
Identification of a Fibroblast-Related Prognostic Model in Glioma Based on Bioinformatics Methods
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Certification of Differentially Expressed Fibroblast-Related Genes (DEFRGs)
2.3. Functional Enrichment Analysis
2.4. Establishment and Validation of the Fibroblast-Related Risk Score Model
2.5. Correlation of Risk Score and Clinical Characteristics
2.6. Independent Prognostic Analysis and Construction of a Nomogram
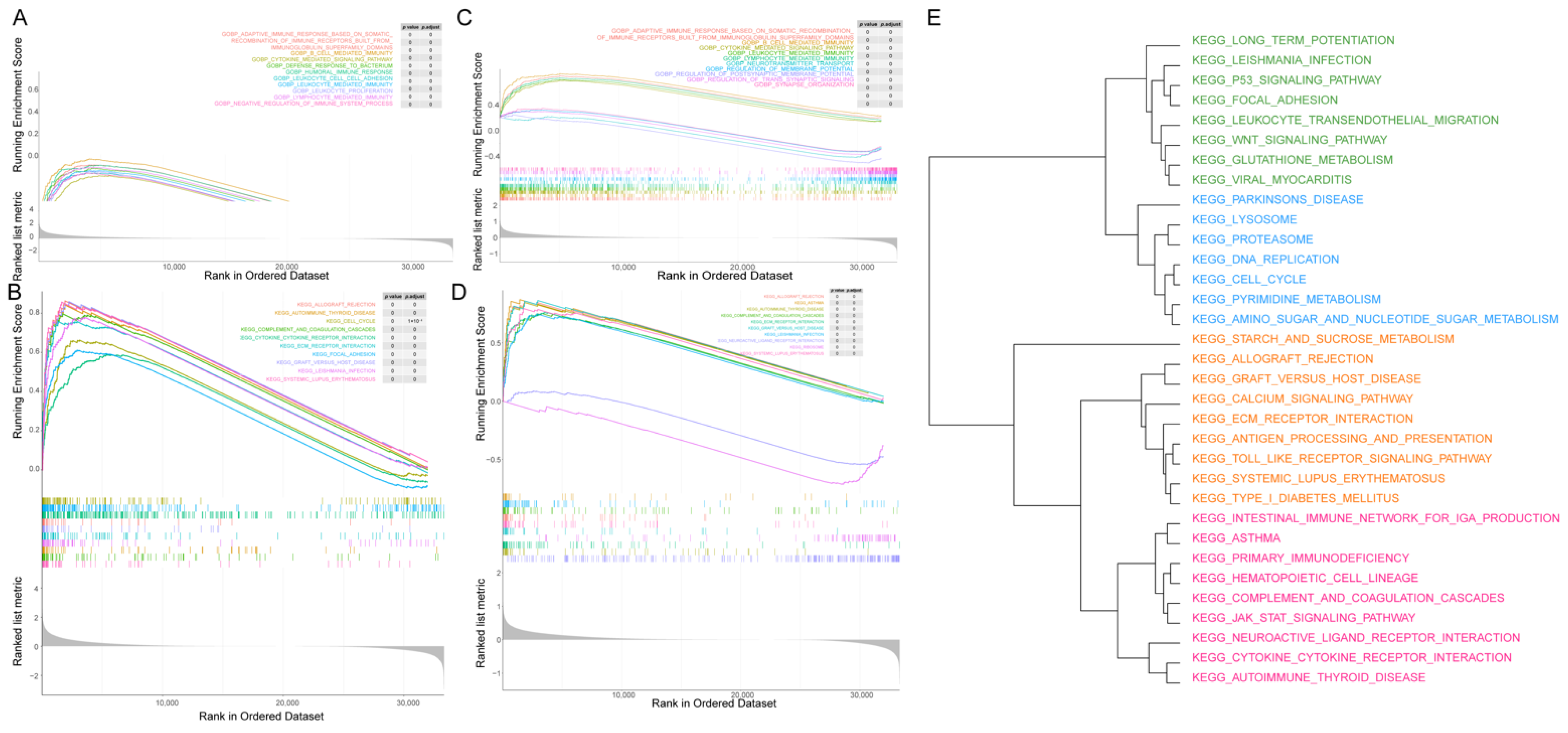
2.7. Gene Set Enrichment Analysis
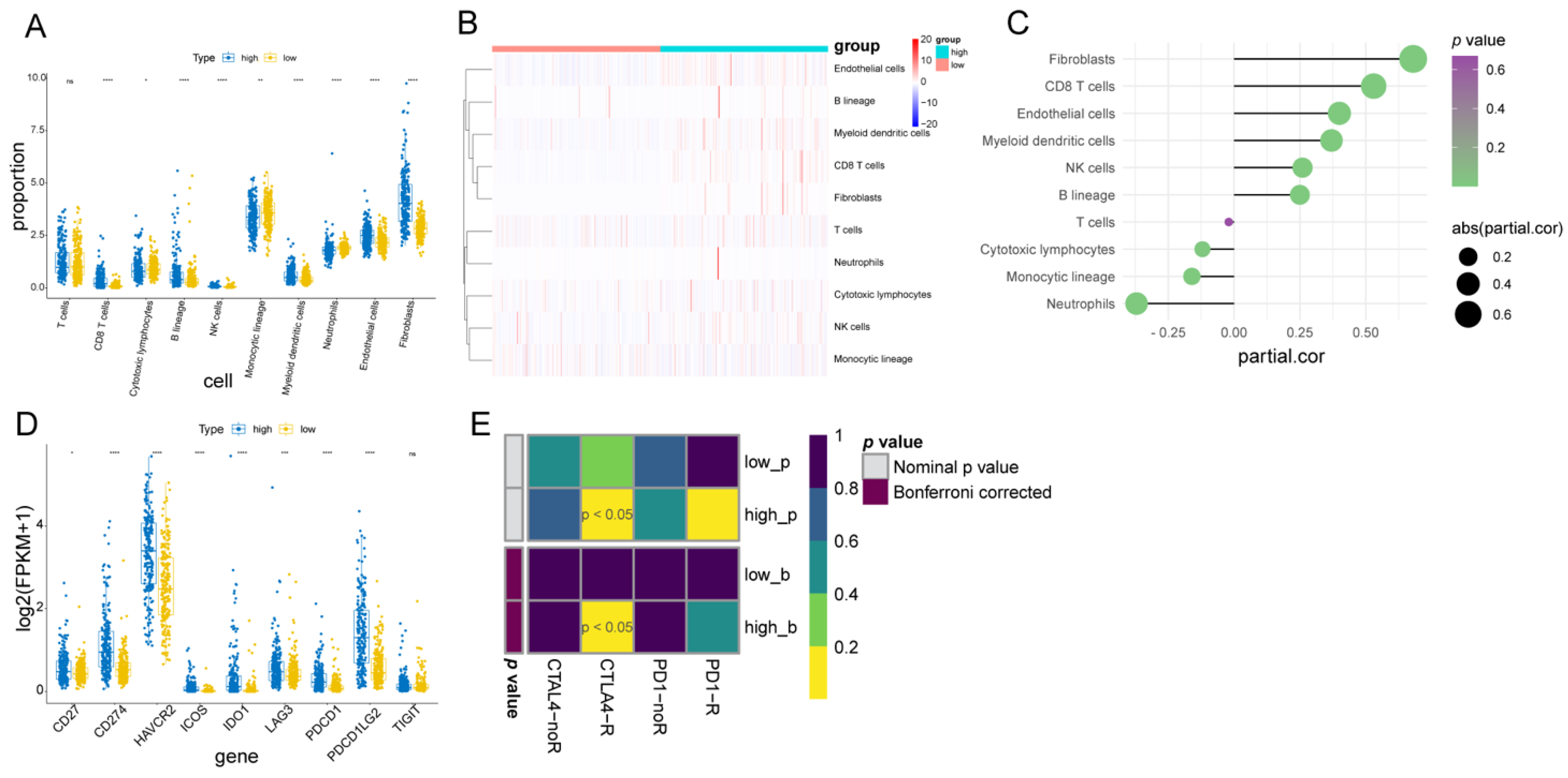
2.8. Assessment of Immune Infiltration Cell and Stromal Cell, and Immunotherapy Response between Two Fibroblast-Related Risk Subgroups
