

A Comprehensive Study of De Novo Mutations on the Protein-Protein Interaction Interfaces Provides New Insights into Developmental Delay
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Preparation of DNM Missenses and PPI Interface Data
2.2. Enrichment of DNM Missenses on Interaction Interfaces
2.3. Significance Test of PPI Interface Mutations and Identification of PsychiPPI
2.4. Interactome Network Analysis of PsychiPPI Identified DD Candidate Proteins
2.5. Haploinsufficiency and Loss of Function Tolerance Analysis of Candidate Genes
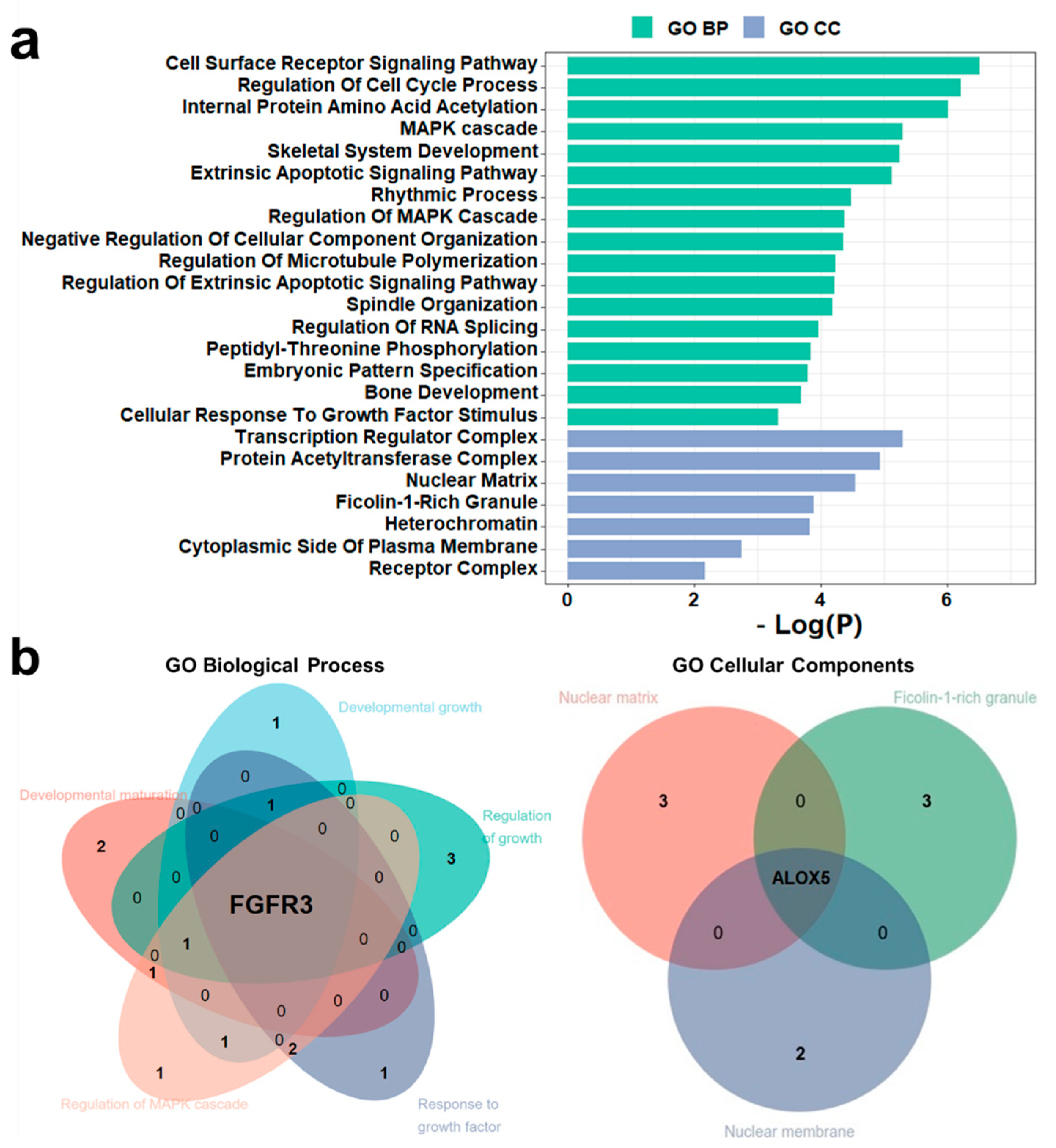
2.6. Gene Ontology and Gene Set Analysis
2.7. Spatio-Temporal Expression Patterns of Candidate Genes
3. Results
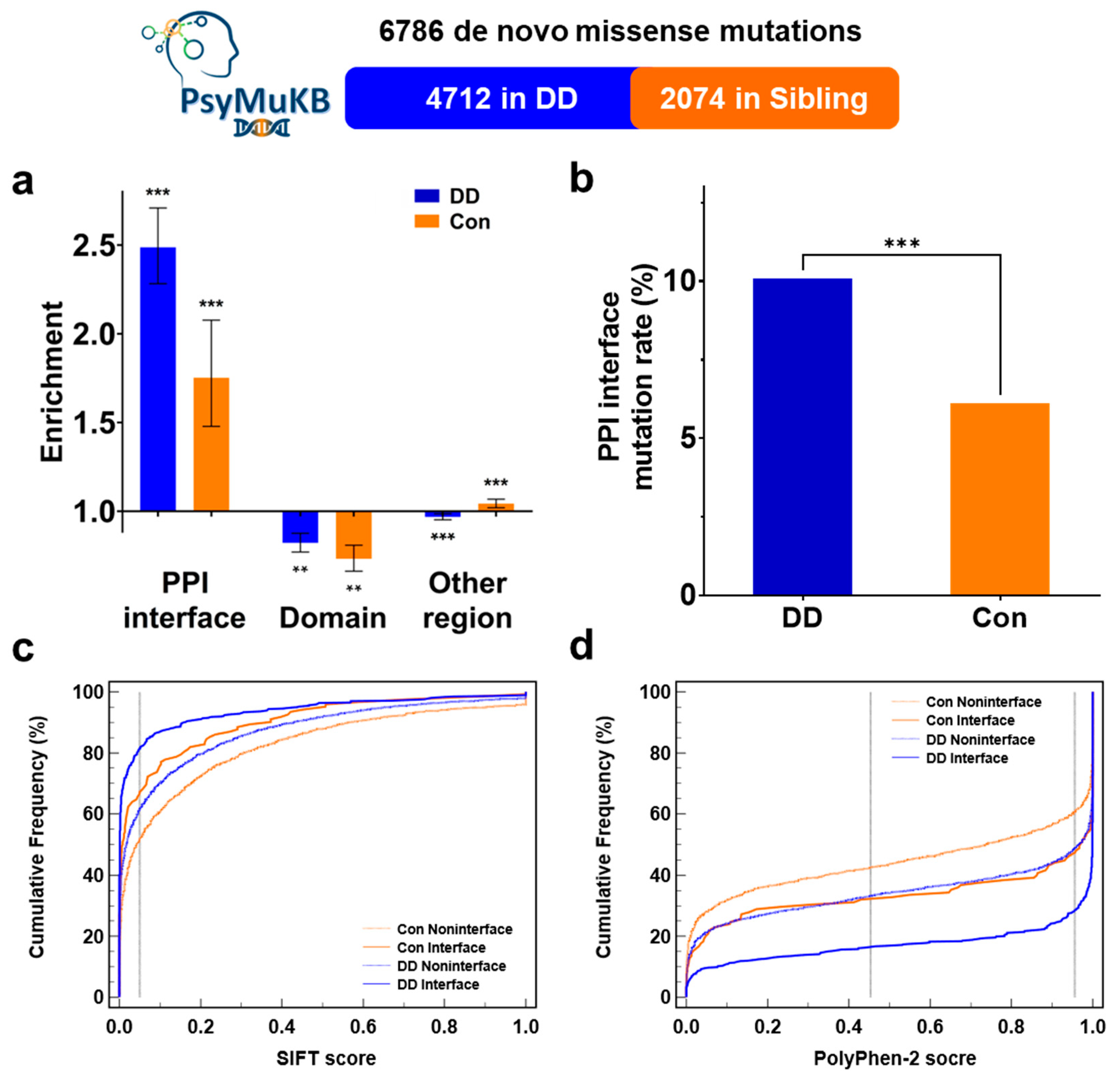
3.1. More De Novo Missense Mutations Were Observed on the Protein-Protein Interaction Interface in DD Patients Than in Controls
3.2. DNM Missenses at PPI Interface Are Significantly More Deleterious in DD Patients Than in Sibling Control
3.3. Extracting Potential DD Genes and DNM Missenses by Identifying PsychiPPI
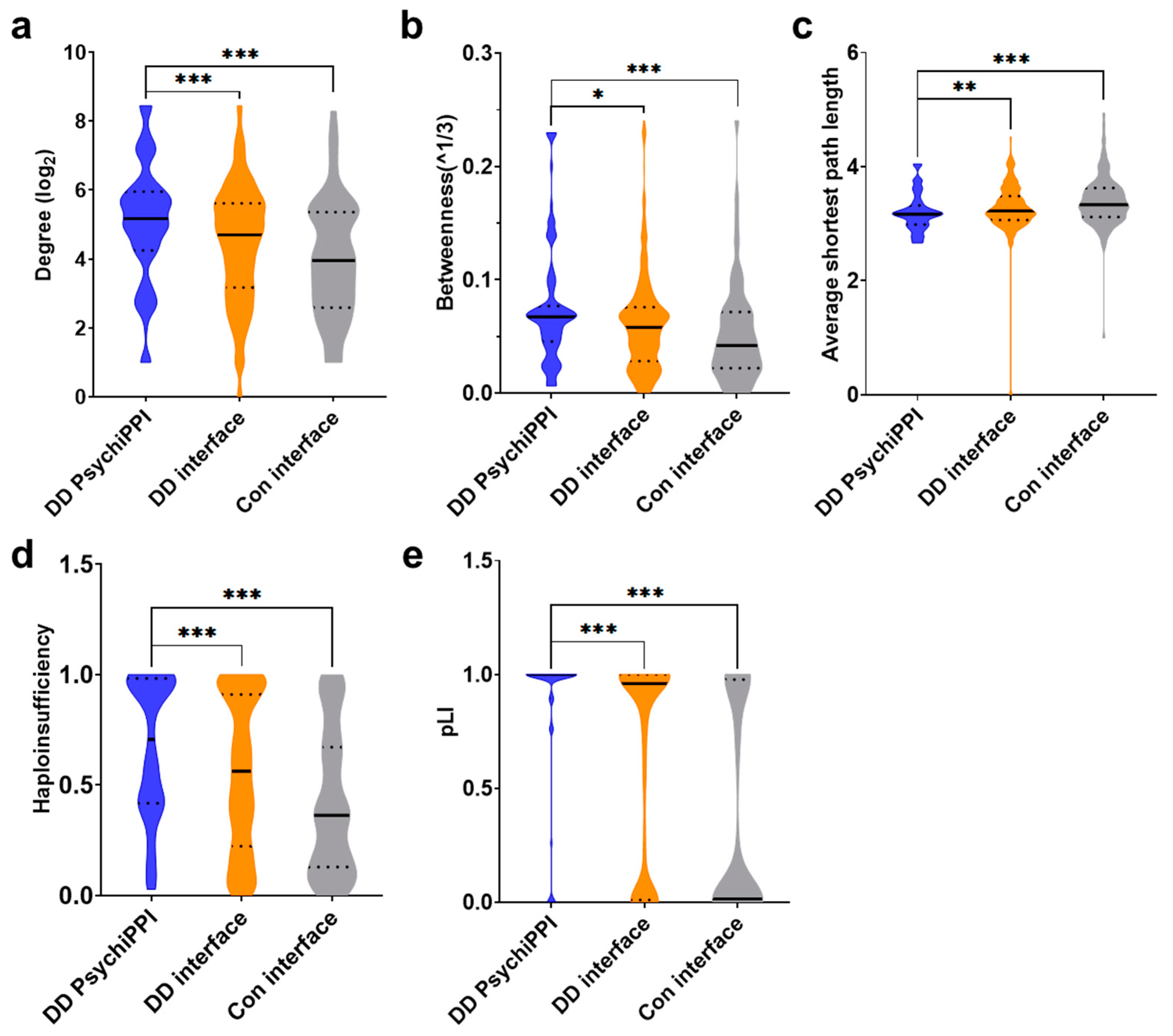
3.4. PsychiPPI Affected Hub Proteins in the Human Interactome
3.5. PsychiPPI-Related DD Candidate Genes Tend to Be Haploinsufficient
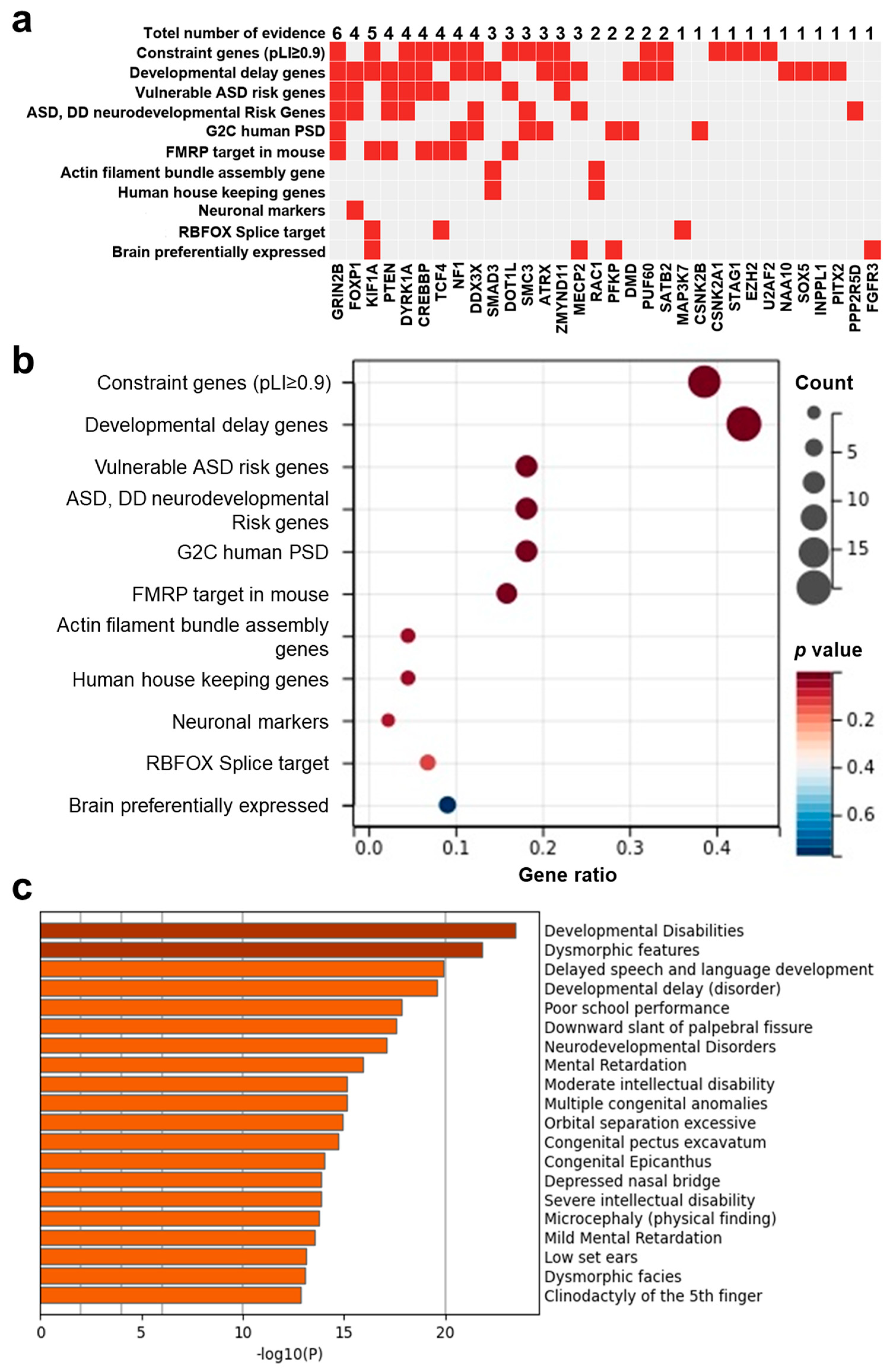
3.6. PsychiPPI-Related DD Candidate Genes Are Enriched in Curated DD-Related Geneset
3.7. Identification of New Potential DD Genes and De Novo Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oberklaid, F.; Efron, D. Developmental delay—Identification and management. Aust. Fam. Physician 2005, 34, 739–742. [Google Scholar]
- Rydz, D.; Srour, M.; Oskoui, M.; Marget, N.; Shiller, M.; Birnbaum, R.; Majnemer, A.; Shevell, M.I. Screening for developmental delay in the setting of a community pediatric clinic: A prospective assessment of parent-report questionnaires. Pediatrics 2006, 118, e1178–e1186. [Google Scholar] [CrossRef]
- Mackrides, P.S.; Ryherd, S.J. Screening for developmental delay. Am. Fam. Physician 2011, 84, 544–549. [Google Scholar]
- Glascoe, F.P. Screening for developmental and behavioral problems. Ment. Retard. Dev. Disabil. Res. Rev. 2005, 11, 173–179. [Google Scholar] [CrossRef]
- Yeo, G.S.; Connie Hung, C.-C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O’Rahilly, S.; Farooqi, I.S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef]
- Takenouchi, T.; Kosaki, R.; Niizuma, T.; Hata, K.; Kosaki, K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am. J. Med. Genet. A 2015, 167A, 2822–2825. [Google Scholar] [CrossRef]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef]
- Tanaka, A.J.; Bai, R.; Cho, M.T.; Anyane-Yeboa, K.; Ahimaz, P.; Wilson, A.L.; Kendall, F.; Hay, B.; Moss, T.; Nardini, M.; et al. De novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000356. [Google Scholar] [CrossRef] [Green Version]
