
SMN Is Physiologically Downregulated at Wild-Type Motor Nerve Terminals but Aggregates Together with Neurofilaments in SMA Mouse Models
 , , , , and
, , , , and 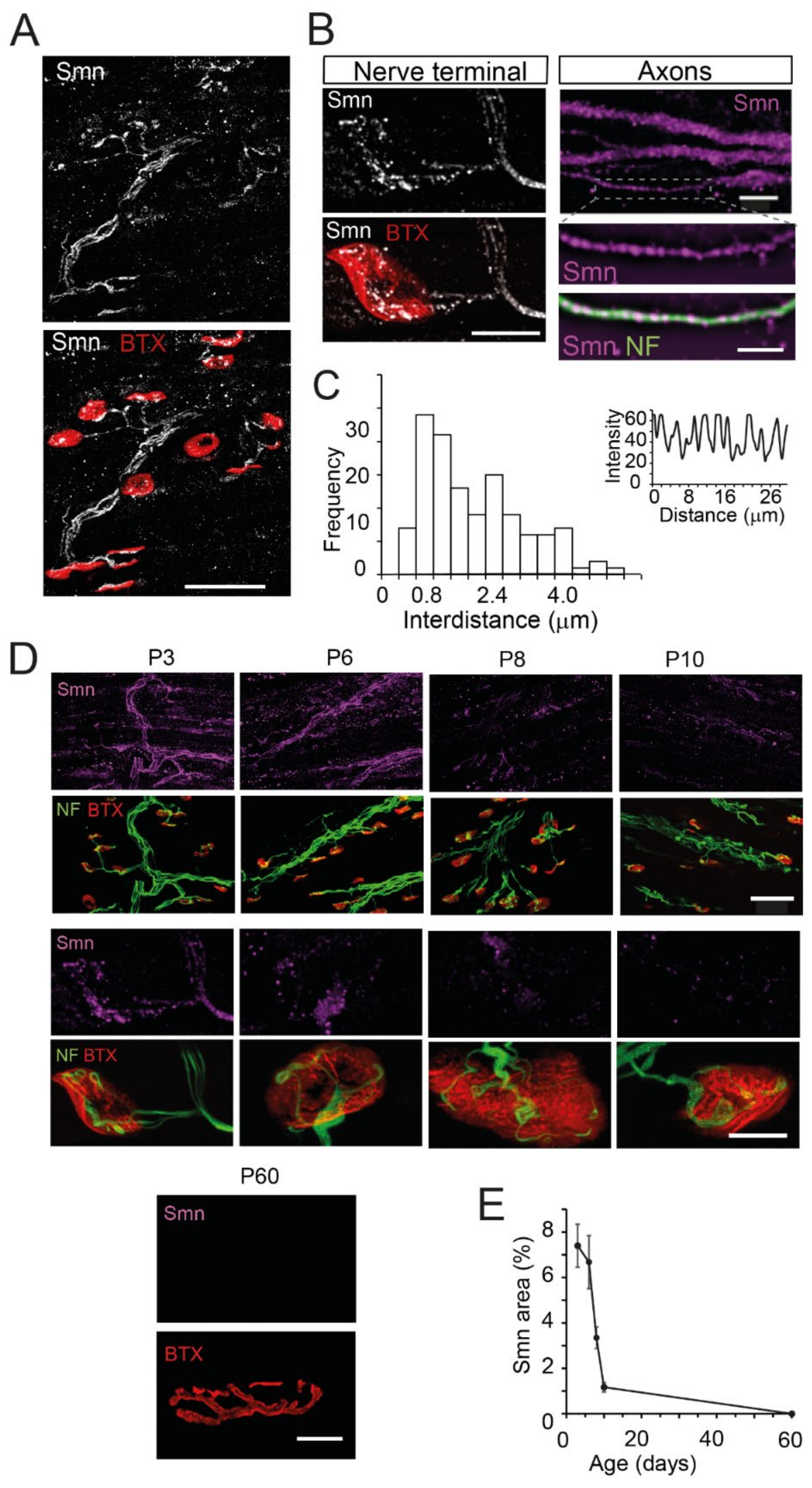{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Models
2.2. Neuromuscular Preparations
2.3. Neuromuscular in Situ Hybridization
2.4. Electron Microscopy
2.5. Western Blot
2.6. Immunohistochemistry
2.7. Image Acquisition and Analysis
2.8. STED Super-Resolution Microscopy
2.9. Statistical Analysis
3. Results
3.1. SMN Is Localized as Granules in Axons and Motor Nerve Terminals
3.2. Smn Progressively Decreases during the Early Postnatal Period in the Axon and the Presynaptic Motor Terminal of Wild-Type Mice
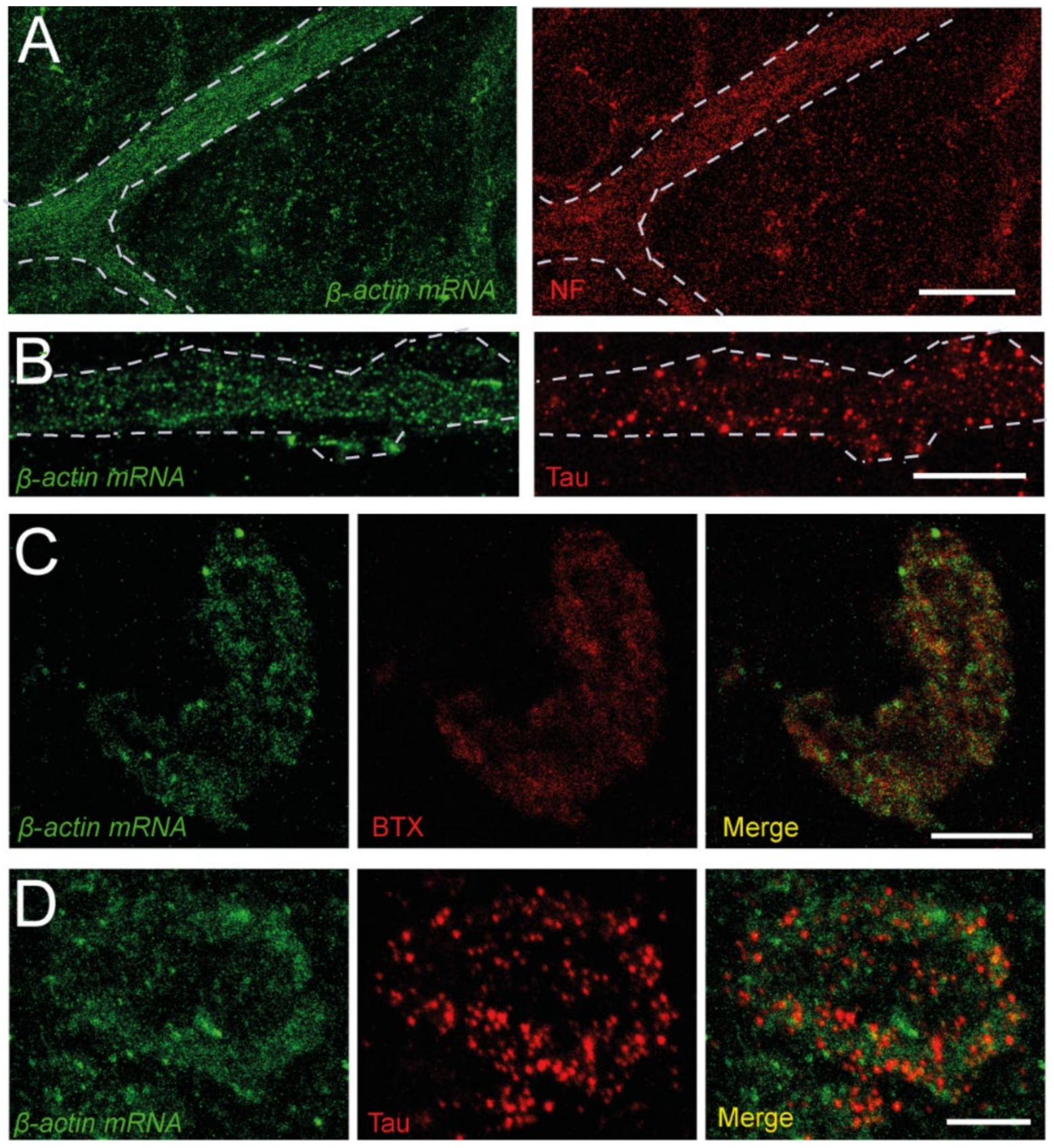
3.3. β-Actin mRNA Transcripts Localize in the Presynaptic Compartment
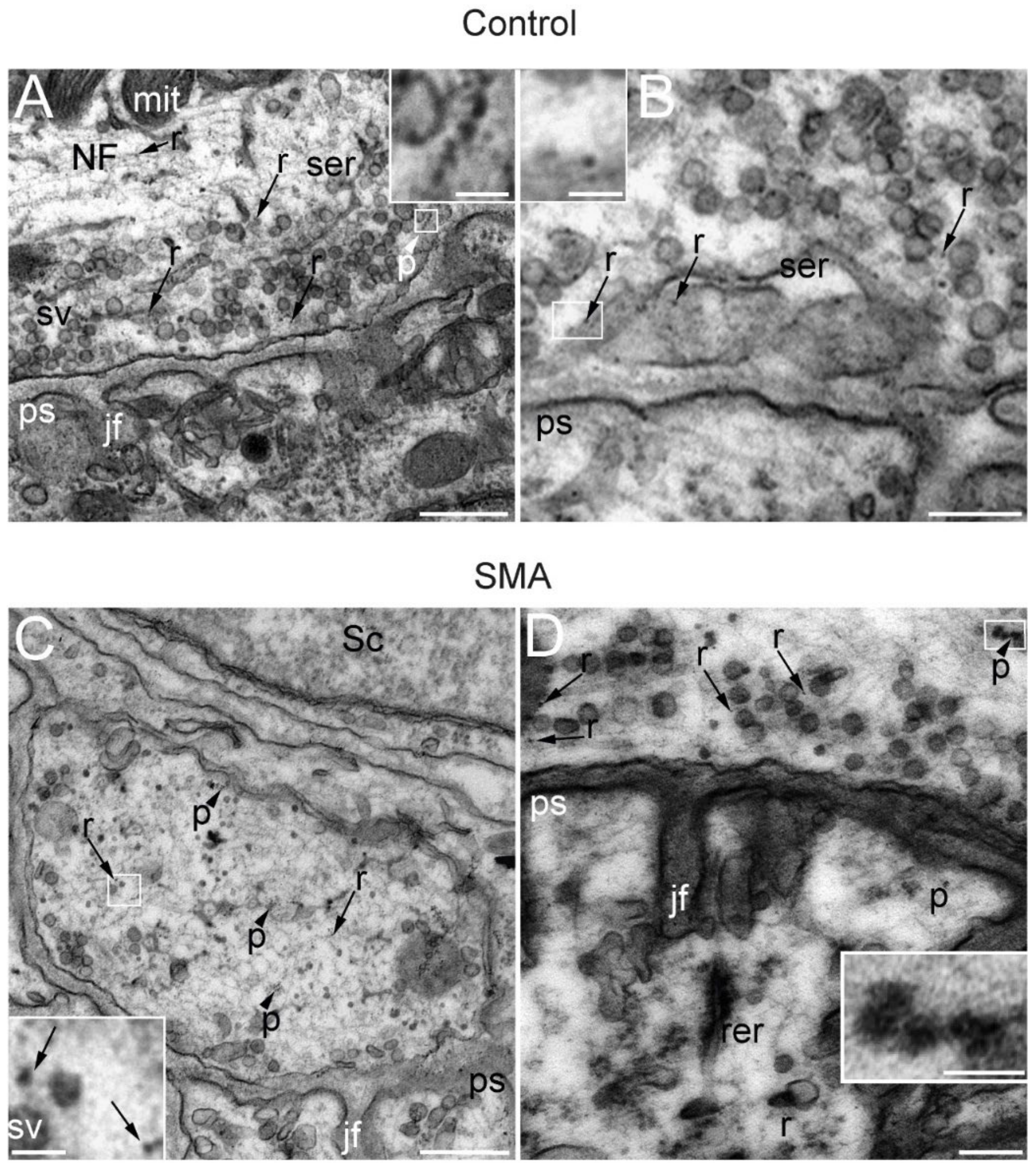
3.4. Ribosomes and Polysomes Are Present in Nerve Terminals
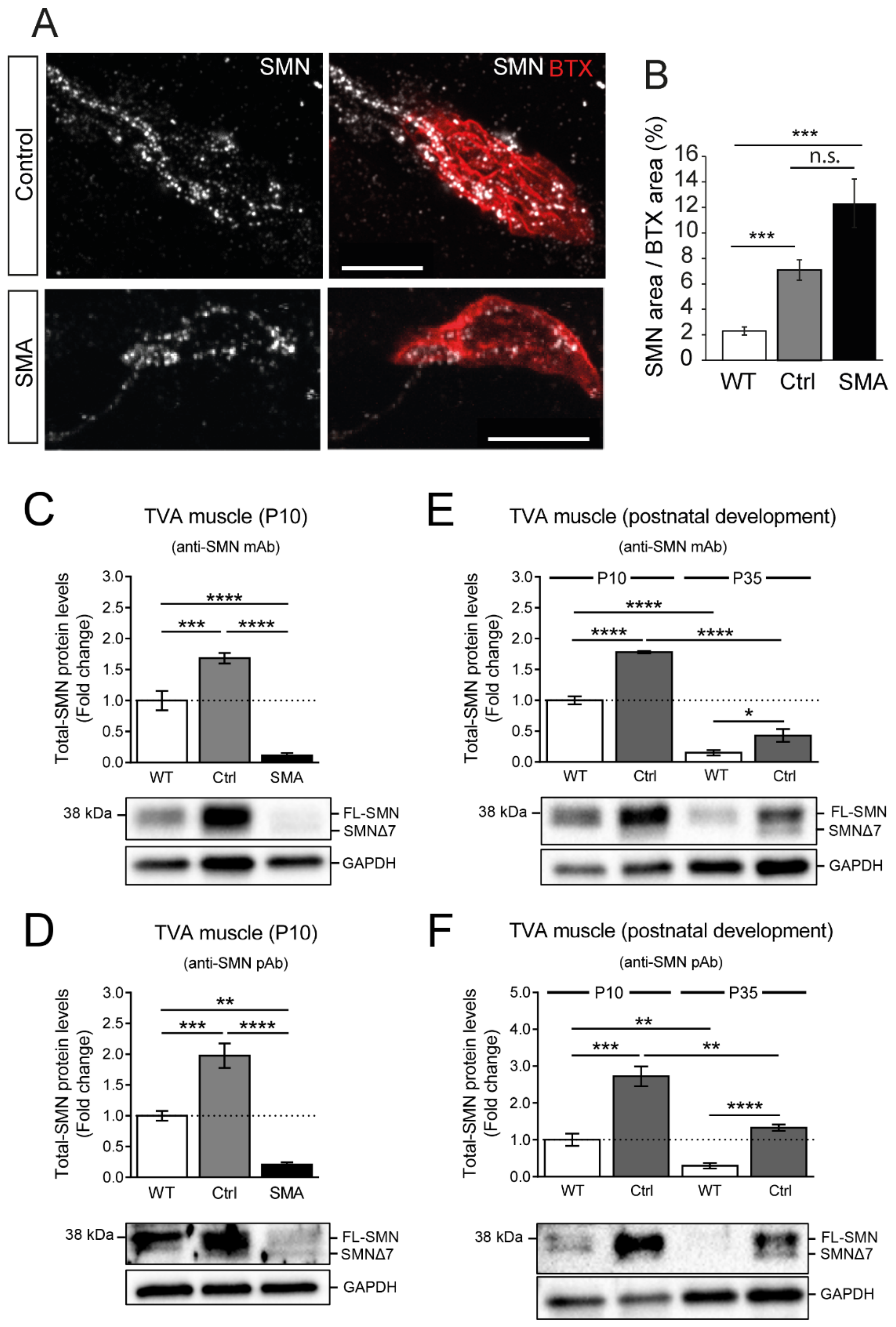
3.5. Axonal and Presynaptic SMN Granules Abundancy Remains High in SMA Models
3.6. SMN Protein Levels Are Elevated in the TVA Muscle of Control Transgenic Mice
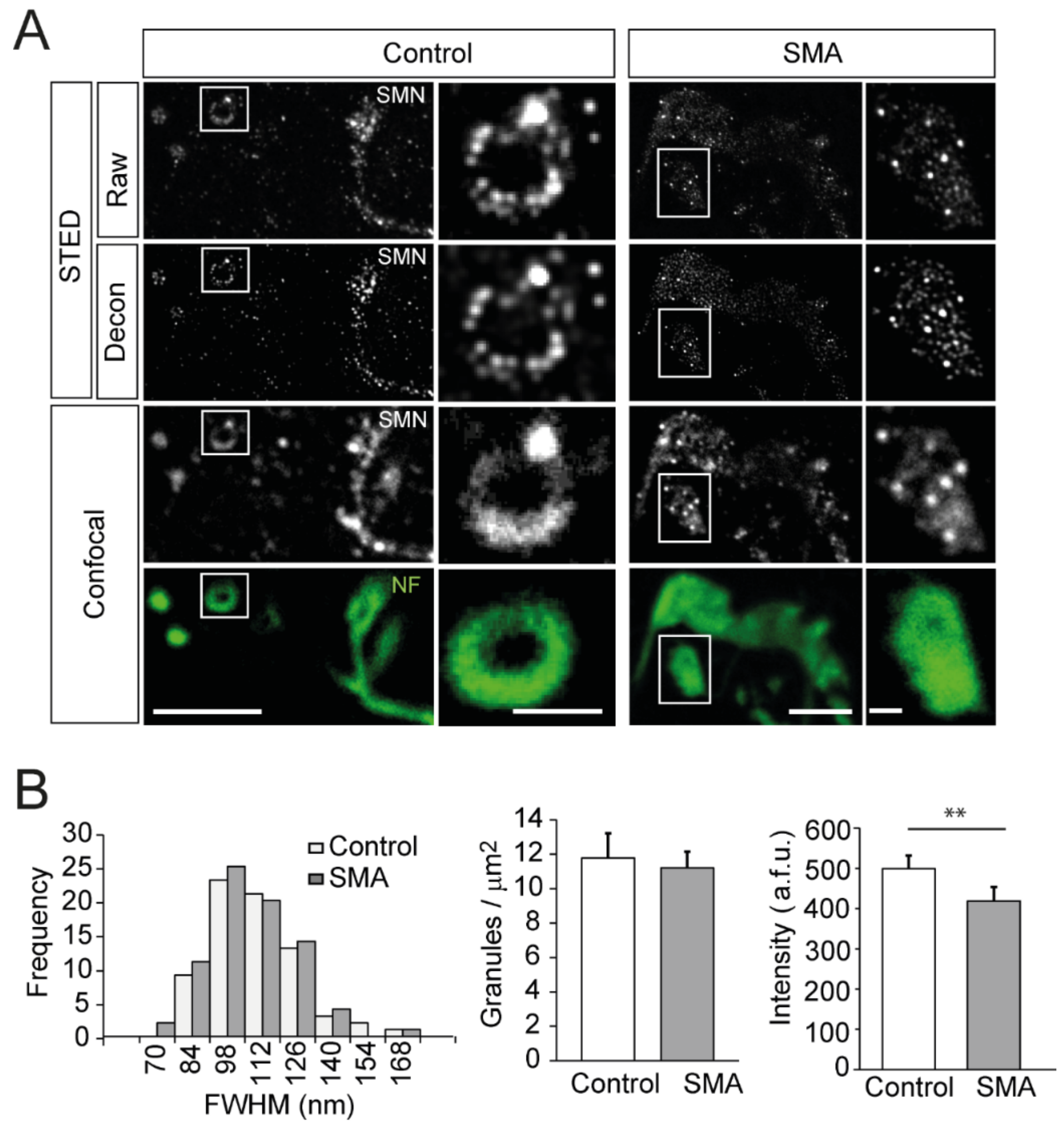
3.7. SMN Granules Visualized at Super-Resolution
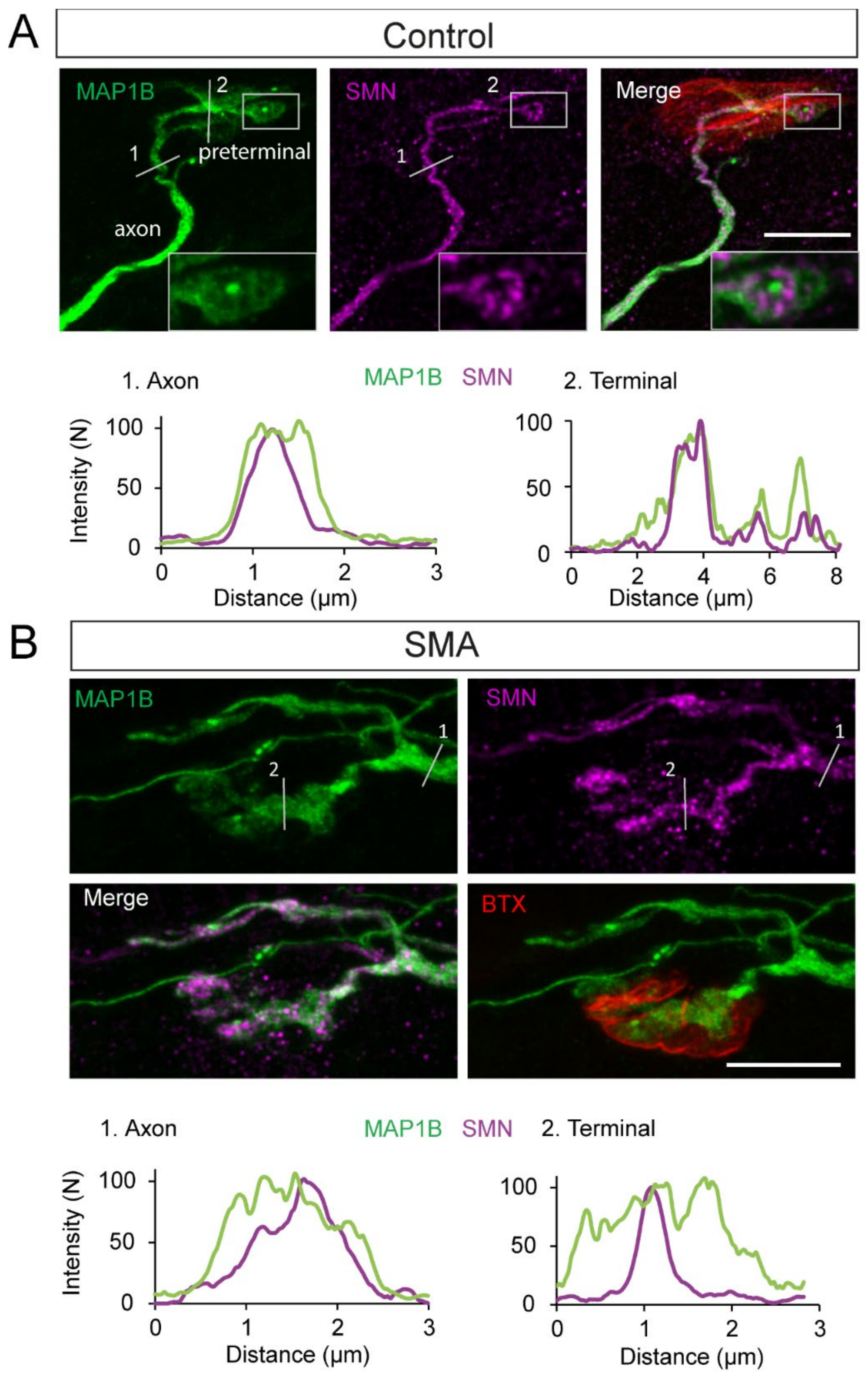
3.8. SMN Is Spatially Associated with MAP1B in SMA and Controls
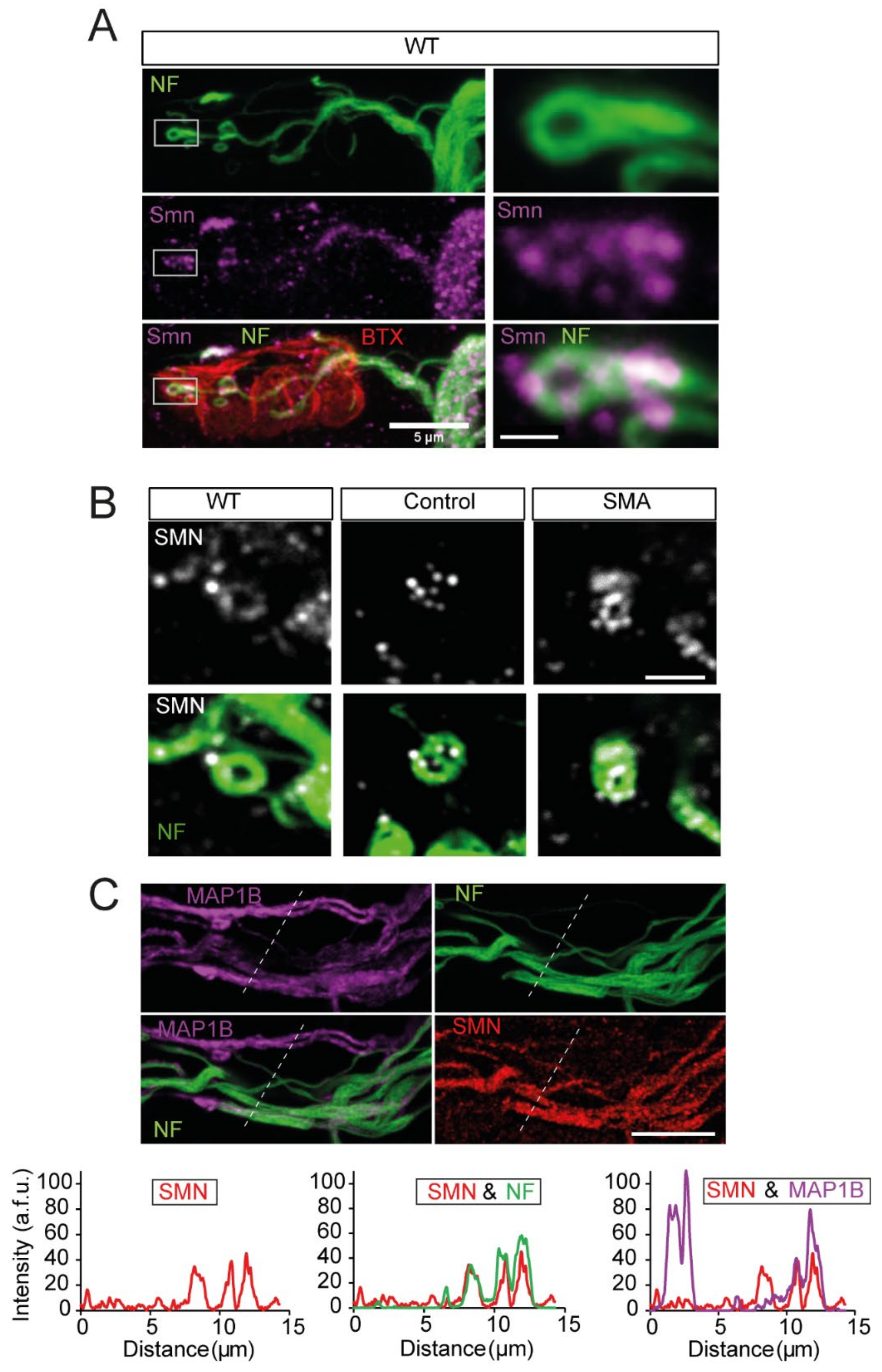
3.9. SMN and NFs Follow the Same Path at Motor Nerve Terminals
3.10. NFs and SMN Aggregate Together
4. Discussion
4.1. Physiological Age-Dependent SMN Expression in Nerve Terminals
4.2. The Physiological Role of SMN Granules at Motor Axons and Presynaptic Terminals
4.3. SMN Granules and Cytoskeleton
4.4. SMN Granules Accumulation Can Contribute to SMA Pathology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AChR—Acetylcholine receptor |
| ANOVA—Analysis of variance |
| BTX—Bungarotoxin |
| BTX—rho-Bungarotoxin-rhodamine |
| Crtl—Control |
| DIG—Digoxigenin |
| EDTA—Ethylenediaminetetraacetic acid |
| FISH—Fluorescent in situ hybridization |
| FL—Full length |
| FWHM—Full-width at half maximum |
| GAP43—Growth Associated Protein 43 |
| GAPDH—Glyceraldehyde 3-phosphatase dehydrogenase |
| hnRNP R—Heterogeneous nuclear ribonucleoprotein R |
| IHC—Immunohistochemistry |
| IMP1—IGF2 mRNA-binding protein 1 |
| LAL—Levator auris longus |
| LNA—Locked nucleic acid |
| mAb—Monoclonal antibody |
| MAP1B—Microtubules-associated protein 1B |
| mRNP—Messenger ribonucleoprotein |
| NF—Neurofilament |
| NMJ—Neuromuscular Junction |
| OA—Obliquus abdominis |
| pAb—Polyclonal antibody |
| PCR—Polimerase Chain-Reaction |
| PBS—Phosphate-Buffered Saline |
| PFA—Paraformaldehyde |
| PLP—Paraformaldehyde lysine phosphate |
| RIPA—Radioimmunoprecipitation assay buffer |
| RT—Room temperature |
| SDS—Sodium dodecyl sulfate |
| SMA—Spinal muscular atrophy |
| SMN—Survival motor neuron |
| snRNA—small nuclear RNA |
| snRNP—Small U-type ribonucleoprotein |
| SSC—Saline-sodium citrate |
| STED—Stimulated emission depletion |
| TBST—Tween 20 and Tris-buffered saline |
| TVA—Transversus abdominis |
| WB—Western blot |
| WT—Wild-type |
References