2.9. Subsection Evaluation of Chemotherapy Sensitivity between Two Fibroblast-Related Risk Subgroups
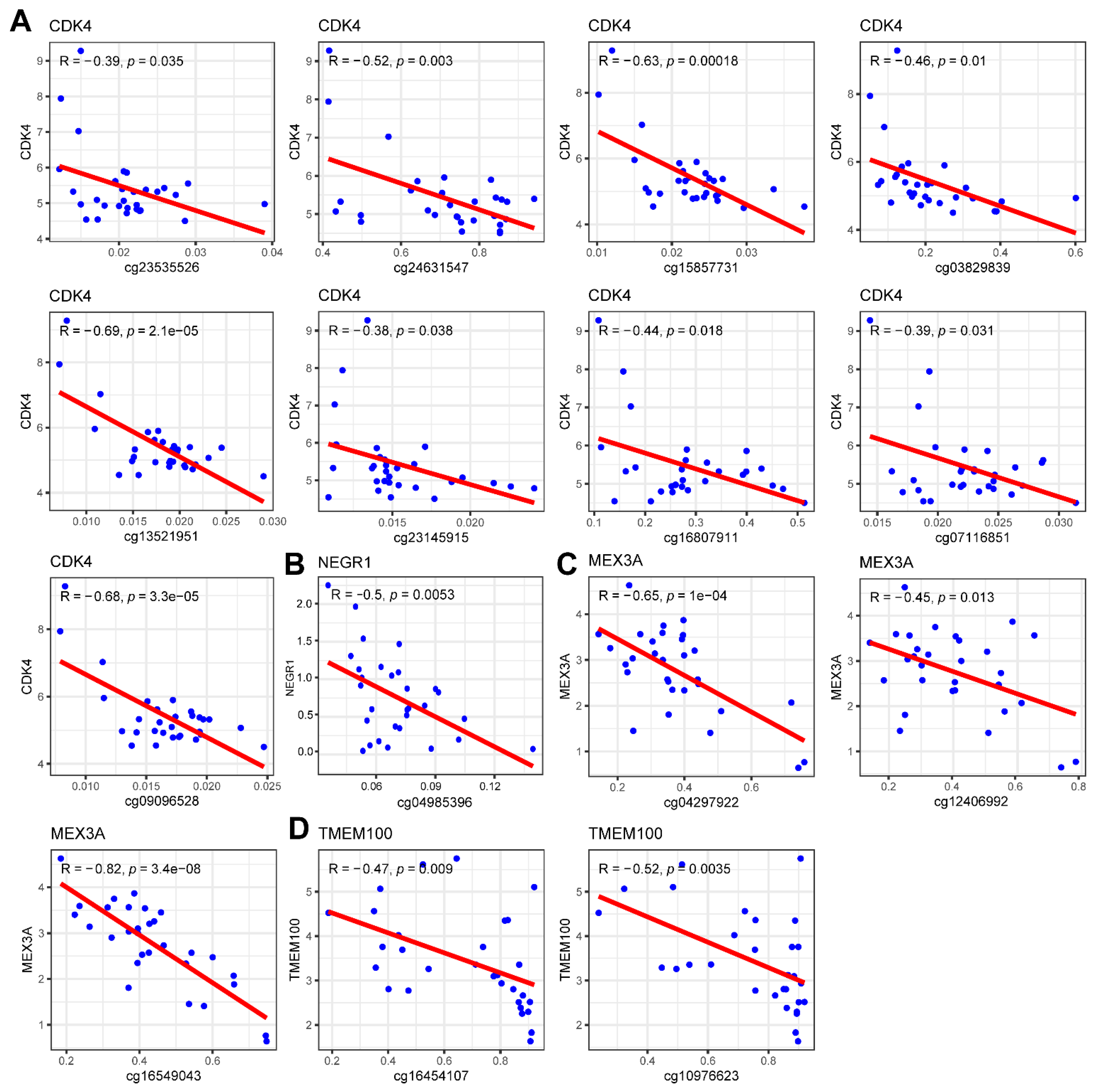
2.10. Methylation Analysis of Prognostic Genes
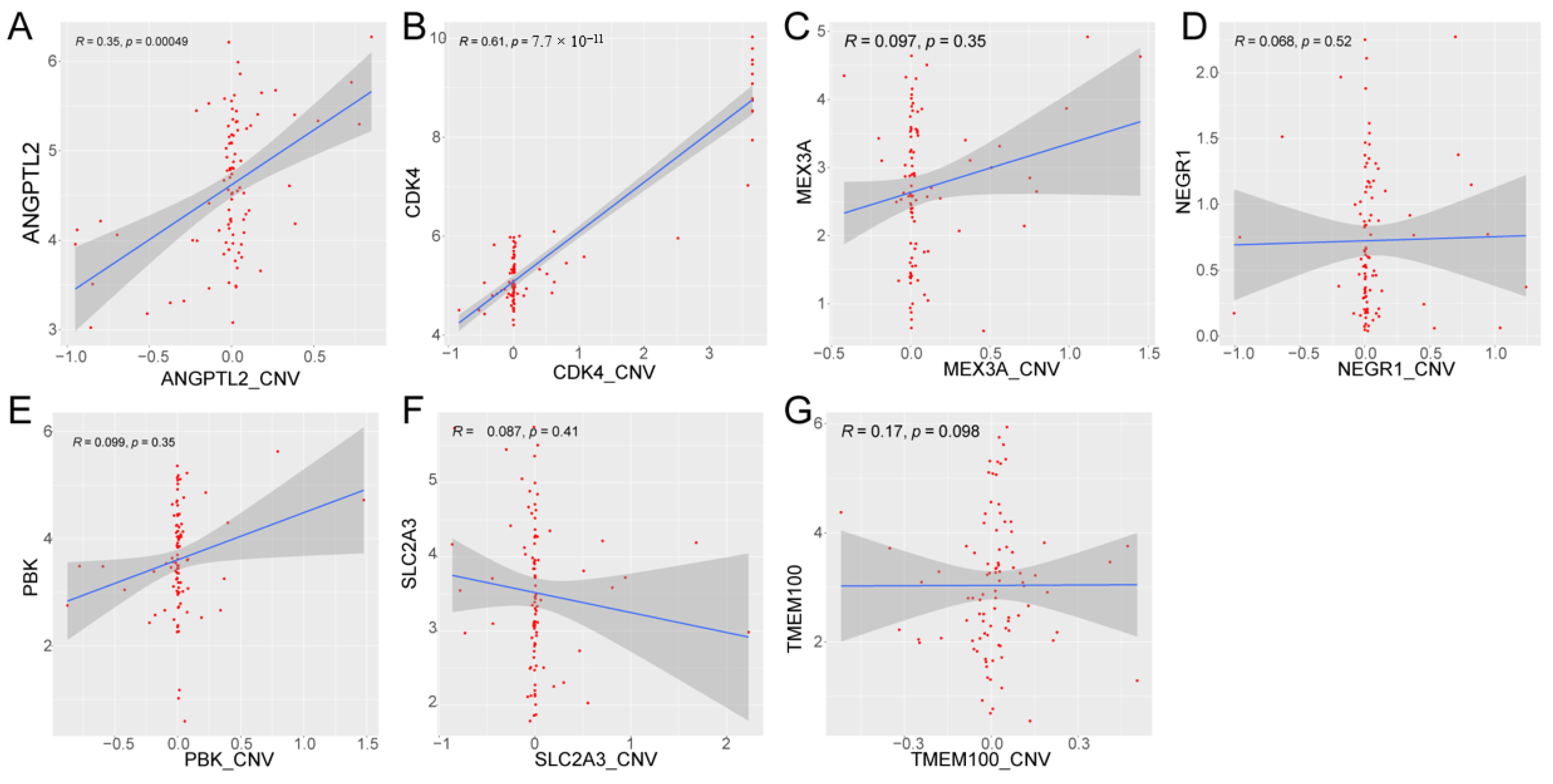
2.11. Correlation Analysis between Prognostic Gene Expression and CNV
2.12. Antibodies
2.13. Immunohistochemistry (IHC)
2.14. Statistical Analysis
3. Results
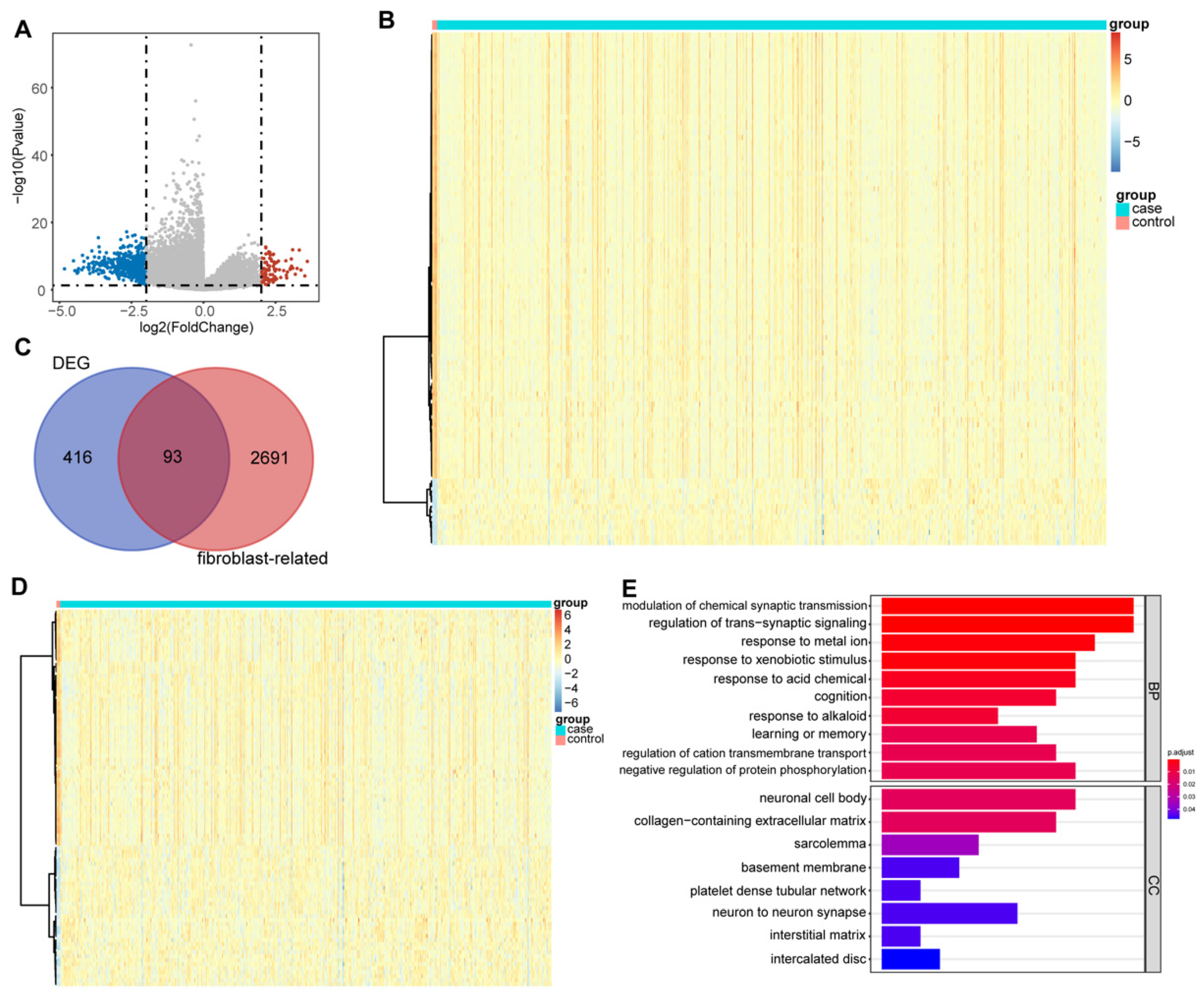
3.1. DEFRGs in Glioma
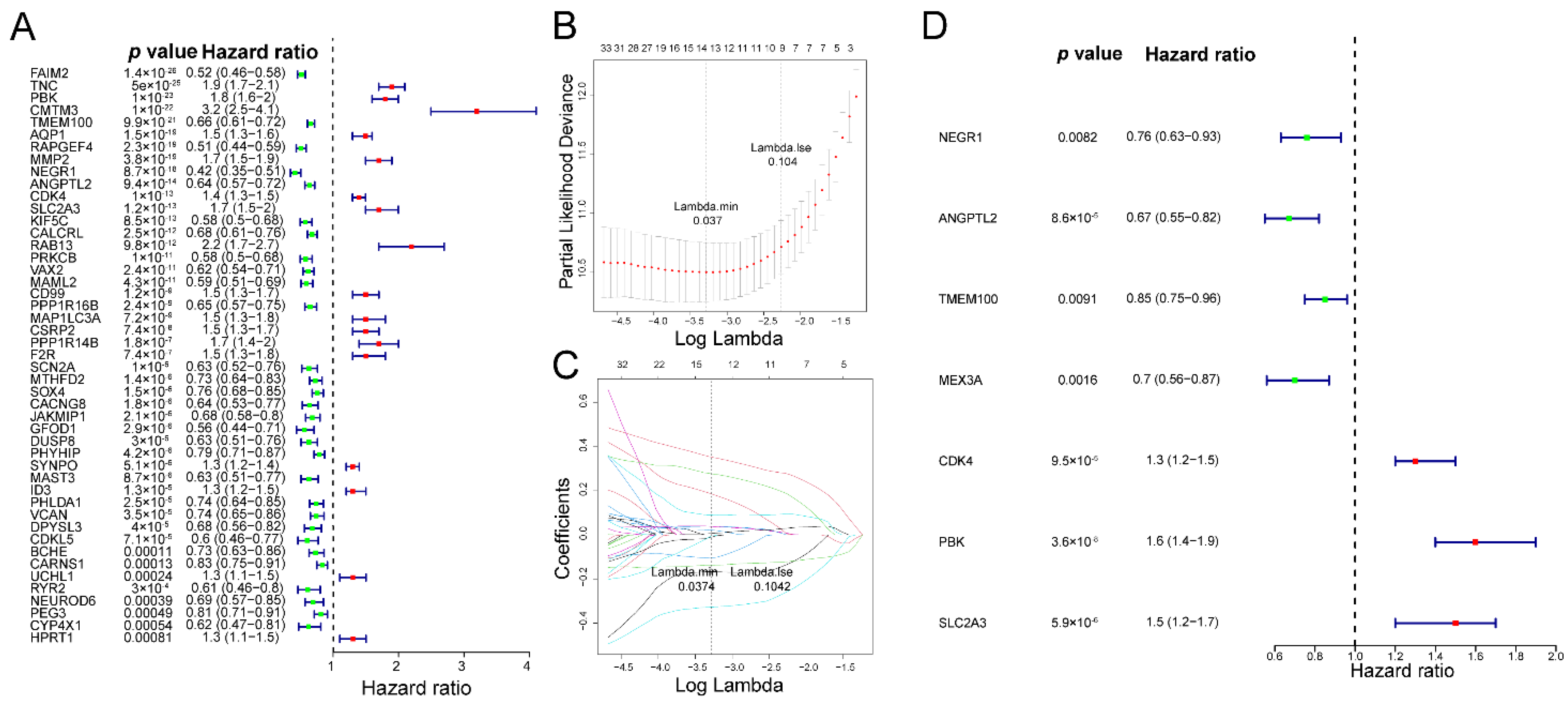
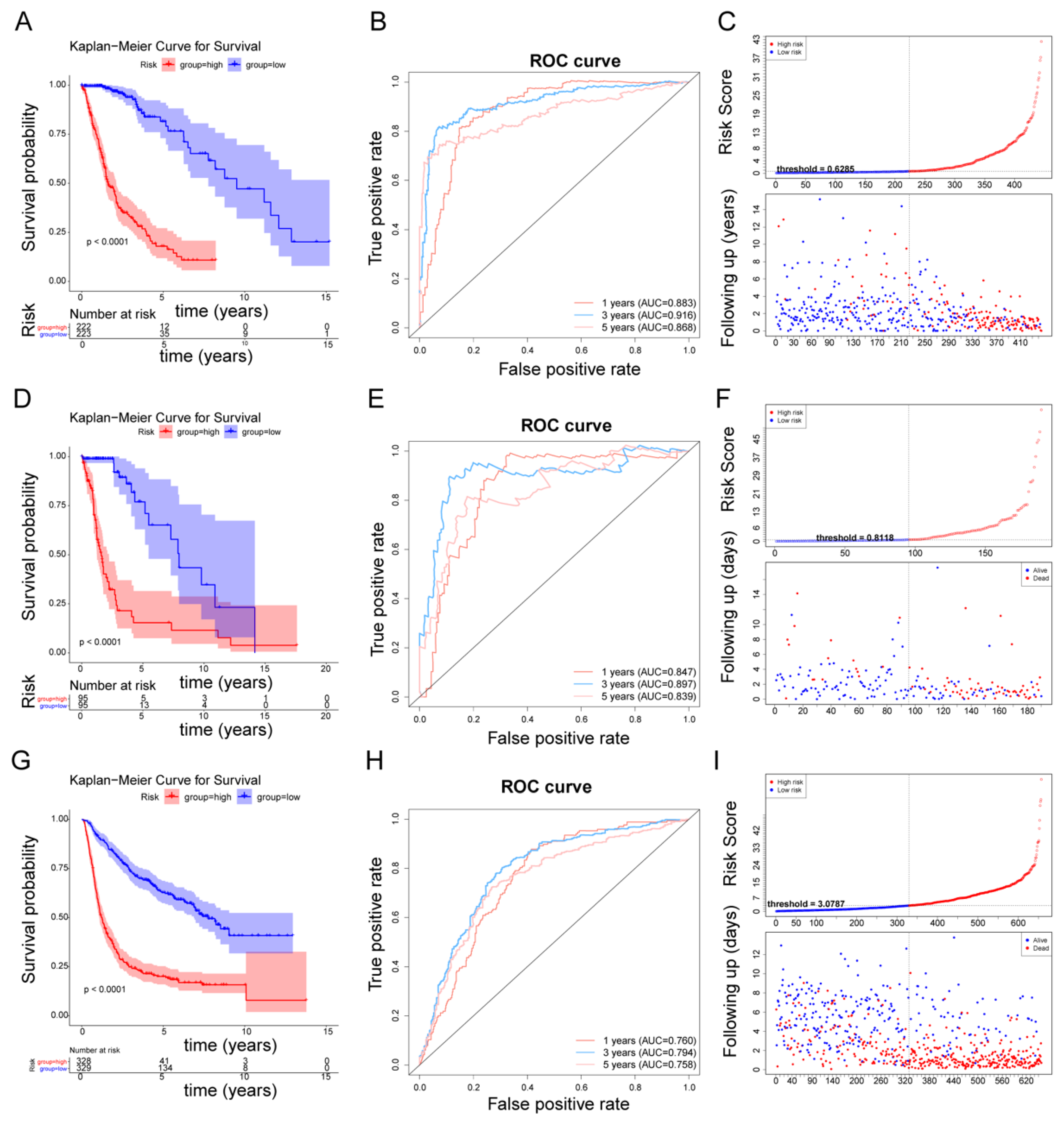
3.2. The Fibroblast-Related Risk Score Model Based on DEFRGs
3.3. Affirmation of the Fibroblast-Related Risk Model Via External Cohort
3.4. Correlation between the FRG-Related Gene Signature and Clinicopathological Characteristics
