
The Potential Role of Polyamines in Epilepsy and Epilepsy-Related Pathophysiological Changes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
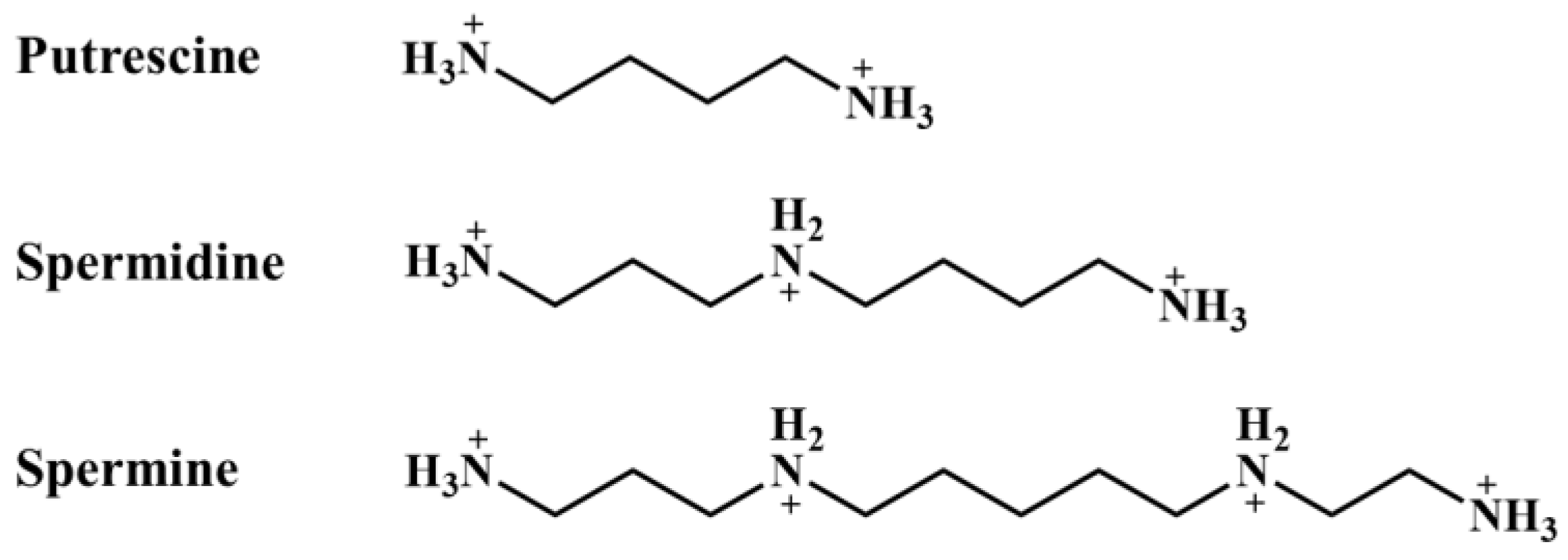
2. Polyamines
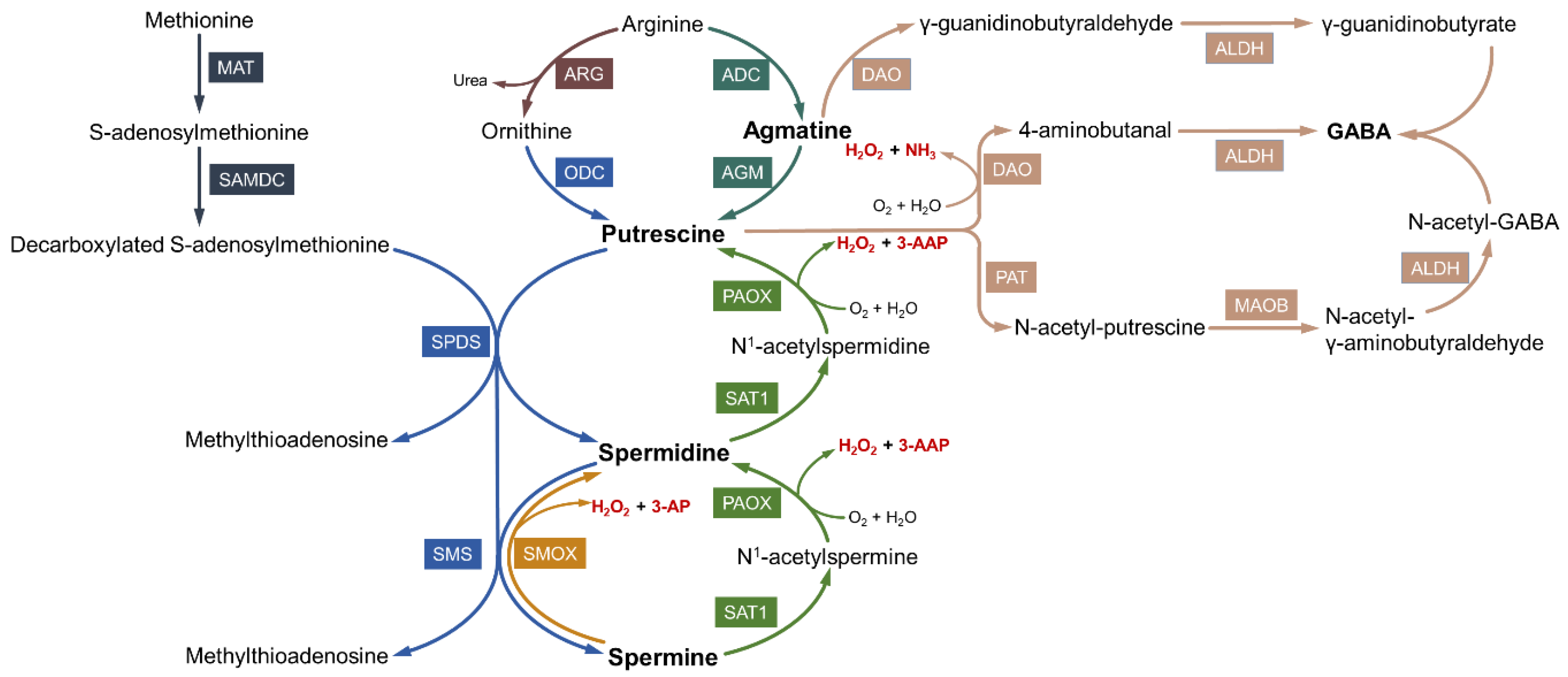
2.1. Biosynthesis of Polyamines
2.2. Catabolism and Interconversion of Polyamines
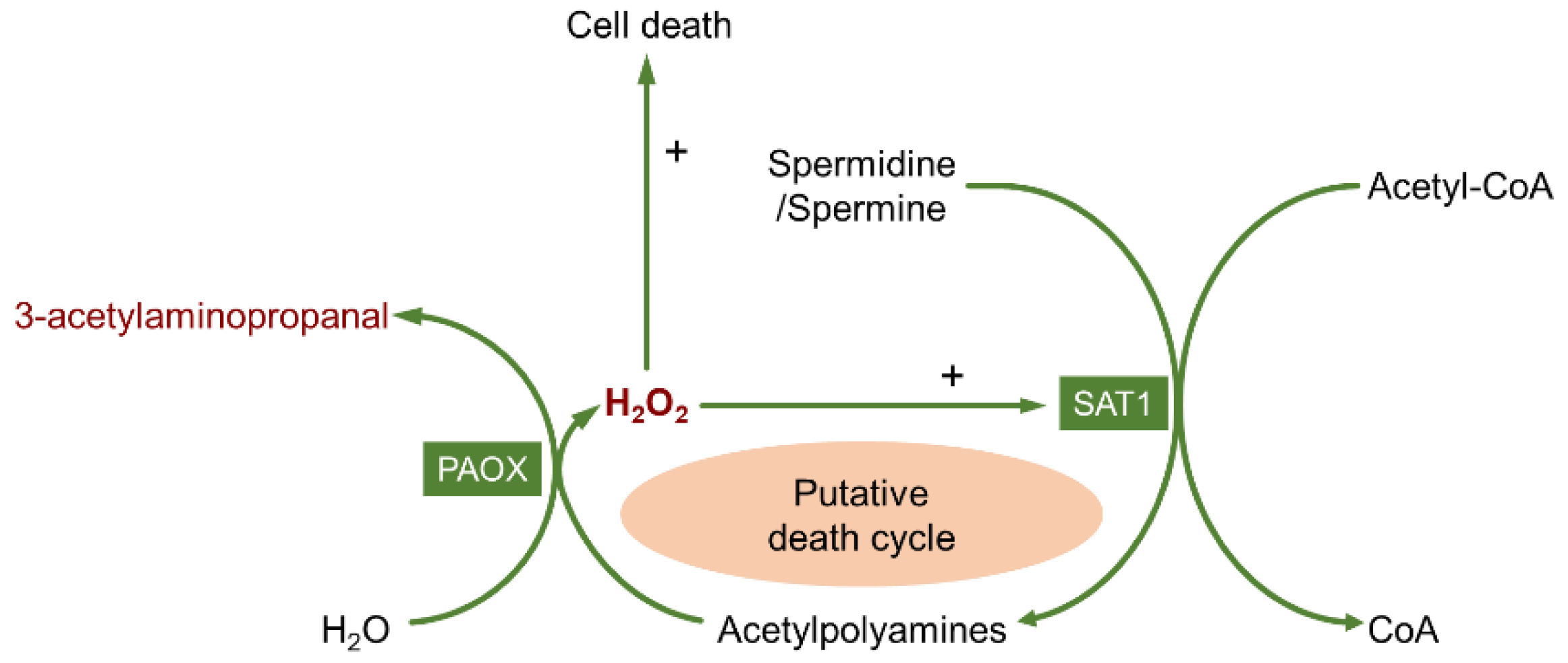
2.3. Toxic Products of Polyamine Oxidation
3. Alteration of the Polyamine System in Epilepsy
3.1. Alteration of Polyamine Metabolism
3.2. Heterogeneity of Polyamine Distribution and Changes in Polyamine Homeostasis
4. Effects of Polyamines on Epileptic Activity: Inhibiting or Promoting?
4.1. The Influence of the Polyamine System on Seizure Susceptibility
4.2. Effects of Polyamines on Epilepsy Progression
5. Polyamines and Pathological Changes in Epilepsy
5.1. The Role of Polyamines in Regulating Neuronal Excitability
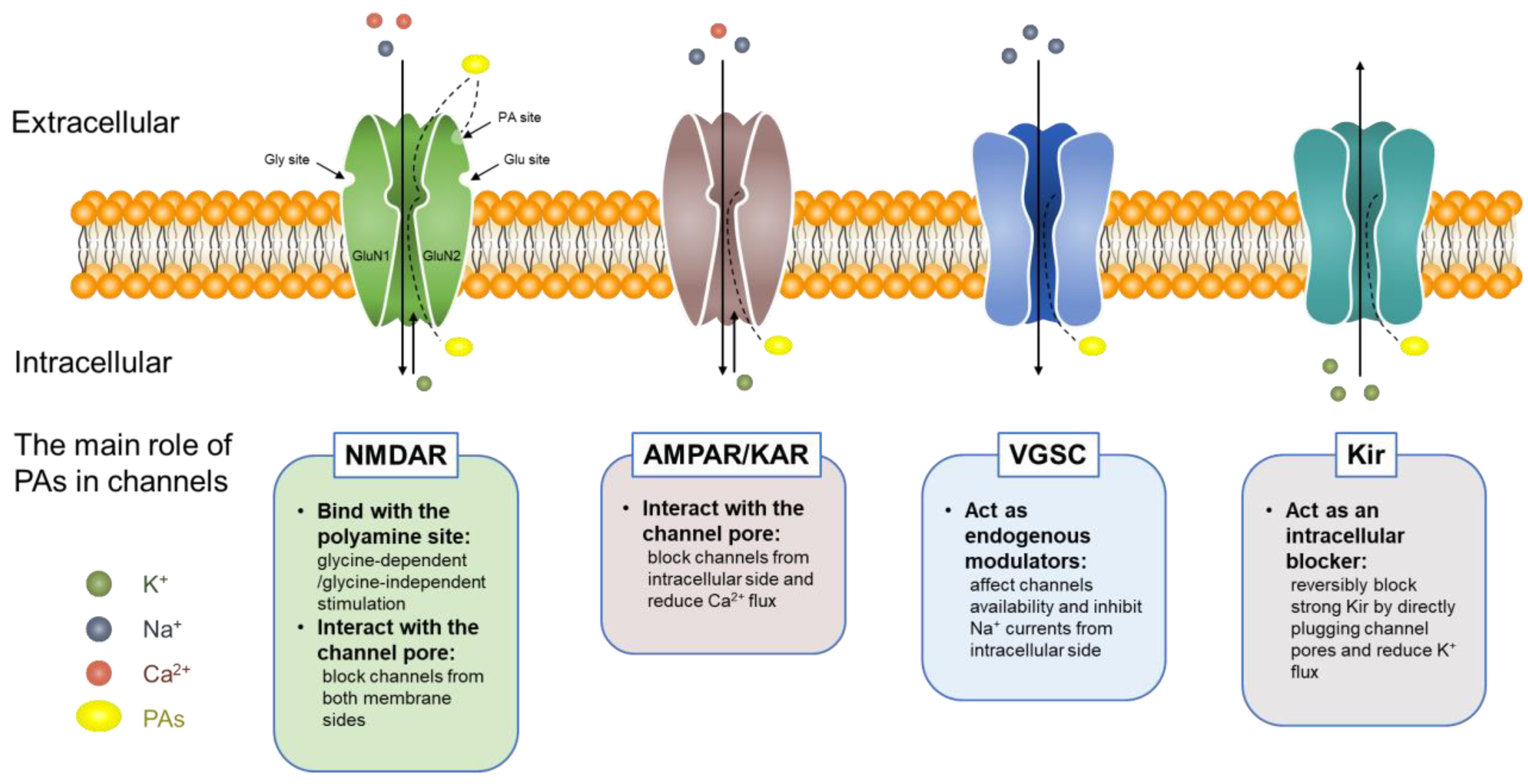
5.1.1. Polyamines and Ion Channels
5.1.2. Polyamines and Astrocytic Transmission
5.2. The Potential Role of Polyamines in Damage and Pathological Changes after Seizures
5.2.1. Hypoperfusion/Hypoxia Following Seizures
5.2.2. Gliosis
5.2.3. Neuroinflammation
5.2.4. Synaptic Plasticity
5.2.5. Cell Damage and Apoptosis
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bae, D.H.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. The old and new biochemistry of polyamines. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. The functional role of polyamines in eukaryotic cells. Int. J. Biochem. Cell Biol. 2019, 107, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Moinard, C.; Cynober, L.; de Bandt, J.P. Polyamines: Metabolism and implications in human diseases. Clin. Nutr. 2005, 24, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Doyle, K.M.; Tatlisumak, T. Polyamines in the brain: Distribution, biological interactions, and their potential therapeutic role in brain ischaemia. Curr. Med. Chem. 2007, 14, 1807–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamwal, S.; Singh, S.; Kaur, N.; Kumar, P. Protective Effect of Spermidine Against Excitotoxic Neuronal Death Induced by Quinolinic Acid in Rats: Possible Neurotransmitters and Neuroinflammatory Mechanism. Neurotox. Res. 2015, 28, 171–184. [Google Scholar] [CrossRef]
- Jamwal, S.; Kumar, P. Spermidine ameliorates 3-nitropropionic acid (3-NP)-induced striatal toxicity: Possible role of oxidative stress, neuroinflammation, and neurotransmitters. Physiol. Behav. 2016, 155, 180–187. [Google Scholar] [CrossRef]
- Laschet, J.; Trottier, S.; Leviel, V.; Guibert, B.; Bansard, J.Y.; Chauvel, P.; Bureau, M. Heterogeneous distribution of polyamines in temporal lobe epilepsy. Epilepsy Res. 1999, 35, 161–172. [Google Scholar] [CrossRef]
- de Vera, N.; Camon, L.; Martinez, E. Cerebral distribution of polyamines in kainic acid-induced models of status epilepticus and ataxia in rats. Overproduction of putrescine and histological damage. Eur. Neuropsychopharmacol. 2002, 12, 397–405. [Google Scholar] [CrossRef]
- Zahedi, K.; Huttinger, F.; Morrison, R.; Murray-Stewart, T.; Casero, R.A.; Strauss, K.I. Polyamine catabolism is enhanced after traumatic brain injury. J. Neurotrauma 2010, 27, 515–525. [Google Scholar] [CrossRef]
- Mahajan, U.V.; Varma, V.R.; Griswold, M.E.; Blackshear, C.T.; An, Y.; Oommen, A.M.; Varma, S.; Troncoso, J.C.; Pletnikova, O.; O’Brien, R.; et al. Dysregulation of multiple metabolic networks related to brain transmethylation and polyamine pathways in Alzheimer disease: A targeted metabolomic and transcriptomic study. PLoS Med. 2020, 17, e1003012. [Google Scholar]
- Morrison, L.D.; Kish, S.J. Brain polyamine levels are altered in Alzheimer’s disease. Neurosci. Lett. 1995, 197, 5–8. [Google Scholar] [CrossRef]
- Baskaya, M.K.; Rao, A.M.; Dogan, A.; Donaldson, D.; Gellin, G.; Dempsey, R.J. Regional brain polyamine levels in permanent focal cerebral ischemia. Brain Res. 1997, 744, 302–308. [Google Scholar] [CrossRef]
- de Vera, N.; Artigas, F.; Serratosa, J.; Martinez, E. Changes in polyamine levels in rat brain after systemic kainic acid administration: Relationship to convulsant activity and brain damage. J. Neurochem. 1991, 57, 1–8. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Avoli, M.; Louvel, J.; Pumain, R.; Kohling, R. Cellular and molecular mechanisms of epilepsy in the human brain. Prog. Neurobiol. 2005, 77, 166–200. [Google Scholar] [CrossRef]
- Pitkanen, A.; Lukasiuk, K. Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 2009, 14 (Suppl. 1), 16–25. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; O’Brien, T.J.; Jette, N.; Scheffer, I.E.; de Curtis, M.; Perucca, P. Epilepsy. Nat. Rev. Dis. Prim. 2018, 4, 18024. [Google Scholar] [CrossRef]
- Hitiris, N.; Mohanraj, R.; Norrie, J.; Brodie, M.J. Mortality in epilepsy. Epilepsy Behav. 2007, 10, 363–376. [Google Scholar] [CrossRef]
- DeGiorgio, C.M.; Curtis, A.; Hertling, D.; Moseley, B.D. Sudden unexpected death in epilepsy: Risk factors, biomarkers, and prevention. Acta Neurol. Scand. 2019, 139, 220–230. [Google Scholar] [CrossRef]
- Tang, F.; Hartz, A.M.S.; Bauer, B. Drug-Resistant Epilepsy: Multiple Hypotheses, Few Answers. Front. Neurol. 2017, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef]
- Halaris, A.; Plietz, J. Agmatine: Metabolic pathway and spectrum of activity in brain. CNS Drugs 2007, 21, 885–900. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Ealick, S.E. Structural biology of S-adenosylmethionine decarboxylase. Amino Acids 2010, 38, 451–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.; Wu, Q.; Li, J.; Sun, S.; Sun, S. S-adenosylmethionine: A metabolite critical to the regulation of autophagy. Cell Prolif. 2020, 53, e12891. [Google Scholar] [CrossRef]
- Mistry, S.K.; Burwell, T.J.; Chambers, R.M.; Rudolph-Owen, L.; Spaltmann, F.; Cook, W.J.; Morris, S.M., Jr. Cloning of human agmatinase. An alternate path for polyamine synthesis induced in liver by hepatitis B virus. Am. J. Physiol. Gastrointest Liver Physiol. 2002, 282, G375–G381. [Google Scholar] [CrossRef]
- Tabor, C.W.; Tabor, H. Polyamines. Annu. Rev. Biochem. 1984, 53, 749–790. [Google Scholar] [CrossRef]
- Moretti, M.; Matheus, F.C.; de Oliveira, P.A.; Neis, V.B.; Ben, J.; Walz, R.; Rodrigues, A.L.; Prediger, R.D. Role of agmatine in neurodegenerative diseases and epilepsy. Front. Biosci.-Elite 2014, 6, 341–359. [Google Scholar] [CrossRef]
- Benitez, J.; Garcia, D.; Romero, N.; Gonzalez, A.; Martinez-Oyanedel, J.; Figueroa, M.; Salas, M.; Lopez, V.; Garcia-Robles, M.; Dodd, P.R.; et al. Metabolic strategies for the degradation of the neuromodulator agmatine in mammals. Metabolism 2018, 81, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Casero, R.A., Jr.; Pegg, A.E. Spermidine/spermine N1-acetyltransferase—The turning point in polyamine metabolism. FASEB J. 1993, 7, 653–661. [Google Scholar] [CrossRef]
