

Lessons Learned from Two Decades of Modeling the Heat-Shock Response

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
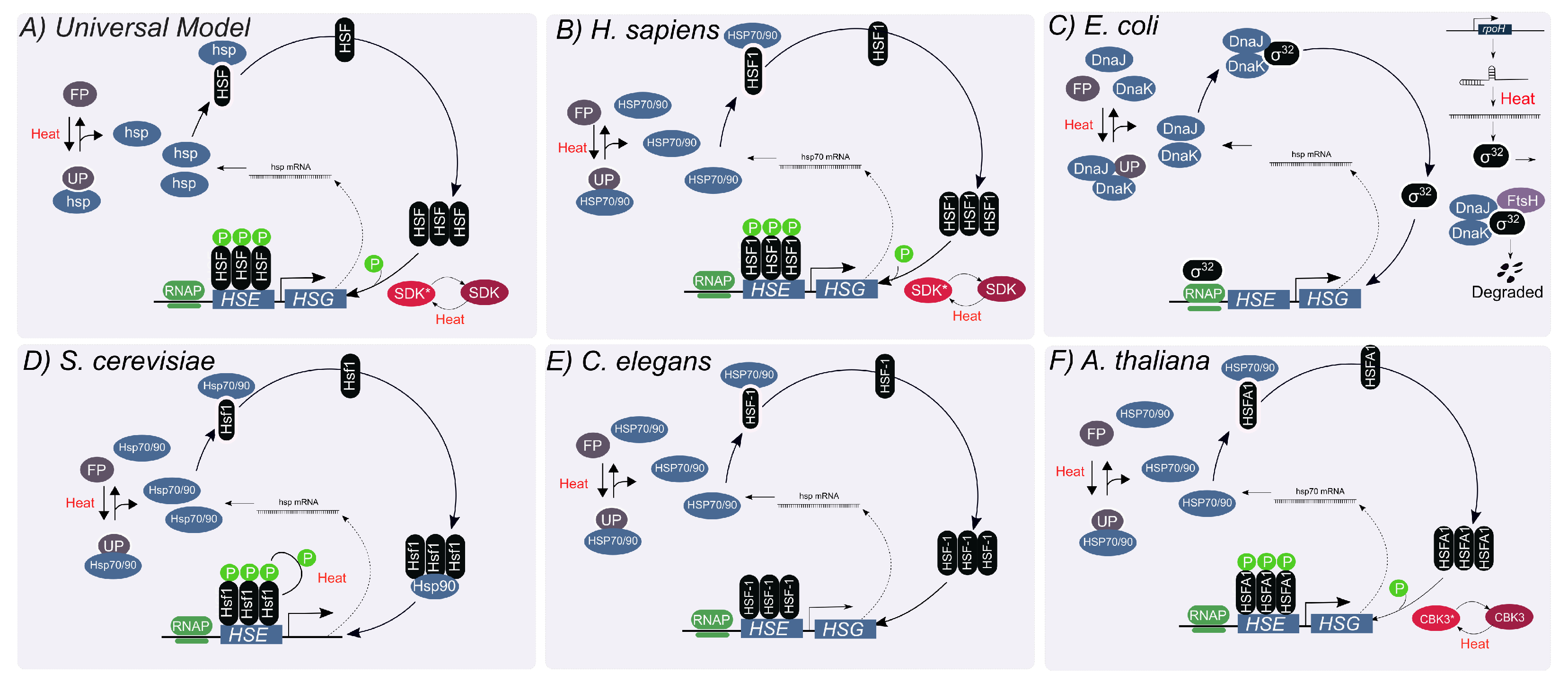
2. The Titration Feedback Is Central to the HSR Regulation
2.1. The HSR in Mammalian Cells
2.2. The HSR in Escherichia coli
2.3. The HSR in S. cerevisiae
2.4. The HSR in C. elegans
2.5. The HSR in A. thaliana
3. Experimental Evidence of the Mechanism and Basis of Models
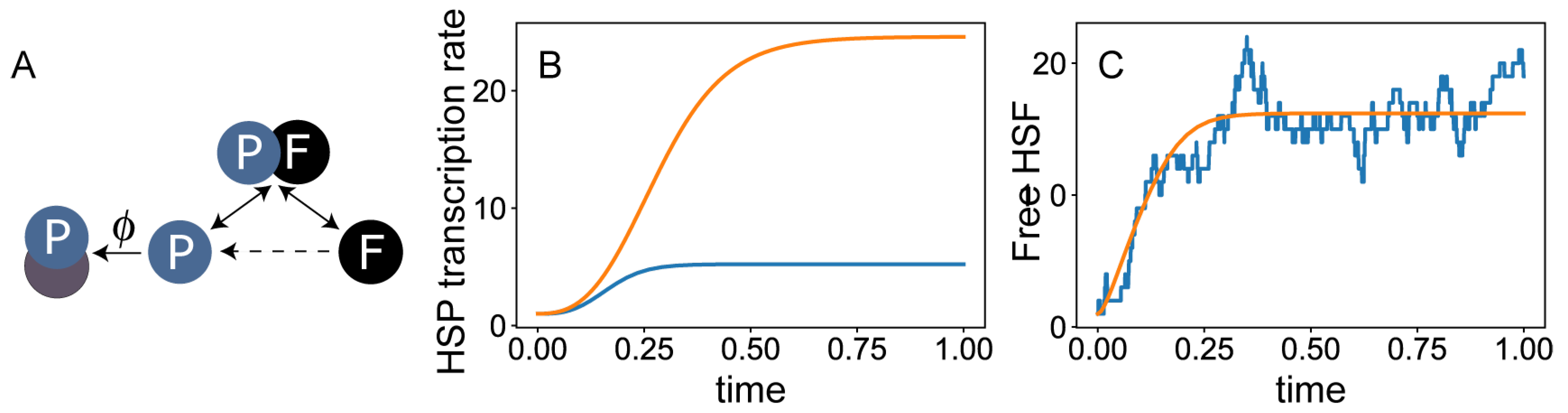
4. Mathematical Modeling of the Heat-Shock Response
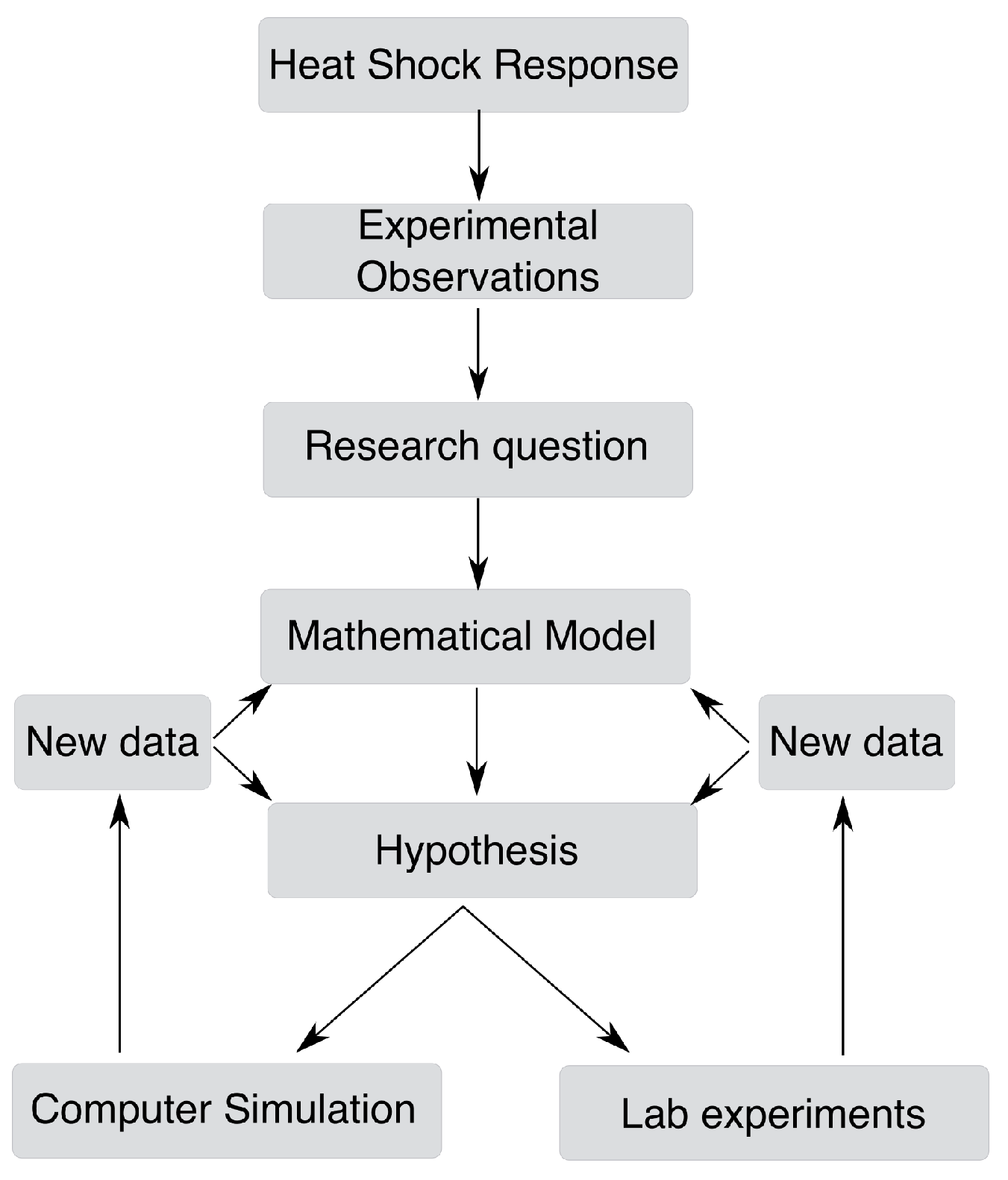
4.1. From a Biological Question to a Mathematical Model
4.2. Alternative Modeling Frameworks
5. Modeling the HSR
5.1. Modeling the HSR in Mammalian Cells
5.2. Modeling the HSR in Yeast
5.3. Modeling the HSR in Bacteria
5.4. Modeling the HSR in Worms
5.5. Modeling the HSR in Plants
6. What Did the Mathematical Models Contribute to Our Understanding of the HSR
7. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomlin, C.J.; Axelrod, J.D. Biology by numbers: Mathematical modelling in developmental biology. Nat. Rev. Genet. 2007, 8, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deisboeck, T.S. Mathematical modeling in cancer drug discovery. Drug Discov. Today 2014, 19, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Sriram, G. Mathematical modeling: Bridging the gap between concept and realization in synthetic biology. J. Biomed. Biotechnol. 2010, 2010, 541609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Zügel, U.; Kaufmann, S.H.E. Role of Heat Shock Proteins in Protection from and Pathogenesis of Infectious Diseases. Clin. Microbiol. Rev. 1999, 12, 19–39. [Google Scholar] [CrossRef] [Green Version]
- van Eden, W.; van der Zee, R.; Prakken, B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005, 5, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. The Heat Shock Response: Systems Biology of Proteotoxic Stress in Aging and Disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2018, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Peper, A.; Grimbergen, C.A.; Spaan, J.A.E.; Souren, J.E.M.; Wijk, R.V. A mathematical model of the hsp70 regulation in the cell. Int. J. Hyperth. 1998, 14, 97–124. [Google Scholar] [CrossRef]
- Zheng, X.; Krakowiak, J.; Patel, N.; Beyzavi, A.; Ezike, J.; Khalil, A.S.; Pincus, D. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. eLife 2016, 5, 3–5. [Google Scholar] [CrossRef]
- Magni, S.; Succurro, A.; Skupin, A.; Ebenhoeh, O. Data-driven dynamical model indicates that the heat shock response in Chlamydomonas reinhardtii is tailored to handle natural temperature variation. J. R. Soc. Interface. 2018, 15, 20170965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.; Peterson, M.S.; Bentley, W.E. Stochastic kinetic analysis of the Escherichia coli stress circuit using σ32-targeted antisense. Biotechnol. Bioeng. 2001, 75, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Kurata, H.; El-Samad, H.; Iwasaki, R.; Ohtake, H.; Doyle, J.C.; Grigorova, I.; Gross, C.A.; Khammash, M. Module-based analysis of robustness trade offs in the heat shock response system. PLoS Comput. Biol. 2006, 2, e59. [Google Scholar] [CrossRef] [Green Version]