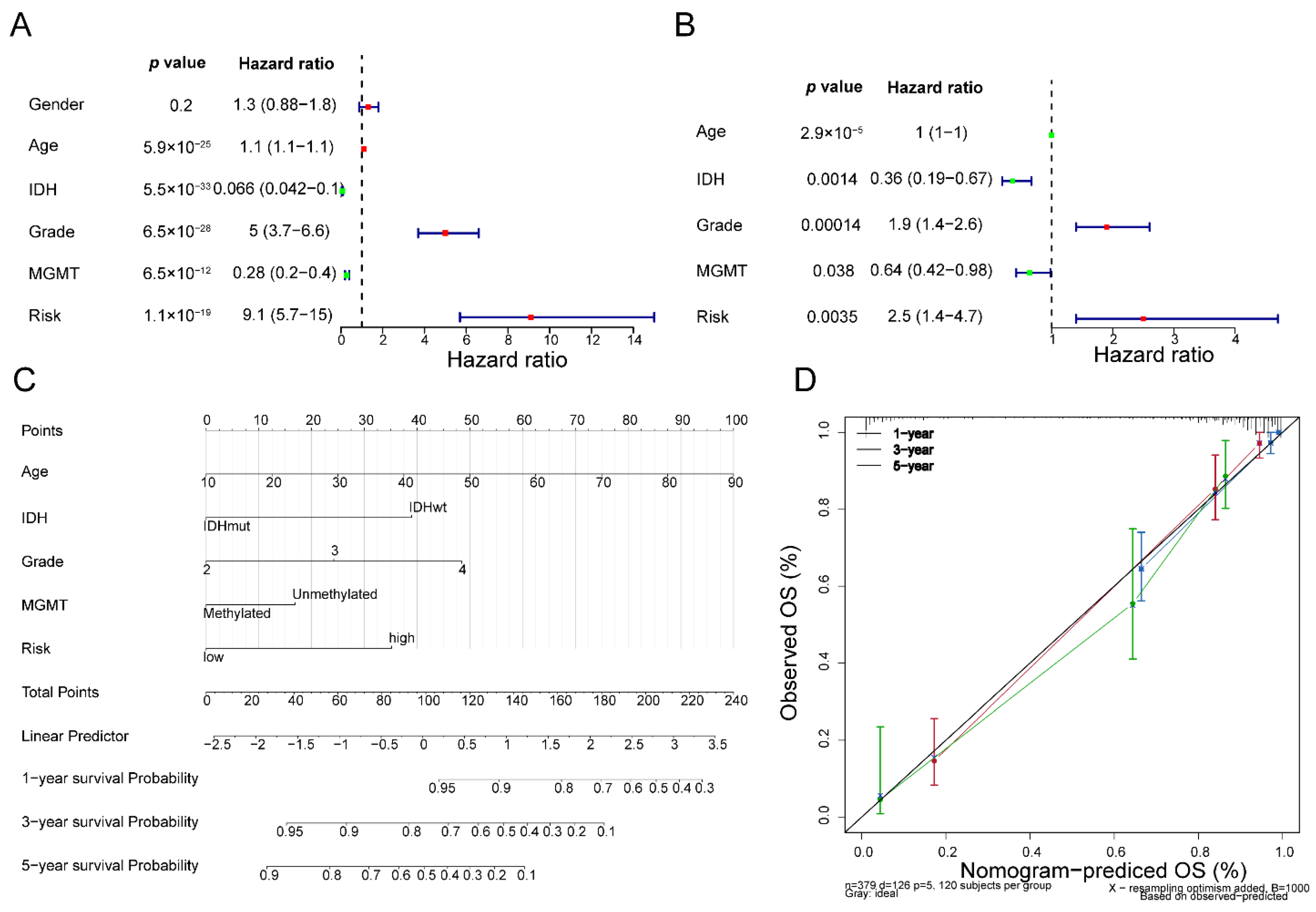
3.5. Fibroblast-Related Risk Signature Is an Independent Predictor for Glioma Prognosis
3.6. GSEA between the Two Fibroblast-Related Risk Subgroups
3.7. Immune Infiltration Cell, Stromal Cell, and Immunotherapy Response between the Two Fibroblast-Related Risk Subgroups
3.8. DEFRGs in Glioma
3.9. Methylation and CNV Analysis of Prognostic Genes
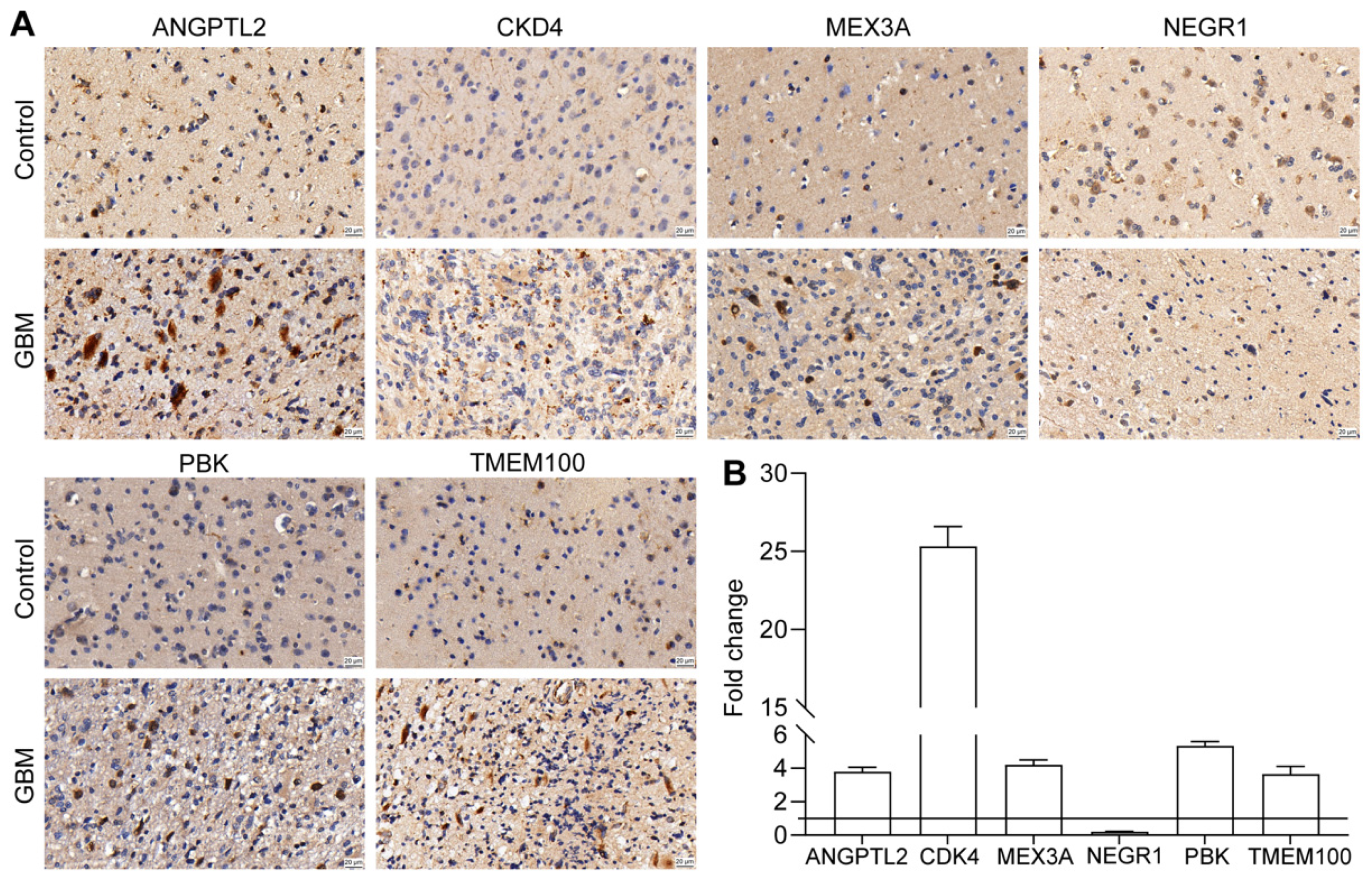
3.10. Validation of Prognostic Gene Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Wang, D.; He, M.Q.; Fan, D.Q. RARRES1 is a novel immune-related biomarker in GBM. Am. J. Transl. Res. 2019, 11, 5655–5663. [Google Scholar] [PubMed]