- Singh, R.N.; Howell, M.D.; Ottesen, E.W.; Singh, N.N. Diverse Role of Survival Motor Neuron Protein. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 299–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Rochette, C.F.; Gilbert, N.; Simard, L.R. SMN Gene Duplication and the Emergence of the SMN2 Gene Occurred in Distinct Hominids: SMN2 Is Unique to Homo sapiens. Hum. Genet. 2001, 108, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A Single Nucleotide in the SMN Gene Regulates Splicing and Is Responsible for Spinal Muscular Atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Gray, K.M.; Kaifer, K.A.; Baillat, D.; Wen, Y.; Bonacci, T.R.; Ebert, A.D.; Raimer, A.C.; Spring, A.M.; ten Have, S.; Glascock, J.J.; et al. Self-Oligomerization Regulates Stability of Survival Motor Neuron Protein Isoforms by Sequestering an SCF Slmb Degron. Mol. Biol. Cell 2018, 29, 96–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruss, O.J.; Meduri, R.; Schilling, M.; Fischer, U. UsnRNP Biogenesis: Mechanisms and Regulation. Chromosoma 2017, 126, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.; Englbrecht, C.; Chari, A. Biogenesis of Spliceosomal Small Nuclear Ribonucleoproteins. Wiley Interdiscip. Rev. RNA 2011, 2, 718–731. [Google Scholar] [CrossRef]
- Jablonka, S.; Bandilla, M.; Wiese, S.; Bühler, D.; Wirth, B.; Sendtner, M.; Fischer, U. Co-Regulation of Survival of Motor Neuron (SMN) Protein and Its Interactor SIP1 during Development and in Spinal Muscular Atrophy. Hum. Mol. Genet. 2001, 10, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Xing, L.; Rossoll, W.; Wichterle, H.; Singer, R.H.; Bassell, G.J. Multiprotein Complexes of the Survival of Motor Neuron Protein SMN with Gemins Traffic to Neuronal Processes and Growth Cones of Motor Neurons. J. Neurosci. 2006, 26, 8622–8632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrel, T.L.; McWhorter, M.L.; Workman, E.; Zhang, H.; Wolstencroft, E.C.; Lorson, C.L.; Bassell, G.J.; Burghes, A.H.M.; Beattie, C.E. Survival Motor Neuron Function in Motor Axons Is Independent of Functions Required for Small Nuclear Ribonucleoprotein Biogenesis. J. Neurosci. 2006, 26, 11014–11022. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Kröning, A.-K.; Ohndorf, U.-M.; Steegborn, C.; Jablonka, S.; Sendtner, M. Specific Interaction of Smn, the Spinal Muscular Atrophy Determining Gene Product, with HnRNP-R and Gry-Rbp/HnRNP-Q: A Role for Smn in RNA Processing in Motor Axons? Hum. Mol. Genet. 2002, 11, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kröning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the Spinal Muscular Atrophy-Determining Gene Product, Modulates Axon Growth and Localization of β-Actin mRNA in Growth Cones of Motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.G.; Morse, R.; Shaw, D.J.; McGinley, S.; Stebbings, H.; Young, P.J. SMN, Gemin2 and Gemin3 Associate with Beta-Actin mRNA in the Cytoplasm of Neuronal Cells in Vitro. J. Mol. Biol. 2010, 401, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Akten, B.; Kye, M.J.; Hao, L.T.; Wertz, M.H.; Singh, S.; Nie, D.; Huang, J.; Merianda, T.T.; Twiss, J.L.; Beattie, C.E.; et