- Dahlstrom, E.; Levine, E. Dynamics of Heat Shock Detection and Response in the Intestine of Caenorhabditis elegans. bioRxiv 2019, 794800, 1. [Google Scholar] [CrossRef]
- Anckar, J.; Sistonen, L. Regulation of HSF1 Function in the Heat Stress Response: Implications in Aging and Disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. [Google Scholar] [CrossRef]
- Jee, H.; Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [Green Version]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.E.; Schuetz, T.J.; Larin, Z. Heat-inducible human factor that binds to a human hsp70 promoter. Mol. Cell. Biol. 1987, 7, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, R.I.; Sarge, K.D.; Abravaya, K. Transcriptional regulation of heat shock genes. A paradigm for inducible genomic responses. J. Biol. Chem. 1992, 267, 21987–21990. [Google Scholar] [CrossRef]
- Shi, Y.; Mosser, D.D.; Morimoto, R.I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998, 12, 654–666. [Google Scholar] [CrossRef]
- Wu, C. Heat Shock Transcription Factors: Structure and Regulation. Annu. Rev. Cell Dev. Biol. 1995, 11, 441–469. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, C.I.; Hietakangas, V.; Mikhailov, A.; Rantanen, J.O.; Kallio, M.; Meinander, A.; Hellman, J.; Morrice, N.; Mackintosh, C.; Morimoto, R.I.; et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat-shock factor 1. EMBO J. 2001, 20, 3800–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straus, D.; Walter, W.; Gross, C. The heat shock response of E. coli is regulated by changes in the concentration of σ32. Nature 1987, 329, 348–351. [Google Scholar] [CrossRef]
- Gamer, J.; Multhaup, G.; Tomoyasu, T.; McCarty, J.S.; Rüdiger, S.; Schönfeld, H.J.; Schirra, C.; Bujard, H.; Bukau, B. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor sigma32. EMBO J. 1996, 15, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Straus, D.; Walter, W.; Gross, C.A. DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of σ32. Genes Dev. 1990, 4, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Guisbert, E.; Herman, C.; Zen Lu, C.; Gross, C.A. A chaperone network controls the heat shock response in E. coli. Genes Dev. 2004, 18, 2812–2821. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Kanemori, M.; Yanagi, H.; Yura, T. Heat-Induced Synthesis of ς32 in Escherichia coli: Structural and Functional Dissection of rpoH mRNA Secondary Structure. J. Bacteriol. 1999, 181, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.T.; Kanemori, M.; Yanagi, H.; Yura, T. Dynamic interplay between antagonistic pathways controlling the sigma 32 level in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5860–5865. [Google Scholar] [CrossRef] [Green Version]