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E995–E1010. [Google Scholar] [CrossRef]
- Seiler, N. Chapter 33 Polyamine oxidase, properties and functions. Prog. Brain Res. 1995, 106, 333–344. [Google Scholar] [CrossRef]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [Green Version]
- Agostinelli, E.; Arancia, G.; Vedova, L.D.; Belli, F.; Marra, M.; Salvi, M.; Toninello, A. The biological functions of polyamine oxidation products by amine oxidases: Perspectives of clinical applications. Amino Acids 2004, 27, 347–358. [Google Scholar] [CrossRef]
- Seiler, N.; Bink, G.; Grove, J. Regulatory interrelations between GABA and polyamines. I. Brain GABA levels and polyamine metabolism. Neurochem. Res. 1979, 4, 425–435. [Google Scholar] [CrossRef]
- Seiler, N. Catabolism of polyamines. Amino Acids 2004, 26, 217–233. [Google Scholar] [CrossRef]
- Chopra, S.; Wallace, H.M. Induction of spermidine/spermine N1-acetyltransferase in human cancer cells in response to increased production of reactive oxygen species. Biochem. Pharm. 1998, 55, 1119–1123. [Google Scholar] [CrossRef]
- Wallace, H.M.; Fraser, A.V.; Hughes, A. A perspective of polyamine metabolism. Biochem. J 2003, 376, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.L.; Khan, M.A.; Moskal, J.R. The concept of “aldehyde load” in neurodegenerative mechanisms: Cytotoxicity of the polyamine degradation products hydrogen peroxide, acrolein, 3-aminopropanal, 3-acetamidopropanal and 4-aminobutanal in a retinal ganglion cell line. Brain Res. 2007, 1145, 150–156. [Google Scholar] [CrossRef]
- Li, W.; Yuan, X.M.; Ivanova, S.; Tracey, K.J.; Eaton, J.W.; Brunk, U.T. 3-Aminopropanal, formed during cerebral ischaemia, is a potent lysosomotropic neurotoxin. Biochem. J. 2003, 371, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Li, W.; Brunk, U.T. 3-Aminopropanal is a lysosomotropic aldehyde that causes oxidative stress and apoptosis by rupturing lysosomes. Apmis 2003, 111, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Uemura, T.; Kashiwagi, K. Acrolein toxicity at advanced age: Present and future. Amino Acids 2018, 50, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Protein-conjugated acrolein as a biochemical marker of brain infarction. Mol. Nutr. Food Res. 2011, 55, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, S.; Sakata, K.; Kashiwagi, K.; Ueda, S.; Iwasaki, S.; Shirahata, A.; Igarashi, K. Polyamine cytotoxicity in the presence of bovine serum amine oxidase. Biochem. Biophys. Res. Commun. 2001, 282, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, M.; Tomitori, H.; Machi, Y.; Hagihara, M.; Higashi, K.; Goda, H.; Ohya, T.; Niitsu, M.; Kashiwagi, K.; Igarashi, K. Acrolein toxicity: Comparison with reactive oxygen species. Biochem. Biophys. Res. Commun. 2009, 378, 313–318. [Google Scholar] [CrossRef]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Uchida, K.; Kanematsu, M.; Morimitsu, Y.; Osawa, T.; Noguchi, N.; Niki, E. Acrolein is a product of lipid peroxidation reaction. Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998, 273, 16058–16066. [Google Scholar] [CrossRef] [Green Version]
- Furuhata, A.; Ishii, T.; Kumazawa, S.; Yamada, T.; Nakayama, T.; Uchida, K. N(epsilon)-(3-methylpyridinium)lysine, a major antigenic adduct generated in acrolein-modified protein. J. Biol. Chem. 2003, 278, 48658–48665. [Google Scholar] [CrossRef] [Green Version]
- Tanel, A.; Averill-Bates, D.A. The aldehyde acrolein induces apoptosis via activation of the mitochondrial pathway. Biochim. Biophys. Acta 2005, 1743, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Lovell, M.A.; Xie, C.S.; Markesbery, W.R. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging 2001, 22, 187–194. [Google Scholar] [CrossRef]
- Uemura, T.; Nakamura, M.; Sakamoto, A.; Suzuki, T.; Dohmae, N.; Terui, Y.; Tomitori, H.; Casero, R.A., Jr.; Kashiwagi, K.; Igarashi, K. Decrease in acrolein toxicity based on the decline of polyamine oxidases. Int. J. Biochem. Cell Biol. 2016, 79, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Wang, T.W.; Lin, Y.Y.; Ho, W.C.; Tsai, H.C.; Chen, S.P.; Lin, A.M.; Liu, T.Y.; Wang, H.T. Acrolein is involved in ischemic stroke-induced neurotoxicity through spermidine/spermine-N1-acetyltransferase activation. Exp. Neurol. 2020, 323, 113066. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Modulation of cellular function by polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Igarashi, K. Polyamines and their metabolites as diagnostic markers of human diseases. Biomol. Ther. 2013, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y. Effects of intra-amygdaloid injections of alpha-Difluoromethylornithine and putrescine on the development of electrical kindling in rats. Brain Res. 1991, 560, 181–185. [Google Scholar] [CrossRef]
- Hayashi, Y.; Hattori, Y.; Hori, Y. Involvement of putrescine in the development of kindled seizure in rats. J. Neurochem. 1992, 58, 562–566. [Google Scholar] [CrossRef]
- Vivo, M.; Camon, L.; de Vera, N.; Martinez, E. Extracellular putrescine content after acute excitotoxic brain damage in the rat. Neurosci. Lett. 2002, 330, 74–78. [Google Scholar] [CrossRef]
- Pajunen, A.E.; Hietala, O.A.; Baruch-Virransalo, E.L.; Piha, R.S. The effect of DL-allylglycine on polyamine and GABA metabolism in mouse brain. J. Neurochem. 1979, 32, 1401–1408. [Google Scholar] [CrossRef]
- Royeck, M.; Kelly, T.; Opitz, T.; Otte, D.M.; Rennhack, A.; Woitecki, A.; Pitsch, J.; Becker, A.; Schoch, S.; Kaupp, U.B.; et al. Downregulation of Spermine Augments Dendritic Persistent Sodium Currents and Synaptic Integration after Status Epilepticus. J. Neurosci. 2015, 35, 15240–15253. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.M.; Hatcher, J.F.; Dogan, A.; Dempsey, R.J. Elevated N-1-acetylspermidine levels in gerbil and rat brains after CNS injury. J. Neurochem. 2000, 74, 1106–1111. [Google Scholar] [CrossRef]
- Henley, C.M.; Wey, K.; Takashima, A.; Mills, C.; Granmayeh, E.; Krishnappa, I.; Robertson, C.S. S-adenosylmethionine decarboxylase activity is decreased in the rat cortex after traumatic brain injury. J. Neurochem. 1997, 69, 259–265. [Google Scholar] [CrossRef]
- Sadeghi, L.; Rizvanov, A.A.; Salafutdinov, I.I.; Dabirmanesh, B.; Sayyah, M.; Fathollahi, Y.; Khajeh, K. Hippocampal asymmetry: Differences in the left and right hippocampus proteome in the rat model of temporal lobe epilepsy. J. Proteom. 2017, 154, 22–29. [Google Scholar] [CrossRef]