- Fukai, R.; Saitsu, H.; Tsurusaki, Y.; Sakai, Y.; Haginoya, K.; Takahashi, K.; Hubshman, M.W.; Okamoto, N.; Nakashima, M.; Tanaka, F.; et al. De novo KCNH1 mutations in four patients with syndromic developmental delay, hypotonia and seizures. J. Hum. Genet. 2016, 61, 381–387. [Google Scholar] [CrossRef]
- Li, N.; Xu, Y.; Li, G.; Yu, T.; Yao, R.E.; Wang, X.; Wang, J. Exome sequencing identifies a de novo mutation of CTNNB1 gene in a patient mainly presented with retinal detachment, lens and vitreous opacities, microcephaly, and developmental delay: Case report and literature review. Medicine 2017, 96, e6914. [Google Scholar] [CrossRef]
- Chong, J.X.; McMillin, M.J.; Shively, K.M.; Beck, A.E.; Marvin, C.T.; Armenteros, J.R.; Buckingham, K.J.; Nkinsi, N.T.; Boyle, E.A.; Berry, M.N.; et al. De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am. J. Hum. Genet. 2015, 96, 462–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, C.W.; Yeung, W.L.; Law, C.Y. Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin. Chim. Acta 2017, 469, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Popp, B.; Stove, S.I.; Endele, S.; Myklebust, L.M.; Hoyer, J.; Sticht, H.; Azzarello-Burri, S.; Rauch, A.; Arnesen, T.; Reis, A. De novo missense mutations in the NAA10 gene cause severe non-syndromic developmental delay in males and females. Eur. J. Hum. Genet. 2015, 23, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, M.; Tohyama, J.; Nakagawa, E.; Watanabe, Y.; Siew, C.G.; Kwong, C.S.; Yamoto, K.; Hiraide, T.; Fukuda, T.; Kaname, T.; et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J. Hum. Genet. 2019, 64, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Masuho, I.; Patil, D.N.; Baumann, H.; Hebert, E.; Steinrucke, S.; Trujillano, D.; Skamangas, N.K.; Dobricic, V.; Huning, I.; et al. Novel GNB1 mutations disrupt assembly and function of G protein heterotrimers and cause global developmental delay in humans. Hum. Mol. Genet. 2017, 26, 1078–1086. [Google Scholar] [CrossRef] [Green Version]
- Ropers, H.H. Genetics of early onset cognitive impairment. Annu. Rev. Genom. Hum. Genet. 2010, 11, 161–187. [Google Scholar] [CrossRef] [Green Version]
- Mefford, H.C.; Batshaw, M.L.; Hoffman, E.P. Genomics, intellectual disability, and autism. N. Engl. J. Med. 2012, 366, 733–743. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.S.; Srinivas, K.; Sujini, G.N.; Kumar, G.N. Protein-protein interaction detection: Methods and analysis. Int. J. Proteom. 2014, 2014, 147648. [Google Scholar] [CrossRef] [Green Version]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot spots in protein-protein interfaces: Towards drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef]