- Verghese, J.; Abrams, J.; Wang, Y.; Morano, K.A. Biology of the heat shock response and protein chaperones: Budding yeast Saccharomyces cerevisiae as a model system. Microbiol. Mol. Biol. Rev. 2012, 76, 115–158. [Google Scholar] [CrossRef] [Green Version]
- Voellmy, R.; Boellmann, F. Chaperone Regulation of the Heat Shock Protein Response. In Molecular Aspects of the Stress Response: Chaperones, Membranes and Networks; Springer: New York, NY, USA, 2007; pp. 89–99. [Google Scholar]
- Sorger, P.K. Heat shock factor and the heat shock response. Cell 1991, 65, 363–366. [Google Scholar] [CrossRef]
- Guisbert, E.; Czyz, D.M.; Richter, K.; McMullen, P.D.; Morimoto, R.I. Identification of a Tissue-Selective Heat Shock Response Regulatory Network. PLoS Genet. 2013, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittenburg, N.; Baumeister, R. Thermal avoidance in Caenorhabditis elegans: An approach to the study of nociception. Proc. Natl. Acad. Sci. USA 1999, 96, 10477–10482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prahlad, V.; Cornelius, T.; Morimoto, R.I. Regulation of the Cellular Heat Shock Response in Caenorhabditis elegans by Thermosensory Neurons. Science 2008, 320, 811–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, F.K.; Prahlad, V. Olfactory experience primes the heat shock transcription factor HSF-1 to enhance the expression of molecular chaperones in C. elegans. Sci. Signal. 2017, 10, eaan4893. [Google Scholar] [CrossRef] [Green Version]
- Tatum, M.C.; Ooi, F.K.; Chikka, M.R.; Chauve, L.; Martinez-Velazquez, L.A.; Steinbusch, H.W.M.; Morimoto, R.I.; Prahlad, V. Neuronal serotonin release triggers the heat shock response in C. elegans in the absence of temperature increase. Curr. Biol. 2015, 25, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional Regulatory Network of Plant Heat Stress Response. Trends Plant Sci. 2017, 22, 53–65. [Google Scholar] [CrossRef]
- Guo, J.; Wu, J.; Ji, Q.; Wang, C.; Luo, L.; Yuan, Y.; Wang, Y.; Wang, J. Genome-wide analysis of heat shock transcription factor families in rice and Arabidopsis. J. Genet. Genom. 2018, 35, 105–118. [Google Scholar] [CrossRef]
- Yang, X.; Zhu, W.; Zhang, H.; Liu, N.; Tian, S. Heat shock factors in tomatoes: Genome-wide identification, phylogenetic analysis and expression profiling under development and heat stress. PeerJ 2016, 4, e1961. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sun, N.; Liu, M.; Liu, J.; Du, B.; Wang, X.; Qi, X. An Autoregulatory Loop Controlling Arabidopsis HsfA2 Expression: Role of Heat Shock-Induced Alternative Splicing. Plant Physiol. 2013, 162, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Fukao, Y.; Hayashi, M.; Fukazawa, M.; Suzuki, I.; Nishimura, M. Cytosolic HSP90 regulates the heat shock response that is responsible for heat acclimation in Arab. thaliana. J. Biol. Chem. 2007, 282, 37794–37804. [Google Scholar] [CrossRef]