al. Interaction of Survival of Motor Neuron (SMN) and HuD Proteins with mRNA Cpg15 Rescues Motor Neuron Axonal Deficits. Proc. Natl. Acad. Sci. USA 2011, 108, 10337–10342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallini, C.; Zhang, H.; Su, Y.; Silani, V.; Singer, R.H.; Rossoll, W.; Bassell, G.J. The Survival of Motor Neuron (SMN) Protein Interacts with the mRNA-Binding Protein HuD and Regulates Localization of Poly(A) mRNA in Primary Motor Neuron Axons. J. Neurosci. 2011, 31, 3914–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubers, L.; Valderrama-Carvajal, H.; Laframboise, J.; Timbers, J.; Sanchez, G.; Côté, J. HuD Interacts with Survival Motor Neuron Protein and Can Rescue Spinal Muscular Atrophy-like Neuronal Defects. Hum. Mol. Genet. 2011, 20, 553–579. [Google Scholar] [CrossRef] [PubMed]
- Fallini, C.; Rouanet, J.P.; Donlin-Asp, P.G.; Guo, P.; Zhang, H.; Singer, R.H.; Rossoll, W.; Bassell, G.J. Dynamics of Survival of Motor Neuron (SMN) Protein Interaction with the mRNA-Binding Protein IMP1 Facilitates Its Trafficking into Motor Neuron Axons. Dev. Neurobiol. 2014, 74, 319–332. [Google Scholar] [CrossRef]
- Fallini, C.; Donlin-Asp, P.G.; Rouanet, J.P.; Bassell, G.J.; Rossoll, W. Deficiency of the Survival of Motor Neuron Protein Impairs mRNA Localization and Local Translation in the Growth Cone of Motor Neurons. J. Neurosci. 2016, 36, 3811–3820. [Google Scholar] [CrossRef] [Green Version]
- Donlin-Asp, P.G.; Fallini, C.; Campos, J.; Chou, C.C.; Merritt, M.E.; Phan, H.C.; Bassell, G.J.; Rossoll, W. The Survival of Motor Neuron Protein Acts as a Molecular Chaperone for MRNP Assembly. Cell Rep. 2017, 18, 1660–1673. [Google Scholar] [CrossRef] [PubMed]
- Saal, L.; Briese, M.; Kneitz, S.; Glinka, M.; Sendtner, M. Subcellular Transcriptome Alterations in a Cell Culture Model of Spinal Muscular Atrophy Point to Widespread Defects in Axonal Growth and Presynaptic Differentiation. RNA 2014, 20, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, G.; Dury, A.Y.; Murray, L.M.; Biondi, O.; Tadesse, H.; El Fatimy, R.; Kothary, R.; Charbonnier, F.; Khandjian, E.W.; Côté, J. A Novel Function for the Survival Motoneuron Protein as a Translational Regulator. Hum. Mol. Genet. 2013, 22, 668–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donlin-Asp, P.G.; Bassell, G.J.; Rossoll, W. A Role for the Survival of Motor Neuron Protein in MRNP Assembly and Transport. Curr. Opin. Neurobiol. 2016, 39, 53–61. [Google Scholar] [CrossRef]
- Fallini, C.; Bassell, G.J.; Rossoll, W. Spinal Muscular Atrophy: The Role of SMN in Axonal mRNA Regulation. Brain Res. 2012, 1462, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; do Carmo Gonçalves, I.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN Regulates Axonal Local Translation via MiR-183/MTOR Pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef] [PubMed]
- Gabanella, F.; Pisani, C.; Borreca, A.; Farioli-Vecchioli, S.; Ciotti, M.T.; Ingegnere, T.; Onori, A.; Ammassari-Teule, M.; Corbi, N.; Canu, N.; et al. SMN Affects Membrane Remodelling and Anchoring of the Protein Synthesis Machinery. J. Cell Sci. 2016, 129, 804–816. [Google Scholar]