- Navío, D.; Rosell, M.; Aguirre, J.; de la Cruz, X.; Fernández-Recio, J. Structural and computational characterization of disease-related mutations involved in protein-protein interfaces. Int. J. Mol. Sci. 2019, 20, 1583. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.J.; Beltran, J.F.; Liang, S.; Fragoza, R.; Rumack, A.; Liang, J.; Wei, X.; Yu, H. Interactome INSIDER: A structural interactome browser for genomic studies. Nat. Methods 2018, 15, 107–114. [Google Scholar] [CrossRef]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wei, X.; Thijssen, B.; Das, J.; Lipkin, S.M.; Yu, H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012, 30, 159–164. [Google Scholar] [CrossRef] [Green Version]
- Jemimah, S.; Gromiha, M.M. Insights into changes in binding affinity caused by disease mutations in protein-protein complexes. Comput. Biol. Med. 2020, 123, 103829. [Google Scholar] [CrossRef]
- Yan, W.; Markegard, E.; Dharmaiah, S.; Urisman, A.; Drew, M.; Esposito, D.; Scheffzek, K.; Nissley, D.V.; McCormick, F.; Simanshu, D.K. Structural insights into the SPRED1-neurofibromin-KRAS complex and disruption of SPRED1-neurofibromin interaction by oncogenic EGFR. Cell Rep. 2020, 32, 107909. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Cicek, E.; Roeder, K.; Yu, H.; Devlin, B. De novo missense variants disrupting protein–protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol. Autism 2020, 11, 76. [Google Scholar] [CrossRef]
- Teppa, E.; Zea, D.J.; Marino-Buslje, C. Protein–protein interactions leave evolutionary footprints: High molecular coevolution at the core of interfaces. Protein Sci. 2017, 26, 2438–2444. [Google Scholar] [CrossRef]
- Fragoza, R.; Das, J.; Wierbowski, S.D.; Liang, J.; Tran, T.N.; Liang, S.; Beltran, J.F.; Rivera-Erick, C.A.; Ye, K.; Wang, T.Y.; et al. Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat. Commun. 2019, 10, 4141. [Google Scholar] [CrossRef] [Green Version]
- Jubb, H.C.; Pandurangan, A.P.; Turner, M.A.; Ochoa-Montano, B.; Blundell, T.L.; Ascher, D.B. Mutations at protein-protein interfaces: Small changes over big surfaces have large impacts on human health. Prog. Biophys. Mol. Biol. 2017, 128, 3–13. [Google Scholar] [CrossRef]
- Chen, S.; Fragoza, R.; Klei, L.; Liu, Y.; Wang, J.; Roeder, K.; Devlin, B.; Yu, H. An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat. Genet. 2018, 50, 1032–1040. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive characterization of protein-protein interactions perturbed by disease mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef]
- Xiong, D.; Lee, D.; Li, L.; Zhao, Q.; Yu, H. Implications of disease-related mutations at protein-protein interfaces. Curr. Opin. Struct. Biol. 2022, 72, 219–225. [Google Scholar] [CrossRef]