- Hahn, A.; Bublak, D.; Schleiff, E.; Scharf, K.D. Crosstalk between Hsp90 and Hsp70 chaperones and heat stress transcription factors in tomato. Plant Cell 2011, 23, 741–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohama, N.; Kusakabe, K.; Mizoi, J.; Zhao, H.; Kidokoro, S.; Koizumi, S.; Takahashi, F.; Ishida, T.; Yanagisawa, S.; Shinozaki, K.; et al. The Transcriptional Cascade in the Heat Stress Response of Arabidopsis Is Strictly Regulated at the Level of Transcription Factor Expression. Plant Cell 2016, 28, 181–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan-Schaminet, K.Y.; Baniwal, S.K.; Bublak, D.; Nover, L.; Scharf, K.D. Specific Interaction between Tomato HsfA1 and HsfA2 Creates Hetero-oligomeric Superactivator Complexes for Synergistic Activation of Heat Stress Gene Expression. J. Biol. Chem. 2009, 284, 20848–20857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, M.; Mitsuda, N.; Ohme-Takagi, M. Arabidopsis HsfB1 and HsfB2b Act as Repressors of the Expression of Heat-Inducible Hsfs But Positively Regulate the Acquired Thermotolerance. Plant Physiol. 2011, 157, 1243–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugio, A.; Dreos, R.; Aparicio, F.; Maule, A.J. The Cytosolic Protein Response as a Subcomponent of the Wider Heat Shock Response in Arabidopsis. Plant Cell 2009, 21, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Thalor, S.K.; Takahashi, Y.; Berberich, T.; Kusano, T. An inhibitory effect of the sequence-conserved upstream open-reading frame on the translation of the main open-reading frame of HsfB1 transcripts in Arabidopsis. Plant Cell Environ. 2012, 35, 2014–2030. [Google Scholar] [CrossRef]
- Hsu, S.F.; Lai, H.C.; Jinn, T.L. Cytosol-Localized Heat Shock Factor-Binding Protein, AtHSBP, Functions as a Negative Regulator of Heat Shock Response by Translocation to the Nucleus and Is Required for Seed Development in Arabidopsis. Plant Physiol. 2010, 153, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Baler, R.; Dahl, G.; Voellmy, R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol. Cell. Biol. 1993, 13, 2486–2496. [Google Scholar] [CrossRef] [Green Version]
- Cotto, J.J.; Kline, M.; Morimoto, R.I. Activation of Heat Shock Factor 1 DNA Binding Precedes Stress-induced Serine Phosphorylation evidence for a multistep pathway of regulation. J. Biol. Chem. 1996, 271, 3355–3358. [Google Scholar] [CrossRef] [Green Version]
- Straus, D.B.; Walter, W.A.; Gross, C.A. The activity of σ32 is reduced under conditions of excess heat shock protein production in Escherichia coli. Genes Dev. 1989, 3, 2003–2010. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Guettouche, T.; Smith, D.F.; Voellmy, R. Repression of Heat Shock Transcription Factor HSF1 Activation by HSP90 (HSP90 Complex) that Forms a Stress-Sensitive Complex with HSF1. Cell 1998, 94, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Tomoyasu, T.; Ogura, T.; Tatsuta, T.; Bukau, B. Levels of DnaK and DnaJ provide tight control of heat shock gene expression and protein repair in Escherichia coli. Mol. Microbiol. 