- Najm, I.; el-Skaf, G.; Massicotte, G.; Vanderklish, P.; Lynch, G.; Baudry, M. Changes in polyamine levels and spectrin degradation following kainate-induced seizure activity: Effect of difluoromethylornithine. Exp. Neurol. 1992, 116, 345–354. [Google Scholar] [CrossRef]
- Paschen, W. Polyamine metabolism in different pathological states of the brain. Mol. Chem. Neuropathol. 1992, 16, 241–271. [Google Scholar] [CrossRef]
- Zawia, N.H.; Bondy, S.C. Electrically stimulated rapid gene expression in the brain: Ornithine decarboxylase and c-fos. Brain Res. Mol. Brain Res. 1990, 7, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, H.G.; Muller, M. The cellular localization of the L-ornithine decarboxylase/polyamine system in normal and diseased central nervous systems. Prog. Neurobiol. 1999, 57, 485–505. [Google Scholar] [CrossRef]
- Baudry, M.; Najm, I. Kainate-Induced seizure activity stimulates the polyamine interconversion pathway in rat brain. Neurosci. Lett. 1994, 171, 151–154. [Google Scholar] [CrossRef]
- Hayashi, Y.; Morizumi, Y.; Hattori, Y.; Tanaka, J. Pentylenetetrazol-induced kindling stimulates the polyamine interconversion pathway in rat brain. Brain Res. 1999, 828, 184–188. [Google Scholar] [CrossRef]
- Dogan, A.; Rao, A.M.; Hatcher, J.; Rao, V.L.; Baskaya, M.K.; Dempsey, R.J. Effects of MDL 72527, a specific inhibitor of polyamine oxidase, on brain edema, ischemic injury volume, and tissue polyamine levels in rats after temporary middle cerebral artery occlusion. J. Neurochem. 1999, 72, 765–770. [Google Scholar] [CrossRef]
- Paschen, W.; Rohn, G.; Meese, C.O.; Djuricic, B.; Schmidt-Kastner, R. Polyamine metabolism in reversible cerebral ischemia: Effect of alpha-difluoromethylornithine. Brain Res. 1988, 453, 9–16. [Google Scholar] [CrossRef]
- Seiler, N. Oxidation of polyamines and brain injury. Neurochem. Res. 2000, 25, 471–490. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Cruz, N.F.; Rosenfeld, S.J. Temporal profiles of proteins responsive to transient ischemia. J. Neurochem. 1985, 44, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Röhn, G.; Schlenker, M.; Paschen, W. Activity of ornithine decarboxylase and S-adenosylmethionine decarboxylase in transient cerebral ischemia: Relationship to the duration of vascular occulusion. Exp. Neurol. 1992, 117, 210–215. [Google Scholar] [CrossRef]
- Pajunen, A.E.; Hietala, O.A.; Virransalo, E.L.; Piha, R.S. Ornithine decarboxylase and adenosylmethionine decarboxylase in mouse brain--effect of electrical stimulation. J. Neurochem. 1978, 30, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Sherwin, A.L.; Carmant, L.; Kish, S.J. Activity of S-adenosylmethionine decarboxylase, a key regulatory enzyme in polyamine biosynthesis, is increased in epileptogenic human cortex. Arch. Neurol. 1994, 51, 581–584. [Google Scholar] [CrossRef]
- Beckonert, N.M.; Opitz, T.; Pitsch, J.; Soares da Silva, P.; Beck, H. Polyamine Modulation of Anticonvulsant Drug Response: A Potential Mechanism Contributing to Pharmacoresistance in Chronic Epilepsy. J. Neurosci. 2018, 38, 5596–5605. [Google Scholar] [CrossRef] [Green Version]
- Morrison, L.D.; Becker, L.; Ang, L.C.; Kish, S.J. Polyamines in human brain: Regional distribution and influence of aging. J. Neurochem. 1995, 65, 636–642. [Google Scholar] [CrossRef]
- Seiler, N.; Schmidt-Glenewinkel, T. Regional distribution of putrescine, spermidine and spermine in relation to the distribution of RNA and DNA in the rat nervous system. J. Neurochem. 1975, 24, 791–795. [Google Scholar] [CrossRef]
- Laschet, J.; Trottier, S.; Grisar, T.; Leviel, V. Polyamine metabolism in epileptic cortex. Epilepsy Res. 1992, 12, 151–156. [Google Scholar] [CrossRef]
- Bondy, S.C.; Mitchell, C.L.; Rahmaan, S.; Mason, G. Regional variation in the response of cerebral ornithine decarboxylase to electroconvulsive shock. Neurochem. Pathol. 1987, 7, 129–141. [Google Scholar] [CrossRef]
- Halonen, T.; Sivenius, J.; Miettinen, R.; Halmekyto, M.; Kauppinen, R.; Sinervirta, R.; Alakuijala, L.; Alhonen, L.; MacDonald, E.; Janne, J.; et al. Elevated seizure threshold and impaired spatial learning in transgenic mice with putrescine overproduction in the brain. Eur. J. Neurosci. 1993, 5, 1233–1239. [Google Scholar] [CrossRef]
- Lukkarinen, J.A.; Kauppinen, R.A.; Grohn, O.H.; Oja, J.M.; Sinervirta, R.; Jarvinen, A.; Alhonen, L.I.; Janne, J. Neuroprotective role of ornithine decarboxylase activation in transient focal cerebral ischaemia: A study using ornithine decarboxylase-overexpressing transgenic rats. Eur. J. Neurosci. 1998, 10, 2046–2055. [Google Scholar] [CrossRef]
- Gerrish, K.E.; Fuller, D.J.; Gerner, E.W.; Gensler, H.L. Inhibition of DFMO-induced audiogenic seizures by chlordiazepoxide. Life Sci. 1993, 52, 1101–1108. [Google Scholar] [CrossRef]
- Kaasinen, K.; Koistinaho, J.; Alhonen, L.; Janne, J. Overexpression of spermidine/spermine N-acetyltransferase in transgenic mice protects the animals from kainate-induced toxicity. Eur. J. Neurosci. 2000, 12, 540–548. [Google Scholar] [CrossRef]
- Kaasinen, S.K.; Grohn, O.H.; Keinanen, T.A.; Alhonen, L.; Janne, J. Overexpression of spermidine/spermine N1-acetyltransferase elevates the threshold to pentylenetetrazol-induced seizure activity in transgenic mice. Exp. Neurol. 2003, 183, 645–652. [Google Scholar] [CrossRef]
- Leonetti, A.; Baroli, G.; Fratini, E.; Pietropaoli, S.; Marcoli, M.; Mariottini, P.; Cervelli, M. Epileptic seizures and oxidative stress in a mouse model over-Expressing spermine oxidase. Amino Acids 2020, 52, 129–139. [Google Scholar] [CrossRef]
- Cervelli, M.; Bellavia, G.; D’Amelio, M.; Cavallucci, V.; Moreno, S.; Berger, J.; Nardacci, R.; Marcoli, M.; Maura, G.; Piacentini, M.; et al. A New Transgenic Mouse Model for Studying the Neurotoxicity of Spermine Oxidase Dosage in the Response to Excitotoxic Injury. PLoS ONE 2013, 8, e64810. [Google Scholar] [CrossRef]
- Pietropaoli, S.; Leonetti, A.; Cervetto, C.; Venturini, A.; Mastrantonio, R.; Baroli, G.; Persichini, T.; Colasanti, M.; Maura, G.; Marcoli, M.; et al. Glutamate Excitotoxicity Linked to Spermine Oxidase Overexpression. Mol. Neurobiol. 2018, 55, 7259–7270. [Google Scholar] [CrossRef]
- Cervetto, C.; Vergani, L.; Passalacqua, M.; Ragazzoni, M.; Venturini, A.; Cecconi, F.; Berretta, N.; Mercuri, N.; D’Amelio, M.; Maura, G.; et al. Astrocyte-Dependent Vulnerability to Excitotoxicity in Spermine Oxidase-Overexpressing Mouse. Neuromolecular Med. 2016, 18, 50–68. [Google Scholar] [CrossRef]
- Lukkarainen, J.; Kauppinen, R.A.; Koistinaho, J.; Halmekyto, M.; Alhonen, L.; Janne, J. Cerebral energy metabolism and immediate early gene induction following severe incomplete ischaemia in transgenic mice overexpressing the human ornithine decarboxylase gene: Evidence that putrescine is not neurotoxic in vivo. Eur. J. Neurosci. 1995, 7, 1840–1849. [Google Scholar] [CrossRef]