- Rossoll, W.; Bassell, G.J. Spinal Muscular Atrophy and a Model for Survival of Motor Neuron Protein Function in Axonal Ribonucleoprotein Complexes. Results Probl. Cell Differ. 2009, 48, 289–326. [Google Scholar]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.R.; Zhang, H.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H.M. SMNDelta7, the Major Product of the Centromeric Survival Motor Neuron (SMN2) Gene, Extends Survival in Mice with Spinal Muscular Atrophy and Associates with Full-Length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Hsieh-Li, H.M.; Chang, J.G.; Jong, Y.J.; Wu, M.H.; Wang, N.M.; Tsai, C.H.; Li, H. A Mouse Model for Spinal Muscular Atrophy. Nat. Genet. 2000, 24, 66–70. [Google Scholar] [CrossRef]
- Dachs, E.; Hereu, M.; Piedrafita, L.; Casanovas, A.; Calderó, J.; Esquerda, J.E. Defective Neuromuscular Junction Organization and Postnatal Myogenesis in Mice with Severe Spinal Muscular Atrophy. J. Neuropathol. Exp. Neurol. 2011, 70, 444–461. [Google Scholar] [CrossRef] [Green Version]
- Tejero, R.; Lopez-Manzaneda, M.; Arumugam, S.; Tabares, L. Synaptotagmin-2, and -1, Linked to Neurotransmission Impairment and Vulnerability in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 4703–4716. [Google Scholar] [CrossRef]
- Jablonka, S.; Beck, M.; Lechner, B.D.; Mayer, C.; Sendtner, M. Defective Ca2+ Channel Clustering in Axon Terminals Disturbs Excitability in Motoneurons in Spinal Muscular Atrophy. J. Cell Biol. 2007, 179, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombert, B.; Sivadasan, R.; Simon, C.M.; Jablonka, S.; Sendtner, M. Presynaptic Localization of SMN and HnRNP R in Axon Terminals of Embryonic and Postnatal Mouse Motoneurons. PLoS ONE 2014, 9, e110846. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.P.; Rajendra, T.K.; Saieva, L.; Fuentes, J.L.; Pellizzoni, L.; Matera, A.G. SMN Complex Localizes to the Sarcomeric Z-Disc and Is a Proteolytic Target of Calpain. Hum. Mol. Genet. 2008, 17, 3399–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berciano, M.T.; Castillo-Iglesias, M.S.; Val-Bernal, J.F.; Lafarga, V.; Rodriguez-Rey, J.C.; Lafarga, M.; Tapia, O. Mislocalization of SMN from the I-Band and M-Band in Human Skeletal Myofibers in Spinal Muscular Atrophy Associates with Primary Structural Alterations of the Sarcomere. Cell Tissue Res. 2020, 381, 461–478. [Google Scholar] [CrossRef]
- Burlet, P.; Huber, C.; Bertrandy, S.; Ludosky, M.A.; Zwaenepoel, I.; Clermont, O.; Roume, J.; Delezoide, A.L.; Cartaud, J.; Munnich, A.; et al. The Distribution of SMN Protein Complex in Human Fetal Tissues and Its Alteration in Spinal Muscular Atrophy. Hum. Mol. Genet. 1998, 7, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Schrank, B.; Kralewski, M.; Rossoll, W.; Sendtner, M. Reduced Survival Motor Neuron (Smn) Gene Dose in Mice Leads to Motor Neuron Degeneration: An Animal Model for Spinal Muscular Atrophy Type III. Hum. Mol. Genet. 2000, 9, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, D.T.; Olson, R.J.; Sly, L.; Swanson, C.J.; Chung, B.; Naryshkin, N.; Narasimhan, J.; Bhattacharyya, A.; Mullenix, M.; Chen, K.S. Utility of Survival Motor Neuron ELISA for Spinal Muscular Atrophy Clinical and Preclinical Analyses. PLoS ONE 2011, 6, e24269. [Google Scholar] [CrossRef]