1998, 30, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, C.; Thévenet, D.; D’Ari, R.; Bouloc, P. Degradation of σ32, the heat shock regulator in Escherichia coli, is governed by HflB. Proc. Natl. Acad. Sci. USA 1995, 92, 3516–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorakis, N.G.; Morimoto, R.I. Post-transcriptional regulation of hsp70 expression in human cells: Effects of heat shock, inhibition of protein synthesis, and adenovirus infection on translation and mRNA stability. Mol. Cell. Biol. 1987, 7, 4357–4368. [Google Scholar] [CrossRef]
- Kline, M.P.; Morimoto, R.I. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol. Cell. Biol. 1997, 17, 2107–2115. [Google Scholar] [CrossRef] [Green Version]
- Stege, G.J.; Brunsting, J.F.; Kampinga, H.H.; Konings, A.W. Thermotolerance and nuclear protein aggregation: Protection against initial damage or better recovery? J. Cell. Physiol. 1995, 164, 579–586. [Google Scholar] [CrossRef]
- Baler, R.; Zou, J.; Voellmy, R. Evidence for a role of Hsp70 in the regulation of the heat shock response in mammalian cells. Cell Stress Chaperones 1996, 1, 33. [Google Scholar] [CrossRef]
- Mosser, D.D.; Theodorakis, N.G.; Morimoto, R.I. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol. Cell. Biol. 1988, 8, 4736–4744. [Google Scholar] [CrossRef] [Green Version]
- Abravaya, K.; Phillips, B.; Morimoto, R.I. Attenuation of the heat shock response in HeLa cells is mediated by the release of bound heat shock transcription factor and is modulated by changes in growth and heat shock temperatures. Genes Dev. 1991, 5, 2117–2127. [Google Scholar] [CrossRef] [Green Version]
- Andrews, G.K.; Harding, M.A.; Calvet, J.P.; Adamson, E.D. The Heat Shock Response in HeLa Cells Is Accompanied by Elevated Expression of the c-fos Proto-Oncogene. Mol. Cell. Biol. 1987, 7, 3452–3458. [Google Scholar]
- Sapareto, S.A.; Dewey, W.C. Thermal dose determination in cancer therapy. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 787–800. [Google Scholar] [CrossRef]
- Yarmolenko, P.S.; Moon, E.J.; Landon, C.; Manzoor, A.; Hochman, D.W.; Viglianti, B.L.; Dewhirst, M.W. Thresholds for thermal damage to normal tissues: An update. Int. J. Hyperth. 2011, 27, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Viglianti, B.L.; Lora-Michiels, M.; Hanson, M.; Hoopes, P.J. Basic principles of thermal dosimetry and thermal thresholds for tissue damage from hyperthermia. Int. J. Hyperth. 2003, 19, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Rieger, T.R.; Morimoto, R.I.; Hatzimanikatis, V. Mathematical Modeling of the Eukaryotic Heat-Shock Response: Dynamics of the hsp70 Promoter. Biophys. J. 2005, 88, 1646–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petre, I.; Mizera, A.; Hyder, C.L.; Meinander, A.; Mikhailov, A.; Morimoto, R.I.; Sistonen, L.; Eriksson, J.E.; Back, R.J. A simple mass-action model for the eukaryotic heat shock response and its mathematical validation. Nat. Comput. 2011, 10, 595–612. [Google Scholar] [CrossRef]