- Lin, G.N.; Guo, S.; Tan, X.; Wang, W.; Qian, W.; Song, W.; Wang, J.; Yu, S.; Wang, Z.; Cui, D.; et al. PsyMuKB: An Integrative De Novo Variant Knowledge Base for Developmental Disorders. Genom. Proteom. Bioinform. 2019, 17, 453–464. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. UniProt C: UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L. ClinGen—The clinical genome resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [Green Version]
- David, A.; Sternberg, M.J. The contribution of missense mutations in core and rim residues of protein–protein interfaces to human disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [Green Version]
- David, A.; Razali, R.; Wass, M.N.; Sternberg, M.J. Protein-protein interaction sites are hot spots for disease-associated nonsynonymous SNPs. Hum. Mutat. 2012, 33, 359–363. [Google Scholar] [CrossRef]
- Kucukkal, T.G.; Petukh, M.; Li, L.; Alexov, E. Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr. Opin. Struct. Biol. 2015, 32, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Menke, L.A.; DDD Study; Gardeitchik, T.; Hammond, P.; Heimdal, K.R.; Houge, G.; Hufnagel, S.B.; Ji, J.; Johansson, S.; Kant, S.G. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein–Taybi syndrome. Am. J. Med. Genet. Part A 2018, 176, 862–876. [Google Scholar] [CrossRef]
- Biswas, D.; Cary, W.; Nolta, J.A. PPP2R5D-Related Intellectual Disability and Neurodevelopmental Delay: A Review of the Current Understanding of the Genetics and Biochemical Basis of the Disorder. Int. J. Mol. Sci. 2020, 21, 1286. [Google Scholar] [CrossRef] [Green Version]
- Low, K.J.; Ansari, M.; Abou Jamra, R.; Clarke, A.; El Chehadeh, S.; FitzPatrick, D.R.; Greenslade, M.; Henderson, A.; Hurst, J.; Keller, K.; et al. PUF60 variants cause a syndrome of ID, short stature, microcephaly, coloboma, craniofacial, cardiac, renal and spinal features. Eur. J. Hum. Genet. 2017, 25, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Ohba, C.; Haginoya, K.; Osaka, H.; Kubota, K.; Ishiyama, A.; Hiraide, T.; Komaki, H.; Sasaki, M.; Miyatake, S.; Nakashima, M.; et al. De novo KIF1A mutations cause intellectual deficit, cerebellar atrophy, lower limb spasticity and visual disturbance. J. Hum. Genet. 2015, 60, 739–742. [Google Scholar] [CrossRef]
- Moskowitz, A.M.; Belnap, N.; Siniard, A.L.; Szelinger, S.; Claasen, A.M.; Richholt, R.F.; De Both, M.; Corneveaux, J.J.; Balak, C.; Piras, I.S. A de novo missense mutation in ZMYND11 is associated with global developmental delay, seizures, and hypotonia. Mol. Case Stud. 2016, 2, a000851. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Kerner, B.; Suit, L.; Thomas, J.; Kleefstra, T.; Sherr, E. DDX3X-related neurodevelopmental disorder. In GeneReviews®; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Ramocki, M.B.; Tavyev, Y.J.; Peters, S.U. The MECP2 duplication syndrome. Am. J. Med. Genet. Part A 2010, 152, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, R.J.; Wada, T.; Fisher, C.A.; Malik, N.; Mitson, M.J.; Steensma, D.P.; Fryer, A.; Goudie, D.R.; Krantz, I.D. Traeger-Synodinos J: Mutations in the chromatin-associated protein ATRX. Hum. Mutat. 2008, 29, 796–802. [Google Scholar] [CrossRef]