- Bell, M.R.; Belarde, J.A.; Johnson, H.F.; Aizenman, C.D. A neuroprotective role for polyamines in a Xenopus tadpole model of epilepsy. Nat. Neurosci. 2011, 14, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Kapfhamer, D.; McKenna, J.; Yoon, C.J.; Murray-Stewart, T.; Casero, R.A.; Gambello, M.J. Ornithine decarboxylase, the rate-limiting enzyme of polyamine synthesis, modifies brain pathology in a mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2020, 29, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- de Vera, N.; Serratosa, J.; Artigas, F.; Martinez, E. Toxic effects of putrescine in rat brain: Polyamines can be involved in the action of excitotoxins. Amino Acids 1992, 3, 261–269. [Google Scholar] [CrossRef] [PubMed]
- De Sarro, G.B.; Bagetta, G.; Spagnolo, C.; Nistico, G. Antagonists of N-methyl-D-Aspartate receptors block seizures induced by putrescine in the deep prepiriform cortex. Neuropharmacology 1993, 32, 43–50. [Google Scholar] [CrossRef]
- Anderson, D.J.; Crossland, J.; Shaw, G.G. The actions of spermidine and spermine on the central nervous system. Neuropharmacology 1975, 14, 571–577. [Google Scholar] [CrossRef]
- Ran, I.; Miura, R.M.; Puil, E. Spermine modulates neuronal excitability and NMDA receptors in juvenile gerbil auditory thalamus. Hear. Res. 2003, 176, 65–79. [Google Scholar] [CrossRef]
- Lerche, H.; Shah, M.; Beck, H.; Noebels, J.; Johnston, D.; Vincent, A. Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 2013, 591, 753–764. [Google Scholar] [CrossRef]
- Ren, E.; Curia, G. Synaptic Reshaping and Neuronal Outcomes in the Temporal Lobe Epilepsy. Int J. Mol. Sci 2021, 22, 3860. [Google Scholar] [CrossRef]
- Johnson, T.D. Modulation of channel function by polyamines. Trends Pharm. Sci 1996, 17, 22–27. [Google Scholar] [CrossRef]
- Williams, K. Interactions of polyamines with ion channels. Biochem. J. 1997, 325 Pt 2, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Limon, A.; Mamdani, F.; Hjelm, B.E.; Vawter, M.P.; Sequeira, A. Targets of polyamine dysregulation in major depression and suicide: Activity-dependent feedback, excitability, and neurotransmission. Neurosci. Biobehav. Rev. 2016, 66, 80–91. [Google Scholar] [CrossRef]
- Huang, C.J.; Moczydlowski, E. Cytoplasmic polyamines as permeant blockers and modulators of the voltage-Gated sodium channel. Biophys. J. 2001, 80, 1262–1279. [Google Scholar] [CrossRef] [Green Version]
- Fleidervish, I.A.; Libman, L.; Katz, E.; Gutnick, M.J. Endogenous polyamines regulate cortical neuronal excitability by blocking voltage-Gated Na+ channels. Proc. Natl. Acad. Sci. USA 2008, 105, 18994–18999. [Google Scholar] [CrossRef] [Green Version]
- Crill, W.E. Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 1996, 58, 349–362. [Google Scholar] [CrossRef]
- Cummins, T.R.; Xia, Y.; Haddad, G.G. Functional properties of rat and human neocortical voltage-sensitive sodium currents. J. Neurophysiol. 1994, 71, 1052–1064. [Google Scholar] [CrossRef]
- Fleidervish, I.A.; Gutnick, M.J. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. J. Neurophysiol. 1996, 76, 2125–2130. [Google Scholar] [CrossRef]
- Enomoto, A.; Han, J.M.; Hsiao, C.F.; Wu, N.; Chandler, S.H. Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J. Neurosci. 2006, 26, 3412–3422. [Google Scholar] [CrossRef] [Green Version]
- Del Negro, C.A.; Koshiya, N.; Butera, R.J., Jr.; Smith, J.C. Persistent sodium current, membrane properties and bursting behavior of pre-botzinger complex inspiratory neurons in vitro. J. Neurophysiol. 2002, 88, 2242–2250. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.; Draguhn, A.; Egorov, A.V. Persistent sodium current modulates axonal excitability in CA1 pyramidal neurons. J. Neurochem. 2018, 146, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Enomoto, A.; Tanaka, S.; Hsiao, C.F.; Nykamp, D.Q.; Izhikevich, E.; Chandler, S.H. Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J. Neurophysiol. 2005, 93, 2710–2722. [Google Scholar] [CrossRef] [Green Version]
- Stuart, G. Voltage-activated sodium channels amplify inhibition in neocortical pyramidal neurons. Nat. Neurosci. 1999, 2, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Stuart, G.; Sakmann, B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron 1995, 15, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.Y.; Zefirov, A.V.; Amakhin, D.V.; Chizhov, A.V. Effect of Persistent Sodium Current on Neuronal Activity. In Advances in Neural Computation, Machine Learning, and Cognitive Research; Springer: Berlin/Heidelberg, Germany, 2018; pp. 193–199. [Google Scholar]
- Hargus, N.J.; Nigam, A.; Bertram, E.H., 3rd; Patel, M.K. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J. Neurophysiol. 2013, 110, 1144–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Xu, X.; Cai, J.; Hu, H.; Sun, W.; He, G.; Shao, D.; Wang, L.; Chen, T.; Shaw, C.; et al. The up-regulation of voltage-gated sodium channels subtypes coincides with an increased sodium current in hippocampal neuronal culture model. Neurochem. Int. 2013, 62, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Su, H.; Yue, C.; Remy, S.; Royeck, M.; Sochivko, D.; Opitz, T.; Beck, H.; Yaari, Y. An increase in persistent sodium current contributes to intrinsic neuronal bursting after status epilepticus. J. Neurophysiol. 2011, 105, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Alonso, A.; Ragsdale, D.S. Increased persistent sodium currents in rat entorhinal cortex layer V neurons in a post-status epilepticus model of temporal lobe epilepsy. Epilepsia 2003, 44, 1601–1604. [Google Scholar] [CrossRef]
- Thompson, C.H.; Hawkins, N.A.; Kearney, J.A.; George, A.L., Jr. CaMKII modulates sodium current in neurons from epileptic Scn2a mutant mice. Proc. Natl. Acad. Sci. USA 2017, 114, 1696–1701. [Google Scholar] [CrossRef] [Green Version]
- Anderson, L.L.; Thompson, C.H.; Hawkins, N.A.; Nath, R.D.; Petersohn, A.A.; Rajamani, S.; Bush, W.S.; Frankel, W.N.; Vanoye, C.G.; Kearney, J.A.; et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 2014, 55, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.M.; Thompson, C.H.; Hawkins, N.A.; Wagnon, J.L.; Wengert, E.R.; Patel, M.K.; George, A.L., Jr.; Meisler, M.H.; Kearney, J.A. The novel sodium channel modulator GS-458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 2018, 59, 1166–1176. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Vegh, V.; Reutens, D.C. Persistent sodium current blockers can suppress seizures caused by loss of low-threshold D-type potassium currents: Predictions from an in silico study of Kv1 channel disorders. Epilepsia Open 2020, 5, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Hargus, N.J.; Merrick, E.C.; Nigam, A.; Kalmar, C.L.; Baheti, A.R.; Bertram, E.H., 3rd; Patel, M.K. Temporal lobe epilepsy induces intrinsic alterations in Na channel gating in layer II medial entorhinal cortex neurons. Neurobiol. Dis. 2011, 41, 361–376. [Google Scholar] [CrossRef]