- Scheff, J.D.; Stallings, J.D.; Reifman, J.; Rakesh, V. Mathematical Modeling of the Heat-Shock Response in HeLa Cells. Biophys. J. 2015, 109, 182–193. [Google Scholar] [CrossRef] [Green Version]
- Rieger, T.R.; Morimoto, R.I.; Hatzimanikatis, V. Bistability Explains Threshold Phenomena in Protein Aggregation both In Vitro and In Vivo. Biophys. J. 2006, 90, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Sharma, R. Transcription factors and chaperone proteins play a role in launching a faster response to heat stress and aggregation. Comput. Biol. Chem. 2021, 93, 107534. [Google Scholar] [CrossRef]
- Sivéry, A.; Courtade, E.; Thommen, Q. A minimal titration model of the mammalian dynamical heat shock response. Phys. Biol. Phys. Biol. 2016, 13, 066008. [Google Scholar] [CrossRef]
- Sorger, P.K.; Lewis, M.J.; Pelham, H.R.B. Heat shock factor is regulated differently in yeast and HeLa cells. Nature 1987, 329, 81–84. [Google Scholar] [CrossRef]
- Krakowiak, J.; Zheng, X.; Patel, N.; Feder, Z.A.; Anandhakumar, J.; Valerius, K.; Gross, D.S.; Khalil, A.S.; Pincus, D. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 2018, 7, e31668. [Google Scholar] [CrossRef] [PubMed]
- Grossman, A.D.; Straus, D.B.; Walter, W.A.; Gross, C.A. σ32 synthesis can regulate the synthesis of heat shock proteins in Escherichia coli. Genes Dev. 1987, 1, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Kurata, H.; El-Samad, H.; Yi, T.; Khammash, M.; Doyle, J. Feedback regulation of the heat shock response in E. coli. In Proceedings of the 40th IEEE Conference on Decision and Control (Cat. No.01CH37228), Orlando, FL, USA, 4–7 December 2001; Volume 1, pp. 837–842. [Google Scholar] [CrossRef] [Green Version]
- El-Samad, H.; Kurata, H.; Doyle, J.C.; Gross, C.A.; Khammash, M. Surviving heat shock: Control strategies for robustness and performance. Proc. Natl. Acad. Sci. USA 2005, 102, 2736–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Gillespie, D.; Petzold, L. Multiscale stochastic simulation algorithm with stochastic partial equilibrium assumption for chemically reacting systems. J. Comput. Phys. 2005, 206, 395–411. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.W. A multiscale approximation in a heat shock response model of E. coli. BMC Syst. Biol. 2012, 6, 143. [Google Scholar] [CrossRef] [Green Version]
- Vertti-Quintero, N.; Berger, S.; Casadevall i Solvas, X.; Statzer, C.; Annis, J.; Ruppen, P.; Stavrakis, S.; Ewald, C.Y.; Gunawan, R.; deMello, A.J. Stochastic and Age-Dependent Proteostasis Decline Underlies Heterogeneity in Heat-Shock Response Dynamics. Small 2021, 17, 2102145. [Google Scholar] [CrossRef] [PubMed]