- Sturm, D.; Pfister, S.M.; Jones, D.T.W. Pediatric Gliomas: Current Concepts on Diagnosis, Biology, and Clinical Management. J. Clin. Oncol. 2017, 35, 2370–2377. [Google Scholar] [CrossRef]
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Feng, E.; Sui, C.; Wang, T.; Sun, G. Temozolomide with or without Radiotherapy in Patients with Newly Diagnosed Glioblastoma Multiforme: A Meta-Analysis. Eur. Neurol. 2017, 77, 201–210. [Google Scholar] [CrossRef]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neurol. Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, D.; Chang, P.; Weinberg, B.D.; Bota, D.A.; Grinband, J.; Filippi, C.G. Imaging Genetic Heterogeneity in Glioblastoma and Other Glial Tumors: Review of Current Methods and Future Directions. AJR Am. J. Roentgenol. 2018, 210, 30–38. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020, 10, 964–979. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, S.; Piccoli, M.; Rizzolio, F.; Giordano, A.; Nitti, D.; Agostini, M. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J. Cell. Physiol. 2017, 232, 967–975. [Google Scholar] [CrossRef]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B-Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jia, A.; Xu, L.; Wang, Y. Venn diagrams in bioinformatics. Brief. Bioinform. 2021, 22, bbab108. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. Drawing Survival Curves Using ‘ggplot2’ [R Package Survminer Version 0.3.1]. 2017. Available online: https://rpkgs.datanovia.com/survminer/ (accessed on 21 September 2022).
- Heagerty, P.J.; Saha-Chaudhuri, P. Time-Dependent ROC Curve Estimation from Censored Survival Data [R Package Survival ROC Version 1.0.3]. 2013. Available online: https://rdrr.io/cran/survivalROC/man/survivalROC.html (accessed on 21 September 2022).
- Kolde, R. Pretty Heatmaps [R Package Pheatmap Version 1.0.10]. 2018. Available online: https://rdrr.io/cran/pheatmap/ (accessed on 21 September 2022).
- Schober, P.; Vetter, T.R. Chi-square Tests in Medical Research. Anesth. Analg. 2019, 129, 1193. [Google Scholar] [CrossRef] [PubMed]
- Harrell, F.E., Jr. Regression Modeling Strategies [R Package rms Version 5.1-2]. 2018. Available online: https://rdrr.io/cran/rms/ (accessed on 21 September 2022).