- Derera, I.D.; Dulla, C.G. THAR SHE BLOWS! The Search for the Great Spermine Whale of Carbamazepine Resistance. Epilepsy Curr. 2019, 19, 59–61. [Google Scholar] [CrossRef] [Green Version]
- Akyuz, E.; Polat, A.K.; Eroglu, E.; Kullu, I.; Angelopoulou, E.; Paudel, Y.N. Revisiting the role of neurotransmitters in epilepsy: An updated review. Life Sci. 2021, 265, 118826. [Google Scholar] [CrossRef]
- Malpica-Nieves, C.J.; Rivera-Aponte, D.E.; Tejeda-Bayron, F.A.; Mayor, A.M.; Phanstiel, O.; Veh, R.W.; Eaton, M.J.; Skatchkov, S.N. The involvement of polyamine uptake and synthesis pathways in the proliferation of neonatal astrocytes. Amino Acids 2020, 52, 1169–1180. [Google Scholar] [CrossRef]
- Dot, J.; Danchev, N.; Blanco, I.; Rodriguez-Alvarez, J. Polyamine uptake is necessary for a normal biochemical maturation of astrocytes in culture. Neuroreport 2002, 13, 1083–1087. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Antonov, S.M.; Eaton, M.J. Glia and glial polyamines. Role in brain function in health and disease. Biochem. Suppl. Ser. A Membr. Cell Biol. 2016, 10, 73–98. [Google Scholar] [CrossRef]
- Laube, G.; Veh, R.d.W. Astrocytes, not neurons, show most prominent staining for spermidine/spermine-like immunoreactivity in adult rat brain. Glia 1997, 19, 171–179. [Google Scholar] [CrossRef]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Jimenez-Gonzalez, C.; Pirttimaki, T.; Cope, D.W.; Parri, H.R. Non-neuronal, slow GABA signalling in the ventrobasal thalamus targets delta-subunit-containing GABA(A) receptors. Eur. J. Neurosci. 2011, 33, 1471–1482. [Google Scholar] [CrossRef] [Green Version]
- Laschet, J.; Grisar, T.; Bureau, M.; Guillaume, D. Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience 1992, 48, 151–157. [Google Scholar] [CrossRef]
- Heja, L.; Nyitrai, G.; Kekesi, O.; Dobolyi, A.; Szabo, P.; Fiath, R.; Ulbert, I.; Pal-Szenthe, B.; Palkovits, M.; Kardos, J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 2012, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.E.; Lee, C.J. GABA as a rising gliotransmitter. Front. Neural Circuits 2014, 8, 141. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291.e9. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.E.; et al. Astrocytes Control Sensory Acuity via Tonic Inhibition in the Thalamus. Neuron 2020, 108, 691–706.e10. [Google Scholar] [CrossRef]
- Kovacs, Z.; Skatchkov, S.N.; Veh, R.W.; Szabo, Z.; Nemeth, K.; Szabo, P.T.; Kardos, J.; Heja, L. Critical Role of Astrocytic Polyamine and GABA Metabolism in Epileptogenesis. Front. Cell Neurosci. 2021, 15, 787319. [Google Scholar] [CrossRef]
- Szabo, Z.; Peter, M.; Heja, L.; Kardos, J. Dual Role for Astroglial Copper-Assisted Polyamine Metabolism during Intense Network Activity. Biomolecules 2021, 11, 604. [Google Scholar] [CrossRef]
- Deshpande, T.; Li, T.; Herde, M.K.; Becker, A.; Vatter, H.; Schwarz, M.K.; Henneberger, C.; Steinhauser, C.; Bedner, P. Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia 2017, 65, 1809–1820. [Google Scholar] [CrossRef]
- Skatchkov, S.N.; Bukauskas, F.F.; Benedikt, J.; Inyushin, M.; Kucheryavykh, Y.V. Intracellular spermine prevents acid-induced uncoupling of Cx43 gap junction channels. Neuroreport 2015, 26, 528–532. [Google Scholar] [CrossRef]
- Benedikt, J.; Inyushin, M.; Kucheryavykh, Y.V.; Rivera, Y.; Kucheryavykh, L.Y.; Nichols, C.G.; Eaton, M.J.; Skatchkov, S.N. Intracellular polyamines enhance astrocytic coupling. Neuroreport 2012, 23, 1021–1025. [Google Scholar] [CrossRef]
- Kucheryavykh, L.Y.; Benedikt, J.; Cubano, L.A.; Skatchkov, S.N.; Bukauskas, F.F.; Kucheryavykh, Y.V. Polyamines preserve connexin 43-mediated gap junctional communication during intracellular hypercalcemia and acidosis. Neuroreport 2017, 28, 208–213. [Google Scholar] [CrossRef]
- Vincze, R.; Peter, M.; Szabo, Z.; Kardos, J.; Heja, L.; Kovacs, Z. Connexin 43 Differentially Regulates Epileptiform Activity in Models of Convulsive and Non-convulsive Epilepsies. Front. Cell Neurosci. 2019, 13, 173. [Google Scholar] [CrossRef]
- Kekesi, O.; Ioja, E.; Szabo, Z.; Kardos, J.; Heja, L. Recurrent seizure-like events are associated with coupled astroglial synchronization. Front. Cell Neurosci. 2015, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Bedner, P.; Dupper, A.; Huttmann, K.; Muller, J.; Herde, M.K.; Dublin, P.; Deshpande, T.; Schramm, J.; Haussler, U.; Haas, C.A.; et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 2015, 138, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Pitkanen, A.; Ekolle Ndode-Ekane, X.; Lapinlampi, N.; Puhakka, N. Epilepsy biomarkers–Toward etiology and pathology specificity. Neurobiol. Dis. 2019, 123, 42–58. [Google Scholar] [CrossRef]
- Farrell, J.S.; Wolff, M.D.; Teskey, G.C. Neurodegeneration and Pathology in Epilepsy: Clinical and Basic Perspectives. Adv. Neurobiol. 2017, 15, 317–334. [Google Scholar] [CrossRef]
- Tran, C.H.T.; George, A.G.; Teskey, G.C.; Gordon, G.R. Seizures elevate gliovascular unit Ca2+ and cause sustained vasoconstriction. JCI Insight 2020, 5, e136469. [Google Scholar] [CrossRef]
- Suh, M.; Ma, H.; Zhao, M.; Sharif, S.; Schwartz, T.H. Neurovascular coupling and oximetry during epileptic events. Mol. Neurobiol. 2006, 33, 181–197. [Google Scholar] [CrossRef]
- Farrell, J.S.; Gaxiola-Valdez, I.; Wolff, M.D.; David, L.S.; Dika, H.I.; Geeraert, B.L.; Rachel Wang, X.; Singh, S.; Spanswick, S.C.; Dunn, J.F.; et al. Postictal behavioural impairments are due to a severe prolonged hypoperfusion/hypoxia event that is COX-2 dependent. eLife 2016, 5, e19352. [Google Scholar] [CrossRef]
- Farrell, J.S.; Colangeli, R.; Wolff, M.D.; Wall, A.K.; Phillips, T.J.; George, A.; Federico, P.; Teskey, G.C. Postictal hypoperfusion/hypoxia provides the foundation for a unified theory of seizure-induced brain abnormalities and behavioral dysfunction. Epilepsia 2017, 58, 1493–1501. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.T.; Gerner, E.W. Polyamine-Mediated post-Transcriptional regulation of COX-2. Biochimie 2002, 84, 815–819. [Google Scholar] [CrossRef]