- Schroda, M.; Blo, D.; Cker, È.; Beck, C.F. The HSP70A promoter as a tool for the improved expression of transgenes in Chlamydomonas. Plant J. 2000, 21, 121–131. [Google Scholar] [CrossRef]
- Schmollinger, S.; Schulz-Raffelt, M.; Strenkert, D.; Veyel, D.; Vallon, O.; Schroda, M. Dissecting the heat stress response in Chlamydomonas by pharmaceutical and RNAi approaches reveals conserved and novel aspects. Mol. Plant 2013, 6, 1795–1813. [Google Scholar] [CrossRef] [Green Version]
- Mizera, A.; Gambin, B. Stochastic modelling of the eukaryotic heat shock response. J. Theor. Biol. 2010, 265, 455–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Mitarai, N.; Trusina, A. Circuit architecture explains functional similarity of bacterial heat shock responses. Phys. Biol. 2012, 9, 066003. [Google Scholar] [CrossRef]
- Szymańska, Z.; Zylicz, M. Mathematical modeling of heat shock protein synthesis in response to temperature change. J. Theor. Biol. 2009, 259, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, C.J.; Lorimer, I.A.J. Modelling the Role of the Hsp70/Hsp90 System in the Maintenance of Protein Homeostasis. PLoS ONE 2011, 6, e22038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cates, J.; Graham, G.C.; Omattage, N.; Pavesich, E.; Setliff, I.; Shaw, J.; Smith, C.L.; Lipan, O. Sensing the Heat Stress by Mammalian Cells. BMC Biophys. 2011, 4, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, M.D.; Tyc, K.M.; Brown, A.J.P.; Klipp, E. Modelling the Regulation of Thermal Adaptation in Candida Albicans, A Major Fungal Pathog. Humans. PLoS ONE 2012, 7, e32467. [Google Scholar] [CrossRef] [Green Version]
- Lipan, O.; Navenot, J.M.; Wang, Z.; Huang, L.; Peiper, S.C. Heat shock response in CHO mammalian cells is controlled by a nonlinear stochastic process. PLoS Comput. Biol. 2007, 3, 1859–1870. [Google Scholar] [CrossRef]
- Pereira, T.; Vilaprinyo, E.; Belli, G.; Herrero, E.; Salvado, B.; Sorribas, A.; Altés, G.; Alves, R. Quantitative Operating Principles of Yeast Metabolism during Adaptation to Heat Stress. Cell Rep. 2018, 22, 2421–2430. [Google Scholar] [CrossRef] [PubMed]
- Prahlad, V.; Morimoto, R.I. Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 14204–14209. [Google Scholar] [CrossRef] [Green Version]
- Edkins, A.L.; Price, J.T.; Pockley, A.G.; Blatch, G.L. Heat shock proteins as modulators and therapeutic targets of chronic disease: An integrated perspective. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Kaul, G.; Thippeswamy, H. Role of heat shock proteins in diseases and their therapeutic potential. Indian J. Microbiol. 2011, 51, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Pockley, A.G. Heat shock proteins, inflammation, and cardiovascular disease. Circulation 2002, 105, 1012–1017. [Google Scholar] [CrossRef]