- Lee, K.-S.; Choi, M.; Kwon, D.-W.; Kim, D.; Choi, J.-M.; Kim, A.-K.; Ham, Y.; Han, S.-B.; Cho, S.; Cheon, C.K. A novel de novo heterozygous DYRK1A mutation causes complete loss of DYRK1A function and developmental delay. Sci. Rep. 2020, 10, 9849. [Google Scholar] [CrossRef]
- Le Fevre, A.K.; Taylor, S.; Malek, N.H.; Horn, D.; Carr, C.W.; Abdul-Rahman, O.A.; O’Donnell, S.; Burgess, T.; Shaw, M.; Gecz, J. FOXP1 mutations cause intellectual disability and a recognizable phenotype. Am. J. Med. Genet. Part A 2013, 161, 3166–3175. [Google Scholar] [CrossRef]
- Lee, R.W.; Bodurtha, J.; Cohen, J.; Fatemi, A.; Batista, D. Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatric. Neurol. 2013, 48, 317–320. [Google Scholar] [CrossRef]
- Forrest, M.P.; Hill, M.J.; Quantock, A.J.; Martin-Rendon, E.; Blake, D.J. The emerging roles of TCF4 in disease and development. Trends Mol. Med. 2014, 20, 322–331. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Fish, J.L. SATB2-associated syndrome: Mechanisms, phenotype, and practical recommendations. Am. J. Med. Genet. Part A 2017, 173, 327–337. [Google Scholar] [CrossRef]
- Mullegama, S.V.; Klein, S.D.; Mulatinho, M.V.; Senaratne, T.N.; Singh, K.; Center, U.C.G.; Nguyen, D.C.; Gallant, N.M.; Strom, S.P.; Ghahremani, S. De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. Part A 2017, 173, 1319–1327. [Google Scholar] [CrossRef]
- Hu, C.; Chen, W.; Myers, S.J.; Yuan, H.; Traynelis, S.F. Human GRIN2B variants in neurodevelopmental disorders. J. Pharmacol. Sci. 2016, 132, 115–121. [Google Scholar] [CrossRef]
- Mirski, K.T.; Crawford, T.O. Motor and cognitive delay in Duchenne muscular dystrophy: Implication for early diagnosis. J. Pediatr. 2014, 165, 1008–1010. [Google Scholar] [CrossRef]
- Hiraide, T.; Tanaka, T.; Masunaga, Y.; Ohkubo, Y.; Nakashima, M.; Fukuda, T.; Ogata, T.; Saitsu, H. Global developmental delay, systemic dysmorphism and epilepsy in a patient with a de novo U2AF2 variant. J. Hum. Genet. 2021, 66, 1185–1187. [Google Scholar] [CrossRef]
- Reijnders, M.R.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W. RAC1 missense mutations in developmental disorders with diverse phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Varga, E.A.; Pastore, M.; Prior, T.; Herman, G.E.; McBride, K.L. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 2009, 11, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W.T.; Hood, R.L.; Zhan, S.H.; Bulman, D.E.; Fejes, A.P.; Moore, R.; Mungall, A.J.; Eydoux, P.; Babul-Hirji, R.; An, J. Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 2012, 90, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Ruggieri, M.; Maynard, J.; Osborn, M.; Hartog, C.; Mudd, S.; Penttinen, M.; Cordeiro, I.; Ponder, M.; Ponder, B. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum. Genet. 1998, 102, 591–597. [Google Scholar] [CrossRef]