- Nilsson, B.O.; Gomez, M.F.; Sward, K.; Hellstrand, P. Regulation of Ca2+ channel and phosphatase activities by polyamines in intestinal and vascular smooth muscle--implications for cellular growth and contractility. Acta Physiol. Scand. 2002, 176, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, M.; Hellstrand, P. Endogenous polyamines modulate Ca2+ channel activity in guinea-pig intestinal smooth muscle. Pflug. Arch. 1999, 438, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Hellstrand, P. Effects of polyamines on voltage-activated calcium channels in guinea-pig intestinal smooth muscle. Pflug. Arch. 1995, 430, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Sim, J.H.; Kim, Y.H.; Kwon, S.C.; Lee, S.J.; Kim, S.R.; Kim, D.W.; Park, S.M.; Youn, S.J.; Lee, S.J.; et al. Effects of polyamines on contractility of guinea-pig gastric smooth muscle. J. Korean Med. Sci. 2007, 22, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, B.O.; Hellstrand, P. Effects of polyamines on intracellular calcium and mechanical activity in smooth muscle of guinea-pig taenia coli. Acta Physiol. Scand. 1993, 148, 37–43. [Google Scholar] [CrossRef]
- Sward, K.; Nilsson, B.O.; Hellstrand, P. Polyamines increase Ca2+ sensitivity in permeabilized smooth muscle of guinea pig ileum. Am. J. Physiol. 1994, 266, C1754–C1763. [Google Scholar] [CrossRef]
- Sward, K.; Pato, M.D.; Nilsson, B.O.; Nordstrom, I.; Hellstrand, P. Polyamines inhibit myosin phosphatase and increase LC20 phosphorylation and force in smooth muscle. Am. J. Physiol. 1995, 269, C563–C571. [Google Scholar] [CrossRef]
- Herman, M.D.; Reuveny, E.; Narahashi, T. The effect of polyamines on voltage-activated calcium channels in mouse neuroblastoma cells. J. Physiol. 1993, 462, 645–660. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Thom, M. Review: Hippocampal sclerosis in epilepsy: A neuropathology review. Neuropathol. Appl. Neurobiol. 2014, 40, 520–543. [Google Scholar] [CrossRef]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhauser, C. Functional changes in astroglial cells in epilepsy. Glia 2006, 54, 358–368. [Google Scholar] [CrossRef]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef]
- Marcoli, M.; Cervetto, C.; Amato, S.; Fiorucci, C.; Maura, G.; Mariottini, P.; Cervelli, M. Transgenic Mouse Overexpressing Spermine Oxidase in Cerebrocortical Neurons: Astrocyte Dysfunction and Susceptibility to Epileptic Seizures. Biomolecules 2022, 12, 204. [Google Scholar] [CrossRef]
- Cervetto, C.; Averna, M.; Vergani, L.; Pedrazzi, M.; Amato, S.; Pelassa, S.; Giuliani, S.; Baldini, F.; Maura, G.; Mariottini, P.; et al. Reactive Astrocytosis in a Mouse Model of Chronic Polyamine Catabolism Activation. Biomolecules 2021, 11, 1274. [Google Scholar] [CrossRef]
- Jeglinski, W.; Skup, M.; Zaremba, M.; Oderfeld-Nowak, B. Difluoromethylornithine counteracts lesion-induced astrogliosis in rat hippocampus: Enhancement of inhibitory effect by combined treatment with GM1 ganglioside. Acta Neurobiol. Exp. 1996, 56, 549–553. [Google Scholar]
- McKenna, J., 3rd; Kapfhamer, D.; Kinchen, J.M.; Wasek, B.; Dunworth, M.; Murray-Stewart, T.; Bottiglieri, T.; Casero, R.A., Jr.; Gambello, M.J. Metabolomic studies identify changes in transmethylation and polyamine metabolism in a brain-specific mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2018, 27, 2113–2124. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, J.P.; Seidler, F.J. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced astrogliosis does not require activation of ornithine decarboxylase. Neurosci. Lett. 1992, 148, 105–108. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Granata, T. Brain inflammation in epilepsy: Experimental and clinical evidence. Epilepsia 2005, 46, 1724–1743. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q.J. Epilepsy and brain inflammation. Exp. Neurol. 2013, 244, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Minami, M.; Kuraishi, Y.; Satoh, M. Effects of kainic acid on messenger RNA levels of IL-1β, IL-6, TNFα and LIF in the rat brain. Biochem. Biophys. Res. Commun. 1991, 176, 593–598. [Google Scholar] [CrossRef]
- Kegler, A.; Caprara, A.L.F.; Pascotini, E.T.; Arend, J.; Gabbi, P.; Duarte, M.; Furian, A.F.; Oliveira, M.S.; Royes, L.F.F.; Fighera, M.R. Apoptotic Markers Are Increased in Epilepsy Patients: A Relation with Manganese Superoxide Dismutase Ala16Val Polymorphism and Seizure Type through IL-1beta and IL-6 Pathways. Biomed Res. Int. 2020, 2020, 6250429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprica, M.; Eriksson, C.; Schultzberg, M. Inflammatory mechanisms associated with brain damage induced by kainic acid with special reference to the interleukin-1 system. J. Cell Mol. Med. 2003, 7, 127–140. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 2017, 23, 478–498. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, X.; Wang, Y.; Zhou, X.; Qian, Y.; Zhang, S. Suppression of Brain Mast Cells Degranulation Inhibits Microglial Activation and Central Nervous System Inflammation. Mol. Neurobiol. 2017, 54, 997–1007. [Google Scholar] [CrossRef]
- Kilinc, E.; Torun, I.E.; Cetinkaya, A.; Tore, F. Mast cell activation ameliorates pentylenetetrazole-induced seizures in rats: The potential role for serotonin. Eur. J. Neurosci. 2021, 55, 2912–2924. [Google Scholar] [CrossRef]
- Kanerva, K.; Lappalainen, J.; Makitie, L.T.; Virolainen, S.; Kovanen, P.T.; Andersson, L.C. Expression of Antizyme Inhibitor 2 in Mast Cells and Role of Polyamines as Selective Regulators of Serotonin Secretion. PLoS ONE 2009, 4, e6858. [Google Scholar] [CrossRef]
- Coffino, P. Regulation of cellular polyamines by antizyme. Nat. Rev. Mol. Cell Biol. 2001, 2, 188–194. [Google Scholar] [CrossRef]
- Lopez-Contreras, A.J.; Ramos-Molina, B.; Cremades, A.; Penafiel, R. Antizyme inhibitor 2: Molecular, cellular and physiological aspects. Amino Acids 2010, 38, 603–611. [Google Scholar] [CrossRef]
- Fan, J.; Chen, M.; Wang, X.; Tian, Z.; Wang, J.; Fan, D.; Zeng, J.; Zhang, K.; Dai, X. Targeting Smox Is Neuroprotective and Ameliorates Brain Inflammation in Cerebral Ischemia/Reperfusion Rats. Toxicol. Sci. 2019, 168, 381–393. [Google Scholar] [CrossRef]
- Alfarhan, M.; Liu, F.; Shan, S.; Pichavaram, P.; Somanath, P.R.; Narayanan, S.P. Pharmacological Inhibition of Spermine Oxidase Suppresses Excitotoxicity Induced Neuroinflammation in Mouse Retina. Int. J. Mol. Sci. 2022, 23, 2133. [Google Scholar] [CrossRef]
- Luo, J.; Shi, R. Acrolein induces axolemmal disruption, oxidative stress, and mitochondrial impairment in spinal cord tissue. Neurochem. Int. 2004, 44, 475–486. [Google Scholar] [CrossRef]
- Zhao, W.Z.; Wang, H.T.; Huang, H.J.; Lo, Y.L.; Lin, A.M. Neuroprotective Effects of Baicalein on Acrolein-induced Neurotoxicity in the Nigrostriatal Dopaminergic System of Rat Brain. Mol. Neurobiol. 2018, 55, 130–137. [Google Scholar] [CrossRef]
- Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [Green Version]
- McElroy, P.B.; Liang, L.P.; Day, B.J.; Patel, M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp. Neurol. 2017, 298, 13–22. [Google Scholar] [CrossRef]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A., Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef] [Green Version]