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Ay, F.; Kellis, M.; Kahveci, T. SubMAP: Aligning metabolic pathways with subnetwork mappings. J. Comput. Biol. 2011, 18, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Ward, J.P.; Gubin, M.M.; Arthur, C.D.; Lee, S.H.; Hundal, J.; Selby, M.J.; Graziano, R.F.; Mardis, E.R.; Korman, A.J.; et al. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol. Res. 2017, 5, 106–117. [Google Scholar] [CrossRef] [Green Version]
- Gyuris, A.; Navarrete-Perea, J.; Jo, A.; Cristea, S.; Zhou, S.; Fraser, K.; Wei, Z.; Krichevsky, A.M.; Weissleder, R.; Lee, H.; et al. Physical and Molecular Landscapes of Mouse Glioma Extracellular Vesicles Define Heterogeneity. Cell Rep. 2019, 27, 3972–3987.e6. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.Y.; Hou, Y.X.; Fu, L.X.; Xi, L.; Yang, D.; Zhao, M.J.; Qin, Y.L.; Sun, K.X.; Teng, Y.; Liu, M.R. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin beta 3-p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef]
- Elkhattouti, A.; Hassan, M.; Gomez, C.R. Stromal fibroblast in age-related cancer: Role in tumorigenesis and potential as novel therapeutic target. Front. Oncol. 2015, 5, 158. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, X.; Kong, S.; Shang, S.; Qi, Y. CDK4/6 inhibition suppresses tumour growth and enhances the effect of temozolomide in glioma cells. J. Cell. Mol. Med. 2020, 24, 5135–5145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joel, M.; Mughal, A.A.; Grieg, Z.; Murrell, W.; Palmero, S.; Mikkelsen, B.; Fjerdingstad, H.B.; Sandberg, C.J.; Behnan, J.; Glover, J.C.; et al. Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo. Mol. Cancer 2015, 14, 121. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.W.; Lu, Q.; Wang, L.X.; Zhao, W.Y.; Cao, Y.Q.; Li, Y.N.; Han, G.S.; Liu, J.M.; Yue, Z.J. Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2A3 in GBM. BMC Cancer 2013, 13, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.K.; Zhu, J.; Chen, Y.H.; Wang, D.L.; Li, H.; Zhang, L.J.; Zhou, J.R.; Liu, W. Knockdown of Angiopoietin-Like Protein 2 Inhibits Proliferation and Invasion in Glioma Cells via Suppressing the ERK/MAPK Signaling Pathway. Oncol. Res. 2017, 25, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhan, H.Q.; Zhao, Y.Q.; Wu, Y.S.; Li, L.S.; Wang, H.P. MEX3A contributes to development and progression of glioma through regulating cell proliferation and cell migration and targeting CCL2. Cell Death Dis. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Song, H.W.; Fu, X.J.; Wu, C.X.; Li, S.W. Aging-related tumor associated fibroblasts changes could worsen the prognosis of GBM patients. Cancer Cell Int. 2020, 20, 1–19. [Google Scholar] [CrossRef]
- Du, J.; Yan, X.; Mi, S.; Li, Y.; Ji, H.; Hou, K.; Ma, S.; Ba, Y.; Zhou, P.; Chen, L.; et al. Identification of Prognostic Model and Biomarkers for Cancer Stem Cell Characteristics in Glioblastoma by Network Analysis of Multi-Omics Data and Stemness Indices. Front. Cell Dev. Biol. 2020, 8, 558961. [Google Scholar] [CrossRef]
- Li, Y.; Deng, G.; Qi, Y.; Zhang, H.; Gao, L.; Jiang, H.; Ye, Z.; Liu, B.; Chen, Q. Bioinformatic Profiling of Prognosis-Related Genes in Malignant Glioma Microenvironment. Med. Sci. Monit. 2020, 26, e924054. [Google Scholar] [CrossRef]
- Zheng, J.; Zhou, Z.; Qiu, Y.; Wang, M.; Yu, H.; Wu, Z.; Wang, X.; Jiang, X. A Pyroptosis-Related Gene Prognostic Index Correlated with Survival and Immune Microenvironment in Glioma. J. Inflamm. Res. 2022, 15, 17–32. [Google Scholar] [CrossRef]
- Xia, P.; Li, Q.; Wu, G.; Huang, Y. An Immune-Related lncRNA Signature to Predict Survival In Glioma Patients. Cell. Mol. Neurobiol. 2021, 41, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Liyanarachchi, S.; Davuluri, R.V.; Auer, H.; Martin, E.W., Jr.; de la Chapelle, A.; Frankel, W.L. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 2004, 23, 7366–7377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Xu, J.; Huang, N.; Tang, J.; Ma, P.; Cheng, Y. Clinical and Biological Significance of a Necroptosis-Related Gene Signature in Glioma. Front. Oncol. 2022, 12, 855434. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, W.; Xiao, Z.; Guan, G.; Liu, X.; Zhuang, M. A risk signature with four autophagy-related genes for predicting survival of glioblastoma multiforme. J. Cell. Mol. Med. 2020, 24, 3807–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Chang, C.Y.; Pan, P.H.; Wu, C.C.; Liao, S.L.; Chen, W.Y.; Kuan, Y.H.; Wang, W.Y.; Chen, C.J. Endoplasmic Reticulum Stress Contributes to Gefitinib-Induced Apoptosis in Glioma. Int. J. Mol. Sci. 2021, 22, 3934. [Google Scholar] [CrossRef]
- Au, K.; Singh, S.K.; Burrell, K.; Sabha, N.; Hawkins, C.; Huang, A.; Zadeh, G. A preclinical study demonstrating the efficacy of nilotinib in inhibiting the growth of pediatric high-grade glioma. J. Neurooncol. 2015, 122, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanashima, Y.; Sano, E.; Sumi, K.; Ozawa, Y.; Yagi, C.; Tatsuoka, J.; Yoshimura, S.; Yamamuro, S.; Ueda, T.; Nakayama, T.; et al. Antitumor effect of lenalidomide in malignant glioma cell lines. Oncol. Rep. 2020, 43, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Awada, G.; Ben Salama, L.; De Cremer, J.; Schwarze, J.K.; Fischbuch, L.; Seynaeve, L.; Du Four, S.; Vanbinst, A.M.; Michotte, A.; Everaert, H.; et al. Axitinib plus avelumab in the treatment of recurrent glioblastoma: A stratified, open-label, single-center phase 2 clinical trial (GliAvAx). J. Immunother. Cancer 2020, 8, 2. [Google Scholar] [CrossRef]