- Zaworski, P.; von Herrmann, K.M.; Taylor, S.; Sunshine, S.S.; McCarthy, K.; Risher, N.; Newcomb, T.; Weetall, M.; Prior, T.W.; Swoboda, K.J.; et al. SMN Protein Can Be Reliably Measured in Whole Blood with an Electrochemiluminescence (ECL) Immunoassay: Implications for Clinical Trials. PLoS ONE 2016, 11, e0150640. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Perenthaler, E.; Courtney, N.L.; Jordan, C.Y.; Shorrock, H.K.; Van Der Hoorn, D.; Huang, Y.T.; Murray, L.M.; Viero, G.; Gillingwater, T.H. Temporal and Tissue-Specific Variability of SMN Protein Levels in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2018, 27, 2851–2862. [Google Scholar] [CrossRef]
- Ramos, D.M.; D’Ydewalle, C.; Gabbeta, V.; Dakka, A.; Klein, S.K.; Norris, D.A.; Matson, J.; Taylor, S.J.; Zaworski, P.G.; Prior, T.W.; et al. Age-Dependent SMN Expression in Disease-Relevant Tissue and Implications for SMA Treatment. J. Clin. Investig. 2019, 129, 4817–4831. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, S.; Sendtner, M. Developmental Regulation of SMN Expression: Pathophysiological Implications and Perspectives for Therapy Development in Spinal Muscular Atrophy. Gene Ther. 2017, 24, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of Enhanced Survival Motoneuron Protein Imposed during Neuromuscular Junction Maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Wang, X.; Choe, D.W.; Polley, M.; Burnett, B.G.; Bosch-Marcé, M.; Griffin, J.W.; Rich, M.M.; Sumner, C.J. Impaired Synaptic Vesicle Release and Immaturity of Neuromuscular Junctions in Spinal Muscular Atrophy Mice. J. Neurosci. 2009, 29, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Park, G.H.; Maeno-Hikichi, Y.; Leykekhman, O.; Lutz, C.; Arkovitz, M.S.; Landmesser, L.T.; Monani, U.R. Reduced SMN Protein Impairs Maturation of the Neuromuscular Junctions in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 2552–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauria, F.; Bernabò, P.; Tebaldi, T.; Groen, E.J.N.; Perenthaler, E.; Maniscalco, F.; Rossi, A.; Donzel, D.; Clamer, M.; Marchioretto, M.; et al. SMN-Primed Ribosomes Modulate the Translation of Transcripts Related to Spinal Muscular Atrophy. Nat. Cell Biol. 2020, 22, 1239–1251. [Google Scholar] [CrossRef]
- Leung, K.-M.; van Horck, F.P.G.; Lin, A.C.; Allison, R.; Standart, N.; Holt, C.E. Asymmetrical β-Actin mRNA Translation in Growth Cones Mediates Attractive Turning to Netrin-1. Nat. Neurosci. 2006, 9, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Giuditta, A.; Jong, T.C.; Eyman, M.; Cefaliello, C.; Bruno, A.P.; Crispino, M. Local Gene Expression in Axons and Nerve Endings: The Glia-Neuron Unit. Physiol. Rev. 2008, 88, 515–555. [Google Scholar] [CrossRef] [Green Version]
- Swanger, S.A.; Bassell, G.J. Making and Breaking Synapses through Local mRNA Regulation. Curr. Opin. Genet. Dev. 2011, 21, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Di Liegro, C.M.; Schiera, G.; Di Liegro, I. Regulation of mRNA Transport, Localization and Translation in the Nervous System of Mammals (Review). Int. J. Mol. Med. 2014, 33, 747–762. [Google Scholar] [CrossRef] [Green Version]