- Elias, D.; Markovits, D.; Reshef, T.; van der Zee, R.; Cohen, I.R. Induction and therapy of autoimmune diabetes in the non-obese diabetic (NOD/Lt) mouse by a 65-kDa heat shock protein. Proc. Natl. Acad. Sci. USA 1990, 87, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Danladi, J.; Sabir, H. Innate immunity, inflammation activation and heat-shock protein in COVID-19 pathogenesis. J. Neuroimmunol. 2021, 358, 577632. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Hershey, J.W. Heat shock-induced translational alterations in HeLa cells. Initiation factor modifications and the inhibition of translation. J. Biol. Chem. 1984, 259, 11882–11889. [Google Scholar] [CrossRef]
- Mahat, D.B.; Salamanca, H.H.; Duarte, F.M.; Danko, C.G.; Lis, J.T. Mammalian Heat Shock Response and Mechanisms Underlying Its Genome-wide Transcriptional Regulation. Mol. Cell 2016, 62, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Biamonti, G.; Vourc’h, C. Nuclear stress bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000695. [Google Scholar] [CrossRef]
- Jolly, C.; Metz, A.; Govin, J.; Vigneron, M.; Turner, B.M.; Khochbin, S.; Vourc’h, C. Stress-induced transcription of satellite III repeats. J. Cell Biol. 2004, 164, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Caspeta, L.; Chen, Y.; Nielsen, J. Thermotolerant yeasts selected by adaptive evolution express heat stress response at 30 degree celsius. Sci. Rep. 2016, 6, 27003. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.R.; Castenholz, R.W. Evolution of Thermotolerance in Hot Spring Cyanobacteria of the Genus Synechococcus. Appl. Environ. Microbiol. 2000, 66, 4222–4229. [Google Scholar] [CrossRef] [Green Version]
- Queitsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Pfeuty, B.; Courtade, E.; Thommen, Q. Fine-tuned control of stress priming and thermotolerance. Phys. Biol. 2021, 18, 04LT02. [Google Scholar] [CrossRef]
- Tomanek, L. Variation in the heat shock response and its implication for predicting the effect of global climate change on species’ biogeographical distribution ranges and metabolic costs. J. Exp. Biol. 2010, 213, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 2009, 459, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Agozzino, L.; Dill, K.A. Protein evolution speed depends on its stability and abundance and on chaperone concentrations. Proc. Natl. Acad. Sci. USA 2018, 115, 9092–9097. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranawade, A.; Sharma, R.; Levine, E. Lessons Learned from Two Decades of Modeling the Heat-Shock Response. Biomolecules 2022, 12, 1645. https://doi.org/10.3390/biom12111645
Ranawade A, Sharma R, Levine E. Lessons Learned from Two Decades of Modeling the Heat-Shock Response. Biomolecules. 2022; 12(11):1645. https://doi.org/10.3390/biom12111645
Chicago/Turabian StyleRanawade, Ayush, Rati Sharma, and Erel Levine. 2022. "Lessons Learned from Two Decades of Modeling the Heat-Shock Response" Biomolecules 12, no. 11: 1645. https://doi.org/10.3390/biom12111645