- Su, J.M.; Kilburn, L.B.; Mansur, D.B.; Krailo, M.; Buxton, A.; Adekunle, A.; Gajjar, A.; Adamson, P.C.; Weigel, B.; Fox, E.; et al. Phase 1/2 Trial of Vorinostat and Radiation and Maintenance Vorinostat in Children with Diffuse Intrinsic Pontine Glioma: A Children’s Oncology Group Report. Neurol. Oncol. 2022, 24, 655–664. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, A.; Kutalik, Z.; Valsesia, A. Copy Number Variation. Methods Mol. Biol. 2018, 1793, 231–258. [Google Scholar] [CrossRef]
- Buchwald, Z.S.; Tian, S.; Rossi, M.; Smith, G.H.; Switchenko, J.; Hauenstein, J.E.; Moreno, C.S.; Press, R.H.; Prabhu, R.S.; Zhong, J.; et al. Genomic copy number variation correlates with survival outcomes in WHO grade IV glioma. Sci. Rep. 2020, 10, 7355. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Training Set | Testing Set | Validation Set | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Total (n = 379) | High Risk (n = 182) | Low Risk (n = 197) | p-Value | Variable | Total (n = 162) | High Risk (n = 77) | Low Risk (n = 85) | p-Value | Variable | Total (n = 463) | High Risk (n = 229) | Low Risk (n = 234) | p-Value |
| Gender | Gender | Gender | ||||||||||||
| Female | 164 (43.3%) | 81 (44.5%) | 83 (42.1%) | 0.717 | Female | 67 (41.4%) | 31 (40.3%) | 36 (42.4%) | 0.912 | Female | 201 (43.4%) | 103 (45.0%) | 97 (42.4%) | 0.638 |
| Male | 215 (56.7%) | 101 (55.5%) | 114 (57.9%) | Male | 95 (58.6%) | 46 (59.7%) | 49 (57.6%) | Male | 262 (56.6%) | 126 (55.0%) | 132 (57.6%) | |||
| Age (years) | Age (years) | Age (years) | ||||||||||||
| ≥60 | 88 (23.2%) | 72 (39.6%) | 16 (8.1%) | <0.001 | ≥60 | 36 (22.2%) | 27 (35.1%) | 9 (10.6%) | <0.001 | ≥60 | 54 (11.7%) | 43 (18.8%) | 11 (4.8%) | <0.001 |
| <60 | 291 (76.8%) | 110 (60.4%) | 181 (91.9%) | <60 | 126 (77.8%) | 50 (64.9%) | 76 (89.4%) | <60 | 409 (88.3%) | 186 (81.2%) | 218 (95.2%) | |||
| IDH | IDH | IDH | ||||||||||||
| IDHmut-codel | 103 (27.2%) | 16 (8.8%) | 87 (44.2%) | <0.001 | IDHmut-codel | 43 (26.5%) | 3 (3.9%) | 40 (47.1%) | <0.001 | Mutant | 250 (54.0%) | 51 (22.3%) | 196 (85.6%) | <0.001 |
| IDHmut-non-codel | 156 (41.2%) | 47 (25.8%) | 109 (55.3%) | IDHmut-non-codel | 57 (35.2%) | 13 (16.9%) | 44 (51.8%) | Wildtype | 213 (46.0%) | 178 (77.7%) | 33 (14.4%) | |||
| IDHwt | 120 (31.7%) | 119 (65.4%) | 1 (0.5%) | IDHwt | 62 (38.3%) | 61 (79.2%) | 1 (1.2%) | Grade | ||||||
| Grade | Grade | WHO II | 110 (23.8%) | 22 (9.6%) | 86 (37.6%) | <0.001 | ||||||||
| G2 | 149 (39.3%) | 38 (20.9%) | 111 (56.3%) | <0.001 | G2 | 56 (34.6%) | 9 (11.7%) | 47 (55.3%) | <0.001 | WHO III | 176 (38.0%) | 60 (26.2%) | 114 (49.8%) | |
| G3 | 159 (42.0%) | 74 (40.7%) | 85 (43.1%) | G3 | 72 (44.4%) | 34 (44.2%) | 38 (44.7%) | WHO IV | 177 (38.2%) | 147 (64.2%) | 29 (12.7%) | |||
| G4 | 71 (18.7%) | 70 (38.5%) | 1 (0.5%) | G4 | 34 (21.0%) | 34 (44.2%) | 0 (0%) | MGMT | ||||||
| MGMT | MGMT | Methylated | 273 (59.0%) | 134 (58.5%) | 137 (59.8%) | 0.849 | ||||||||
| Methylated | 285 (75.2%) | 101 (55.5%) | 184 (93.4%) | <0.001 | Methylated | 118 (72.8%) | 36 (46.8%) | 82 (96.5%) | <0.001 | Unmethylated | 190 (41.0%) | 95 (41.5%) | 92 (40.2%) | |
| Unmethylated | 94 (24.8%) | 81 (44.5%) | 13 (6.6%) | Unmethylated | 44 (27.2%) | 41 (53.2%) | 3 (3.5%) | Histology | ||||||
| A | 53 (11.4%) | 13 (5.7%) | 39 (17.0%) | <0.001 | ||||||||||
| AA | 63 (13.6%) | 23 (10.0%) | 40 (17.5%) | |||||||||||
| AO | 31 (6.7%) | 4 (1.7%) | 27 (11.8%) | |||||||||||
| AOA | 4 (0.9%) | 3 (1.3%) | 1 (0.4%) | |||||||||||
| GBM | 108 (23.3%) | 87 (38.0%) | 21 (9.2%) | |||||||||||
| O | 29 (6.3%) | 0 (0%) | 29 (12.7%) | |||||||||||
| OA | 2 (0.4%) | 0 (0%) | 2 (0.9%) | |||||||||||
| rA | 16 (3.5%) | 5 (2.2%) | 10 (4.4%) | |||||||||||
| rAA | 55 (11.9%) | 26 (11.4%) | 28 (12.2%) | |||||||||||
| rAO | 23 (5.0%) | 4 (1.7%) | 18 (7.9%) | |||||||||||
| rGBM | 69 (14.9%) | 60 (26.2%) | 8 (3.5%) | |||||||||||
| rO | 10 (2.2%) | 4 (1.7%) | 6 (2.6%) | |||||||||||
| Radio_status | ||||||||||||||
| Radio_treated | 366 (79.0%) | 188 (82.1%) | 174 (76.0%) | 0.136 | ||||||||||
| Radio_untreated | 97 (21.0%) | 41 (17.9%) | 55 (24.0%) | |||||||||||
| Chemo_status | ||||||||||||||
| Chemo_treated | 358 (77.3%) | 187 (81.7%) | 169 (73.8%) | 0.0562 | ||||||||||
| Chemo_untreated | 105 (22.7%) | 42 (18.3%) | 60 (26.2%) | |||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Huang, Y.; Yang, E.; Gao, X.; Zou, P.; Sun, J.; Tian, Z.; Bao, M.; Liao, D.; Ge, J.; et al. Identification of a Fibroblast-Related Prognostic Model in Glioma Based on Bioinformatics Methods. Biomolecules 2022, 12, 1598. https://doi.org/10.3390/biom12111598
Zhang H, Huang Y, Yang E, Gao X, Zou P, Sun J, Tian Z, Bao M, Liao D, Ge J, et al. Identification of a Fibroblast-Related Prognostic Model in Glioma Based on Bioinformatics Methods. Biomolecules. 2022; 12(11):1598. https://doi.org/10.3390/biom12111598
Chicago/Turabian StyleZhang, Haofuzi, Yutao Huang, Erwan Yang, Xiangyu Gao, Peng Zou, Jidong Sun, Zhicheng Tian, Mingdong Bao, Dan Liao, Junmiao Ge, and et al. 2022. "Identification of a Fibroblast-Related Prognostic Model in Glioma Based on Bioinformatics Methods" Biomolecules 12, no. 11: 1598. https://doi.org/10.3390/biom12111598