- Goh, K.-I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.-L. The human disease network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [Green Version]
- Feldman, I.; Rzhetsky, A.; Vitkup, D. Network properties of genes harboring inherited disease mutations. Proc. Natl. Acad. Sci USA 2008, 105, 4323–4328. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, Y. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics 2006, 22, 2800–2805. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, P.F.; Bates, P.A. Global topological features of cancer proteins in the human interactome. Bioinformatics 2006, 22, 2291–2297. [Google Scholar] [CrossRef] [Green Version]
- Veitia, R.A. Exploring the etiology of haploinsufficiency. Bioessays 2002, 24, 175–184. [Google Scholar] [CrossRef]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Ronemus, M.; Iossifov, I.; Levy, D.; Wigler, M. The role of de novo mutations in the genetics of autism spectrum disorders. Nat. Rev. Genet. 2014, 15, 133–141. [Google Scholar] [CrossRef]
- Anney, R.J.; Kenny, E.M.; O’Dushlaine, C.; Yaspan, B.L.; Parkhomenka, E.; Buxbaum, J.D.; Sutcliffe, J.; Gill, M.; Gallagher, L.; The Autism Genome Project; et al. Gene-ontology enrichment analysis in two independent family-based samples highlights biologically plausible processes for autism spectrum disorders. Eur. J. Hum. Genet. 2011, 19, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Correa, T.; Venancio, A.C.; Galera, M.F.; Riegel, M. Candidate Genes Associated with Delayed Neuropsychomotor Development and Seizures in a Patient with Ring Chromosome 20. Case Rep. Genet. 2020, 2020, 5957415. [Google Scholar] [CrossRef]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld, M.A.; Kleefstra, T.; Kramer, J.M. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Leventhal, B.L. Developmental Delay; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rockel, A.J.; Hiorns, R.W.; Powell, T.P. The basic uniformity in structure of the neocortex. Brain 1980, 103, 221–244. [Google Scholar] [CrossRef]
- Marr, D. A theory of cerebellar cortex. J. Physiol. 1969, 202, 437–470. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, C.J. The Cerebellum and Neurodevelopmental Disorders. Cerebellum 2016, 15, 34–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habibullah, H.; Albaradie, R.; Bashir, S. MRI evaluation of global developmental delay: A retrospective study. Dubai Med. J. 2020, 3, 1–4. [Google Scholar] [CrossRef]
- Heide, M.; Huttner, W.B.; Mora-Bermudez, F. Brain organoids as models to study human neocortex development and evolution. Curr. Opin. Cell Biol. 2018, 55, 8–16. [Google Scholar] [CrossRef]
