 0.792 | 0.918 | 0.866 | 0.931 | 0.941 | --------- |
| AUPR | ||||||||
| T1 | 0.451 ± 0.002 | 0.502 ± 0.004 | 0.550 ± 0.002 | 0.546 ± 0.001 | 0.579 ± 0.002 | 0.574 ± 0.001 | 0.578 ± 0.001 | 0.593 ± 0.003 |
| T2 | 0.379 ± 0.002 | 0.047 ± 0.002 | 0.049 ± 0.003 | 0.492 ± 0.001 | 0.138 ± 0.004 | 0.475 ± 0.001 | 0.501 ± 0.003 | 0.526 ± 0.002 |
| Cdataset | 0.574 ± 0.003 | 0.445 ± 0.002 | 0.060 ± 0.002 | 0.679 ± 0.001 | 0.194 ± 0.002 | 0.655 ± 0.002 | 0.688 ± 0.002 | --------- |
| Fdataset | 0.475 ± 0.006 | 0.354 ± 0.005 | 0.062 ± 0.002 | 0.601 ± 0.001 | 0.134 ± 0.002 | 0.566 ± 0.002 | 0.604 ± 0.002 | --------- |
| Avg | 0.470 | 0.337 | 0.180 | 0.580 | 0.261 | 0.568 | 0.593 | --------- |
| Dataset | Methods | AUPR | AUC | RE | ACC | F1 |
|---|---|---|---|---|---|---|
| T1 | LAGCN | 0.228 | 0.678 | 0.503 | 0.756 | 0.325 |
| NIMCGCN | 0.234 | 0.625 | 0.438 | 0.757 | 0.289 | |
| SCMFDD | 0.372 | 0.763 | 0.531 | 0.845 | 0.417 | |
| DRRS | 0.212 | 0.650 | 0.555 | 0.662 | 0.295 | |
| BNNR | 0.217 | 0.625 | 0.580 | 0.582 | 0.269 | |
| DRHGCN | 0.328 | 0.748 | 0.539 | 0.788 | 0.368 | |
| EMPHCN | 0.396 | 0.806 | 0.547 | 0.859 | 0.442 | |
| T2 | LAGCN | 0.053 | 0.630 | 0.098 | 0.986 | 0.094 |
| NIMCGCN | 0.009 | 0.562 | 0.474 | 0.689 | 0.022 | |
| SCMFDD | 0.018 | 0.559 | 0.067 | 0.987 | 0.072 | |
| DRRS | 0.065 | 0.741 | 0.249 | 0.981 | 0.156 | |
| BNNR | 0.073 | 0.805 | 0.165 | 0.987 | 0.153 | |
| DRHGCN | 0.051 | 0.696 | 0.167 | 0.985 | 0.141 | |
| EMPHCN | 0.092 | 0.845 | 0.261 | 0.986 | 0.178 |
| Disease | Rank | Candidate Drug | Evidence |
|---|---|---|---|
| Breast carcinoma | 1 | Topotecan | [44] |
| 2 | Gemcitabine | [47] | |
| 3 | Carboplatin | [45] | |
| 4 | Bleomycin | [46] | |
| 5 | Cisplatin | [48] | |
| 6 | Hydroxyurea | [49] | |
| 7 | Methotrexate | [50] | |
| 8 | Melphalan | [51] | |
| 9 | Thiotepa | [52] | |
| 10 | Trabectedin | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.; Li, Z.; Kang, Y.; Ye, X.; Feng, W. Drug Repositioning Based on the Enhanced Message Passing and Hypergraph Convolutional Networks. Biomolecules 2022, 12, 1666. https://doi.org/10.3390/biom12111666
Huang W, Li Z, Kang Y, Ye X, Feng W. Drug Repositioning Based on the Enhanced Message Passing and Hypergraph Convolutional Networks. Biomolecules. 2022; 12(11):1666. https://doi.org/10.3390/biom12111666
Chicago/Turabian StyleHuang, Weihong, Zhong Li, Yanlei Kang, Xinghuo Ye, and Wenming Feng. 2022. "Drug Repositioning Based on the Enhanced Message Passing and Hypergraph Convolutional Networks" Biomolecules 12, no. 11: 1666. https://doi.org/10.3390/biom12111666