- Belle, N.A.; Dalmolin, G.D.; Fonini, G.; Rubin, M.A.; Rocha, J.B. Polyamines reduces lipid peroxidation induced by different pro-oxidant agents. Brain Res. 2004, 1008, 245–251. [Google Scholar] [CrossRef]
- Matkovics, B.; Kecskemeti, V.; Varga, S.I.; Novak, Z.; Kertesz, Z. Antioxidant properties of di- and polyamines. Comp. Biochem. Physiol. B 1993, 104, 475–479. [Google Scholar] [CrossRef]
- Sava, I.G.; Battaglia, V.; Rossi, C.A.; Salvi, M.; Toninello, A. Free radical scavenging action of the natural polyamine spermine in rat liver mitochondria. Free Radic Biol. Med. 2006, 41, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Mei, Y.H.; Wilson, T. A proposed function for spermine and spermidine: Protection of replicating DNA against damage by singlet oxygen. Proc. Natl. Acad. Sci. USA 1992, 89, 11426–11427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, J.E.; Hacker, A.; Mackintosh, C.A.; Pegg, A.E.; Woster, P.M.; Casero, R.A., Jr. Spermine and spermidine mediate protection against oxidative damage caused by hydrogen peroxide. Amino Acids 2007, 33, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Yager, J.D.; Woster, P.A.; Casero, R.A., Jr. Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun. 1998, 244, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Kobylarek, D.; Iwanowski, P.; Lewandowska, Z.; Limphaibool, N.; Szafranek, S.; Labrzycka, A.; Kozubski, W. Advances in the Potential Biomarkers of Epilepsy. Front. Neurol. 2019, 10, 685. [Google Scholar] [CrossRef] [Green Version]
- Serrano, G.E.; Lelutiu, N.; Rojas, A.; Cochi, S.; Shaw, R.; Makinson, C.D.; Wang, D.; FitzGerald, G.A.; Dingledine, R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J. Neurosci. 2011, 31, 14850–14860. [Google Scholar] [CrossRef]
- Lopantsev, V.; Both, M.; Draguhn, A. Rapid plasticity at inhibitory and excitatory synapses in the hippocampus induced by ictal epileptiform discharges. Eur. J. Neurosci. 2009, 29, 1153–1164. [Google Scholar] [CrossRef]
- Ergina, J.L.; Amakhin, D.V.; Postnikova, T.Y.; Soboleva, E.B.; Zaitsev, A.V. Short-Term Epileptiform Activity Potentiates Excitatory Synapses but Does Not Affect Intrinsic Membrane Properties of Pyramidal Neurons in the Rat Hippocampus In Vitro. Biomedicines 2021, 9, 1374. [Google Scholar] [CrossRef]
- Sperk, G.; Drexel, M.; Pirker, S. Neuronal plasticity in animal models and the epileptic human hippocampus. Epilepsia 2009, 50 (Suppl. 12), 29–31. [Google Scholar] [CrossRef]
- Ben-Ari, Y. Cell death and synaptic reorganizations produced by seizures. Epilepsia 2001, 42 (Suppl. 3), 5–7. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Saiki, R.; Yoshizawa, Y.; Imamura, M.; Higashi, K.; Ishii, I.; Toida, T.; Williams, K.; Kashiwagi, K.; Igarashi, K. Spermidine and Ca(2+), but not Na(+), can permeate NMDA receptors consisting of GluN1 and GluN2A or GluN2B in the presence of Mg(2+). Biochem. Biophys. Res. Commun. 2015, 463, 1190–1195. [Google Scholar] [CrossRef]
- Cull-Candy, S.; Kelly, L.; Farrant, M. Regulation of Ca2+-permeable AMPA receptors: Synaptic plasticity and beyond. Curr. Opin. Neurobiol. 2006, 16, 288–297. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 196, 108540. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Chen, W.; Harnett, M.T.; Smith, S.M. Modulation of neuronal voltage-activated calcium and sodium channels by polyamines and pH. Channels 2007, 1, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Cestaro, B. Effects of arginine, S-adenosylmethionine and polyamines on nerve regeneration. Acta Neurol. Scand. Suppl. 1994, 154, 32–41. [Google Scholar] [CrossRef]
- Abe, K.; Chu, P.; Shirahata, A.; Samejima, K.; Saito, H. Structural requirement for axonal regeneration-promoting effect of polyamines in cultured rat hippocampal neurons. Brain Res. 1997, 766, 281–284. [Google Scholar] [CrossRef]
- Chu, P.J.; Saito, H.; Abe, K. Polyamines promote regeneration of injured axons of cultured rat hippocampal neurons. Brain Res. 1995, 673, 233–241. [Google Scholar] [CrossRef]
- Giménez-Llort, L.; Martínez, E.; Camón, L.; de Vera, N. Long-term effects of status epilepticus induced by kainic acid on hippocampal polyamines. NeuroReport 1998, 9, 937–941. [Google Scholar] [CrossRef] [PubMed]
- Oble, D.A.; Burton, L.; Maxwell, K.; Hassard, T.; Nathaniel, E.J. A comparison of thyroxine- and polyamine-mediated enhancement of rat facial nerve regeneration. Exp. Neurol. 2004, 189, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Kimura, A.; Guo, X.; Azuchi, Y.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis. 2015, 6, e1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, H.S.; Tsai, M.J.; Huang, M.C.; Chiu, C.W.; Tsai, C.Y.; Lee, M.J.; Huang, W.C.; Lin, Y.L.; Kuo, W.C.; Cheng, H. Acid fibroblast growth factor and peripheral nerve grafts regulate Th2 cytokine expression, macrophage activation, polyamine synthesis, and neurotrophin expression in transected rat spinal cords. J. Neurosci. 2011, 31, 4137–4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarero-Basulto, J.J.; Gasca-Martinez, Y.; Rivera-Cervantes, M.C.; Urena-Guerrero, M.E.; Feria-Velasco, A.I.; Beas-Zarate, C. Interactions Between Epilepsy and Plasticity. Pharmaceuticals 2018, 11, 17. [Google Scholar] [CrossRef] [Green Version]
- Henshall, D.C.; Murphy, B.M. Modulators of neuronal cell death in epilepsy. Curr. Opin. Pharm. 2008, 8, 75–81. [Google Scholar] [CrossRef]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Clarkson, A.N.; Liu, H.; Pearson, L.; Kapoor, M.; Harrison, J.C.; Sammut, I.A.; Jackson, D.M.; Appleton, I. Neuroprotective effects of spermine following hypoxic-ischemic-induced brain damage: A mechanistic study. FASEB J. 2004, 18, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Gilad, G.M.; Gilad, V.H. Polyamines can protect against ischemia-induced nerve cell death in gerbil forebrain. Exp. Neurol. 1991, 111, 349–355. [Google Scholar] [CrossRef]
- Raghavendra Rao, V.L.; Dogan, A.; Bowen, K.K.; Dempsey, R.J. Ornithine decarboxylase knockdown exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Cereb. Blood Flow Metab. 2001, 21, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Lukkarinen, J.; Grohn, O.H.; Sinervirta, R.; Jarvinen, A.; Kauppinen, R.A.; Janne, J.; Alhonen, L.I. Transgenic rats as models for studying the role of ornithine decarboxylase expression in permanent middle cerebral artery occlusion. Stroke 1997, 28, 639–645. [Google Scholar] [CrossRef]