- Shigeoka, T.; Jung, H.; Jung, J.; Turner-Bridger, B.; Ohk, J.; Lin, J.Q.; Amieux, P.S.; Holt, C.E. Dynamic Axonal Translation in Developing and Mature Visual Circuits. Cell 2016, 166, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir-Yilmaz, O.E.; Segal, R.A. There and Back Again: Coordinated Transcription, Translation and Transport in Axonal Survival and Regeneration. Curr. Opin. Neurobiol. 2016, 39, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lein, E.; Borm, L.E.; Linnarsson, S. The Promise of Spatial Transcriptomics for Neuroscience in the Era of Molecular Cell Typing. Science 2017, 358, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Briese, M.; Saal, L.; Appenzeller, S.; Moradi, M.; Baluapuri, A.; Sendtner, M. Whole Transcriptome Profiling Reveals the RNA Content of Motor Axons. Nucleic Acids Res. 2015, 44, e33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cajigas, I.J.; Tushev, G.; Will, T.J.; tom Dieck, S.; Fuerst, N.; Schuman, E.M. The Local Transcriptome in the Synaptic Neuropil Revealed by Deep Sequencing and High-Resolution Imaging. Neuron 2012, 74, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Sivadasan, R.; Saal, L.; Lüningschrör, P.; Dombert, B.; Rathod, R.J.; Dieterich, D.C.; Blum, R.; Sendtner, M. Send Differential Roles of α, β-, and γ-Actin in Axon Growth. J. Cell Biol. 2017, 216, 793–814. [Google Scholar] [CrossRef] [Green Version]
- Bernabò, P.; Tebaldi, T.; Groen, E.J.N.; Lane, F.M.; Perenthaler, E.; Mattedi, F.; Newbery, H.J.; Zhou, H.; Zuccotti, P.; Potrich, V.; et al. In Vivo Translatome Profiling in Spinal Muscular Atrophy Reveals a Role for SMN Protein in Ribosome Biology. Cell Rep. 2017, 21, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.; Palay, S.L.; Def, H. The Fine Structure of the Nervous System: The Neurons and Supporting Cells, 2nd revised ed.; Saunders: Philadelphia, PA, USA, 1976. [Google Scholar]
- Court, F.A.; Álvarez, J. Slow Axoplasmic Transport under Scrutiny. Biol. Res. 2011, 44, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.M.; Comley, L.H.; Thomson, D.; Parkinson, N.; Talbot, K.; Gillingwater, T.H. Selective Vulnerability of Motor Neurons and Dissociation of Pre- and Post-Synaptic Pathology at the Neuromuscular Junction in Mouse Models of Spinal Muscular Atrophy. Hum. Mol. Genet. 2008, 17, 949–962. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.M.; Lee, S.; Bäumer, D.; Parson, S.H.; Talbot, K.; Gillingwater, T.H. Pre-Symptomatic Development of Lower Motor Neuron Connectivity in a Mouse Model of Severe Spinal Muscular Atrophy. Hum. Mol. Genet. 2010, 19, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, R.; Casañas, J.J.; Torres-Benito, L.; Cano, R.; Tabares, L. Altered Intracellular Ca2+ Homeostasis in Nerve Terminals of Severe Spinal Muscular Atrophy Mice. J. Neurosci. 