- Ma, X.; Tan, J.; Jiang, L.; Wang, X.; Cheng, B.; Xie, P.; Li, Y.; Wang, J.; Li, S. Aberrant Structural and Functional Developmental Trajectories in Children With Intellectual Disability. Front. Psychiatry 2021, 12, 634170. [Google Scholar] [CrossRef]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H. GeneCards Version 3: The human gene integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef] [Green Version]
- Harada, D.; Yamanaka, Y.; Ueda, K.; Tanaka, H.; Seino, Y. FGFR3-related dwarfism and cell signaling. J. Bone Miner. Metab. 2009, 27, 9–15. [Google Scholar] [CrossRef]
- Šerý, O.; Hlinecká, L.; Povová, J.; Bonczek, O.; Zeman, T.; Janout, V.; Ambroz, P.; Khan, N.A.; Balcar, V.J. Arachidonate 5-lipoxygenase (ALOX5) gene polymorphism is associated with Alzheimer’s disease and body mass index. J. Neurol. Sci. 2016, 362, 27–32. [Google Scholar] [CrossRef]
- Sun, Q.-Y.; Zhou, H.-H.; Mao, X.-Y. Emerging roles of 5-lipoxygenase phosphorylation in inflammation and cell death. Oxidative Med. Cell. Longev. 2019, 2019, 2749173. [Google Scholar] [CrossRef]
- Kim, S.-W.; Kim, Y.; Kim, S.E.; An, J.-Y. Ferroptosis-related genes in neurodevelopment and central nervous system. Biology 2021, 10, 35. [Google Scholar] [CrossRef]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kahler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Krebs, B.B.; Miskus, M.L.; Russell, M.L.; Duffy, E.P.; Graf, J.M.; Lu, H.C. Enhanced FGFR3 activity in postmitotic principal neurons during brain development results in cortical dysplasia and axonal tract abnormality. Sci. Rep. 2020, 10, 18508. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Vargas, A.; Hafner, C.; Perez-Rodriguez, A.G.; Rodriguez-Rojas, L.X.; Gonzalez-Esqueda, P.; Stoehr, R.; Hernandez-Torres, M.; Happle, R. An epidermal nevus syndrome with cerebral involvement caused by a mosaic FGFR3 mutation. Am. J. Med. Genet A 2008, 146, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Joshi, Y.B.; Giannopoulos, P.F.; Chu, J.; Sperow, M.; Kirby, L.G.; Abood, M.E.; Pratico, D. Absence of ALOX5 gene prevents stress-induced memory deficits, synaptic dysfunction and tauopathy in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2014, 23, 6894–6902. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
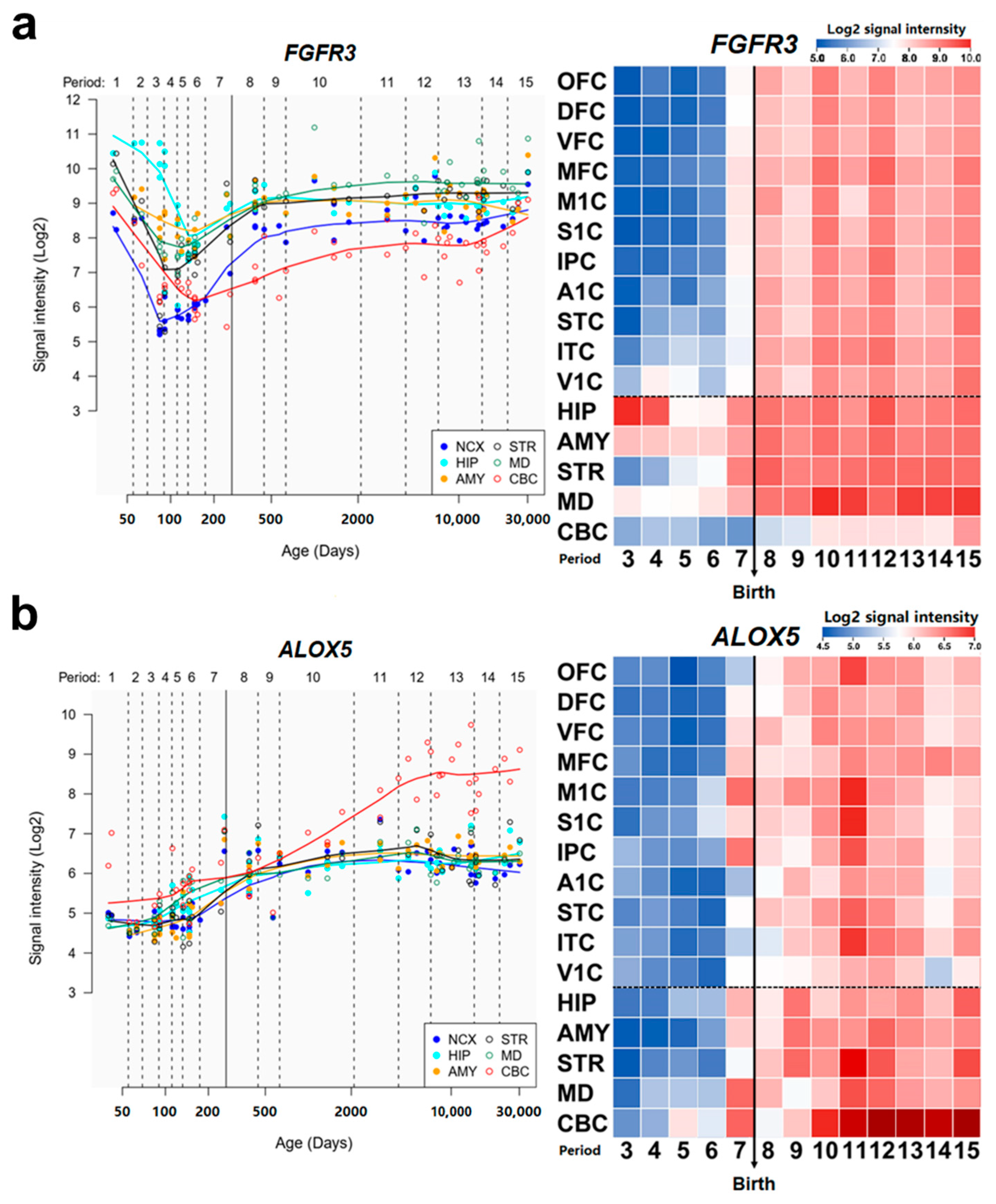{kind=link}
| Gene | Interface/ Mutation | Protein Change | SIFT | Polyphen2 | Ref | ||
|---|---|---|---|---|---|---|---|
| CREBBP | 6/10 | p.C1788W p.H1791D | p.C1370W p.N1551K | p.Y613C p.Y651C | D | D | [49] |
| PPP2R5D | 14/16 | p.E198K | p.P201R | D | D, P | [50] | |
| PUF60 | 3/3 | p.E138K | p.D142N | p.G448E | D, T | D, B | [51] |
| KIF1A | 4/8 | p.R307P | p.R465W | p.R307Q | D | D | [52] |
| NAA10 | 4/5 | p.R82Q p.H114P | p.H120P | p.R83C | D | D, P | [13] |
| ZMYND11 | 5/6 | p.R546W p.C520R | p.C489R p.R583W | p.V493I | D, T | D, B | [53] |