- Lukkarinen, J.A.; Grohn, O.H.J.; Alhonen, L.I.; Janne, J.; Kauppinen, R.A. Enhanced ornithine decarboxylase activity is associated with attenuated rate of damage evolution and reduction of infarct volume in transient middle cerebral artery occlusion in the rat. Brain Res. 1999, 826, 325–329. [Google Scholar] [CrossRef]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef]
- Harada, J.; Sugimoto, M. Polyamines prevent apoptotic cell death in cultured cerebellar granule neurons. Brain Res. 1997, 753, 251–259. [Google Scholar] [CrossRef]
- Farbiszewski, R.; Bielawska, A.; Szymanska, M.; Skrzydlewska, E. Spermine partially normalizes in vivo antioxidant defense potential in certain brain regions in transiently hypoperfused rat brain. Neurochem. Res. 1996, 21, 1497–1503. [Google Scholar] [CrossRef]
- Farbiszewski, R.; Bielawski, K.; Bielawska, A.; Sobaniec, W. Spermine protects in vivo the antioxidant enzymes in transiently hypoperfused rat brain. Acta Neurobiol. Exp. 1995, 55, 253–258. [Google Scholar]
- Otsuki, M.; Davidson, M.; Goodenough, S.; Wilce, P.A.; Tase, C.; Matsumoto, I. In vivo pharmacological study of spermine-induced neurotoxicity. Neurosci. Lett. 1995, 196, 81–84. [Google Scholar] [CrossRef]
- Goodenough, S.; Davidson, M.; Kidd, G.; Matsumoto, I.; Wilce, P. Cell death and immunohistochemistry of p53, c-Fos and c-Jun after spermine injection into the rat striatum. Exp. Brain Res. 2000, 131, 126–134. [Google Scholar] [CrossRef]
- Sparapani, M.; Dall’Olio, R.; Gandolfi, O.; Ciani, E.; Contestabile, A. Neurotoxicity of polyamines and pharmacological neuroprotection in cultures of rat cerebellar granule cells. Exp. Neurol. 1997, 148, 157–166. [Google Scholar] [CrossRef]
- de Vera, N.; Martinez, E.; Sanfeliu, C. Spermine induces cell death in cultured human embryonic cerebral cortical neurons through N-methyl-D-aspartate receptor activation. J. Neurosci. Res. 2008, 86, 861–872. [Google Scholar] [CrossRef]
- Bourdiol, F.; Fage, D.; Serrano, A.; Carter, C.; Benavides, J.; Scatton, B. Neurotoxic effects of the intrastriatal injection of spermine and spermidine: Lack of involvement of NMDA receptors. Brain Res. 1992, 596, 183–188. [Google Scholar] [CrossRef]
- Pegg, A.E. Toxicity of polyamines and their metabolic products. Chem. Res. Toxicol. 2013, 26, 1782–1800. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Nakamura, Y.; Yoneda, Y. Microglial cell death induced by a low concentration of polyamines. Neuroscience 2003, 120, 961–967. [Google Scholar] [CrossRef]
- Takano, K.; Ogura, M.; Yoneda, Y.; Nakamura, Y. Oxidative metabolites are involved in polyamine-induced microglial cell death. Neuroscience 2005, 134, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Mandal, A.; Park, M.H. Depletion of the polyamines spermidine and spermine by overexpression of spermidine/spermine N(1)-acetyltransferase 1 (SAT1) leads to mitochondria-mediated apoptosis in mammalian cells. Biochem. J. 2015, 468, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Igarashi, K.; Yamamoto, N. Polyamine depletion induces apoptosis through mitochondria-mediated pathway. Exp. Cell Res. 2002, 276, 120–128. [Google Scholar] [CrossRef]
- Duan, B.; Wang, Y.Z.; Yang, T.; Chu, X.P.; Yu, Y.; Huang, Y.; Cao, H.; Hansen, J.; Simon, R.P.; Zhu, M.X.; et al. Extracellular spermine exacerbates ischemic neuronal injury through sensitization of ASIC1a channels to extracellular acidosis. J. Neurosci. 2011, 31, 2101–2112. [Google Scholar] [CrossRef] [Green Version]
- Seiler, N.; Raul, F. Polyamines and apoptosis. J. Cell Mol. Med. 2005, 9, 623–642. [Google Scholar] [CrossRef]
- Leal-Campanario, R.; Alarcon-Martinez, L.; Rieiro, H.; Martinez-Conde, S.; Alarcon-Martinez, T.; Zhao, X.; LaMee, J.; Popp, P.J.; Calhoun, M.E.; Arribas, J.I.; et al. Abnormal Capillary Vasodynamics Contribute to Ictal Neurodegeneration in Epilepsy. Sci. Rep. 2017, 7, 43276. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.H.; Sutton, K.G.; Dolphin, A.C. Interactions of polyamines with neuronal ion channels. Trends Neurosci. 1993, 16, 153–160. [Google Scholar] [CrossRef]
- Bailey, D.; Kirby, B.P.; Atkinson, J.; Fixon-Owoo, S.; Henman, M.C.; Shaw, G.G.; Doyle, K.M. Hydroxycinnamic acid amide derivatives of polyamines reverse spermine-induced CNS excitation. Pharm. Biochem. Behav. 2015, 133, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Liu, R.; Schreiber, S.S.; Baudry, M. Role of polyamine metabolism in kainic acid excitotoxicity in organotypic hippocampal slice cultures. J. Neurochem. 2001, 79, 976–984. [Google Scholar] [CrossRef] [Green Version]
- Kirby, B.P.; Ryder, S.A.; Seiler, N.; Renault, J.; Shaw, G.G. N1-dansyl-spermine: A potent polyamine antagonist. Brain Res. 2004, 1011, 69–73. [Google Scholar] [CrossRef]
- Kirby, B.P.; Shaw, G.G. Effect of spermine and N1-dansyl-spermine on epileptiform activity in mouse cortical slices. Eur. J. Pharm. 2005, 524, 53–59. [Google Scholar] [CrossRef]
- Li, J.; Henman, M.C.; Doyle, K.M.; Strbian, D.; Kirby, B.P.; Tatlisumak, T.; Shaw, G.G. The pre-ischaemic neuroprotective effect of a novel polyamine antagonist, N1-dansyl-spermine in a permanent focal cerebral ischaemia model in mice. Brain Res. 2004, 1029, 84–92. [Google Scholar] [CrossRef]
- Allen, A.R.; Eilertson, K.; Sharma, S.; Baure, J.; Allen, B.; Leu, D.; Rosi, S.; Raber, J.; Huang, T.T.; Fike, J.R. Delayed administration of alpha-difluoromethylornithine prevents hippocampus-dependent cognitive impairment after single and combined injury in mice. Radiat. Res. 2014, 182, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Wirth, M.; Benson, G.; Schwarz, C.; Kobe, T.; Grittner, U.; Schmitz, D.; Sigrist, S.J.; Bohlken, J.; Stekovic, S.; Madeo, F.; et al. The effect of spermidine on memory performance in older adults at risk for dementia: A randomized controlled trial. Cortex 2018, 109, 181–188. [Google Scholar] [CrossRef]
- Schwarz, C.; Stekovic, S.; Wirth, M.; Benson, G.; Royer, P.; Sigrist, S.J.; Pieber, T.; Dammbrueck, C.; Magnes, C.; Eisenberg, T.; et al. Safety and tolerability of spermidine supplementation in mice and older adults with subjective cognitive decline. Aging 2018, 10, 19–33. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Yu, Z.; Maimaiti, B.; Meng, Q.; Meng, H. The Potential Role of Polyamines in Epilepsy and Epilepsy-Related Pathophysiological Changes. Biomolecules 2022, 12, 1596. https://doi.org/10.3390/biom12111596
Liu J, Yu Z, Maimaiti B, Meng Q, Meng H. The Potential Role of Polyamines in Epilepsy and Epilepsy-Related Pathophysiological Changes. Biomolecules. 2022; 12(11):1596. https://doi.org/10.3390/biom12111596
Chicago/Turabian StyleLiu, Jiayu, Zhi Yu, Buajieerguli Maimaiti, Qian Meng, and Hongmei Meng. 2022. "The Potential Role of Polyamines in Epilepsy and Epilepsy-Related Pathophysiological Changes" Biomolecules 12, no. 11: 1596. https://doi.org/10.3390/biom12111596