2010, 30, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Torres-Benito, L.; Neher, M.F.; Cano, R.; Ruiz, R.; Tabares, L. SMN Requirement for Synaptic Vesicle, Active Zone and Microtubule Postnatal Organization in Motor Nerve Terminals. PLoS ONE 2011, 6, e26164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Benito, L.; Ruiz, R.; Tabares, L. Synaptic Defects in Spinal Muscular Atrophy Animal Models. Dev. Neurobiol. 2012, 72, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Giesemann, T.; Rathke-Hartlieb, S.; Rothkegel, M.; Bartsch, J.W.; Buchmeier, S.; Jockusch, B.M.; Jockusch, H. A Role for Polyproline Motifs in the Spinal Muscular Atrophy Protein SMN. Profilins Bind to and Colocalize with SMN in Nuclear Gems. J. Biol. Chem. 1999, 274, 37908–37914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nölle, A.; Zeug, A.; van Bergeijk, J.; Tönges, L.; Gerhard, R.; Brinkmann, H.; Al rayes, S.; Hensel, N.; Schill, Y.; Apkhazava, D.; et al. The Spinal Muscular Atrophy Disease Protein SMN Is Linked to the Rho-Kinase Pathway via Profilin. Hum. Mol. Genet. 2011, 20, 4865–4878. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Lambrechts, A.; Hao, L.T.; Le, T.T.; Sewry, C.A.; Ampe, C.; Burghes, A.H.M.; Morris, G.E. A Role for Complexes of Survival of Motor Neurons (SMN) Protein with Gemins and Profilin in Neurite-like Cytoplasmic Extensions of Cultured Nerve Cells. Exp. Cell Res. 2005, 309, 185–197. [Google Scholar] [CrossRef]
- Wen, H.L.; Lin, Y.T.; Ting, C.H.; Lin-Chao, S.; Li, H.; Hsieh-Li, H.M. Stathmin, a Microtubule-Destabilizing Protein, Is Dysregulated in Spinal Muscular Atrophy. Hum. Mol. Genet. 2010, 19, 1766–1778. [Google Scholar] [CrossRef] [Green Version]
- Villarroel-Campos, D.; Gonzalez-Billault, C. The MAP1B Case: An Old MAP That Is New Again. Dev. Neurobiol. 2014, 74, 953–971. [Google Scholar] [CrossRef]
- Godena, V.K.; Romano, G.; Romano, M.; Appocher, C.; Klima, R.; Buratti, E.; Baralle, F.E.; Feiguin, F. TDP-43 Regulates Drosophila Neuromuscular Junctions Growth by Modulating Futsch/MAP1B Levels and Synaptic Microtubules Organization. PLoS ONE 2011, 6, e17808. [Google Scholar] [CrossRef] [Green Version]
- Bora, G.; Hensel, N.; Rademacher, S.; Koyunoğlu, D.; Sunguroğlu, M.; Aksu-Mengeş, E.; Balcı-Hayta, B.; Claus, P.; Erdem-Yurter, H. Microtubule-Associated Protein 1B Dysregulates Microtubule Dynamics and Neuronal Mitochondrial Transport in Spinal Muscular Atrophy. Hum. Mol. Genet. 2021, 29, 3935–3944. [Google Scholar] [CrossRef]
- Pagliardini, S.; Giavazzi, A.; Setola, V.; Lizier, C.; Di Luca, M.; DeBiasi, S.; Battaglia, G.S. Subcellular Localization and Axonal Transport of the Survival Motor Neuron (SMN) Protein in the Developing Rat Spinal Cord. Hum. Mol. Genet. 2000, 9, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. High-Efficiency Transfection of Cultured Primary Motor Neurons to Study Protein Localization, Trafficking, and Function. Mol. Neurodegener. 2010, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifuentes-Diaz, C.; Nicole, S.; Velasco, M.E.; Borra-Cebrian, C.; Panozzo, C.; Frugier, T.; Millet, G.; Roblot, N.; Joshi, V.; Melki, J. Neurofilament Accumulation at the Motor Endplate and Lack of Axonal Sprouting in a Spinal Muscular Atrophy Mouse Model. Hum. Mol. Genet. 2002, 11, 1439–1447. [Google Scholar] [CrossRef]