| DDX3X | 8/15 | p.C325R p.R360C p.A217V | p.P568L p.C452Y | p.V535I p.R475G | D | P, D, B | [54] |
| CSNK2A1 | 6/8 | p.K62R p.I38M | p.F61I | p.R191Q | D | D, P | [14] |
| MECP2 | 6/9 | p.R145C | p.R133H | p.T158M | D | D | [55] |
| ATRX | 1/2 | p.L191F | D | D | [56] | ||
| DYRK1A | 5/7 | p.L169P p.S337P | p.L198P p.A277P | p.D287V | D | D | [57] |
| FOXP1 | 5/7 | p.A534E, | p.F500L | p.A533E | D | D | [58] |
| SOX5 | 2/2 | p.A561P | p.A596P | D | D | [59] | |
| TCF4 | 2/4 | p.R576W | p.R579W | D | D | [60] | |
| CSNK2B | 1/2 | p.R86C | D | D | [14] | ||
| SATB2 | 5/5 | p.E402K p.R389L | p.R389C p.R399H | p.G515S | D | D, P | [61] |
| SMC3 | 3/4 | p.G1188A | p.Q1147E | D | D | [62] | |
| GRIN2B | 3/8 | p.E807K | p.S628F | p.N615K | NA | D | [63] |
| DMD | 1/1 | p.D97N | D | B | [64] | ||
| U2AF2 | 1/4 | p.T252I | D | D | [65] | ||
| RAC1 | 2/3 | p.Y64D | p.P73L | D | P, D | [66] | |
| PTEN | 2/2 | p.L313F | p.D268E | D, T | D, B | [67] | |
| EZH2 | 2/2 | p.R679C | p.R679H | D | D | [68] | |
| NF1 | 1/2 | p.R1830C | D | D | [69] | ||
| STAG1 | 2/2 | p.R216G | D | D | NA | ||
| PTPN3 | 1/1 | p.K708N | D | D | NA | ||
| PITX2 | 1/1 | p.R115G | D | D | NA | ||
| MAP3K7 | 1/2 | p.R238Q | D, T | B | NA | ||
| FGFR3 | 1/1 | p.G380R | D | D | NA | ||
| TOR1AIP1 | 1/1 | p.R438H | D | B | NA | ||
| ING4 | 1/1 | p.Y195C | D | D | NA | ||
| NFAT5 | 1/1 | p.E462D | D | D | NA | ||
| DOT1L | 1/1 | p.R292C | D | B | NA | ||
| INPPL1 | 1/2 | p.R581Q | D | D | NA | ||
| PFKP | 2/2 | p.I473V | p.I651V | D | B | NA | |
| RBM12 | 1/1 | p.V867M | D | D | NA | ||
| FGFR4 | 2/2 | p.D507N | p.R567G | D | D, P | NA | |
| ALOX5 | 1/1 | p.G528S | D | D | NA | ||
| SMAD3 | 1/1 | p.G249C | D | D | NA | ||
| TDRD7 | 1/1 | p.T6I | D | D | NA | ||
| ANKRD28 | 1/1 | p.G247E | D | D | NA | ||
| H2BC3 | 1/2 | p.S56L | NA | B | NA | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maharjan, D.T.; Song, W.; Liu, Z.; Wang, W.; Cai, W.; Chen, J.; Xu, F.; Ying, W.; Lin, G.N. A Comprehensive Study of De Novo Mutations on the Protein-Protein Interaction Interfaces Provides New Insights into Developmental Delay. Biomolecules 2022, 12, 1643. https://doi.org/10.3390/biom12111643
Maharjan DT, Song W, Liu Z, Wang W, Cai W, Chen J, Xu F, Ying W, Lin GN. A Comprehensive Study of De Novo Mutations on the Protein-Protein Interaction Interfaces Provides New Insights into Developmental Delay. Biomolecules. 2022; 12(11):1643. https://doi.org/10.3390/biom12111643
Chicago/Turabian StyleMaharjan, Dhruba Tara, Weichen Song, Zhe Liu, Weidi Wang, Wenxiang Cai, Jue Chen, Fei Xu, Weihai Ying, and Guan Ning Lin. 2022. "A Comprehensive Study of De Novo Mutations on the Protein-Protein Interaction Interfaces Provides New Insights into Developmental Delay" Biomolecules 12, no. 11: 1643. https://doi.org/10.3390/biom12111643