- Price, P.L.; Morderer, D.; Rossoll, W. RNP Assembly Defects in Spinal Muscular Atrophy. Adv. Neurobiol. 2018, 20, 143–171. [Google Scholar] [PubMed]
- Khalil, B.; Morderer, D.; Price, P.L.; Liu, F.; Rossoll, W. mRNP Assembly, Axonal Transport, and Local Translation in Neurodegenerative Diseases. Brain Res. 2018, 1693, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.-M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.T.; Burghes, A.H.M.; Androphy, E.J. SMN Oligomerization Defect Correlates with Spinal Muscular Atrophy Severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The Spinal Muscular Atrophy Disease Gene Product, SMN, and Its Associated Protein SIP1 Are in a Complex with Spliceosomal SnRNP Proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN Mutants of Spinal Muscular Atrophy Patients Are Defective in Binding to SnRNP Proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Dreyfuss, G. A Degron Created by SMN2 Exon 7 Skipping Is a Principal Contributor to Spinal Muscular Atrophy Severity. Genes Dev. 2010, 24, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praveen, K.; Wen, Y.; Gray, K.M.; Noto, J.J.; Patlolla, A.R.; Van Duyne, G.D.; Matera, A.G. SMA-Causing Missense Mutations in Survival Motor Neuron (Smn) Display a Wide Range of Phenotypes When Modeled in Drosophila. PLoS Genet. 2014, 10, e1004489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.; Wen, Y.; Ninan, N.S.; Raimer, A.C.; Sharp, R.; Spring, A.M.; Sarachan, K.L.; Johnson, M.C.; Van Duyne, G.D.; Matera, A.G. Assembly of Higher-Order SMN Oligomers Is Essential for Metazoan Viability and Requires an Exposed Structural Motif Present in the YG Zipper Dimer. Nucleic Acids Res. 2021, 49, 7644–7664. [Google Scholar] [CrossRef]
- Li, D.K.; Tisdale, S.; Lotti, F.; Pellizzoni, L. SMN Control of RNP Assembly: From Post-Transcriptional Gene Regulation to Motor Neuron Disease. Semin. Cell Dev. Biol. 2014, 32, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Zhou, J. Survival Motor Neuron Protein Facilitates Assembly of Stress Granules. FEBS Lett. 2004, 572, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the Spinal Muscular Atrophy Phenotype in a Mouse Model by Early Postnatal Delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowes, L.P.; Alfano, L.N.; Arnold, W.D.; Shell, R.; Prior, T.W.; McColly, M.; Lehman, K.J.; Church, K.; Sproule, D.M.; Nagendran, S.; et al. Impact of Age and Motor Function in a Phase 1/2A Study of Infants with SMA Type 1 Receiving Single-Dose Gene Replacement Therapy. Pediatr. Neurol. 2019, 98, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranello, G.; Darras, B.T.; Day, J.W.; Deconinck, N.; Klein, A.; Masson, R.; Mercuri, E.; Rose, K.; El-Khairi, M.; Gerber, M.; et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N. Engl. J. Med. 2021, 384, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Van Alstyne, M.; Tattoli, I.; Delestrée, N.; Recinos, Y.; Workman, E.; Shihabuddin, L.S.; Zhang, C.; Mentis, G.Z.; Pellizzoni, L. Gain of Toxic Function by Long-Term AAV9-Mediated SMN Overexpression in the Sensorimotor Circuit. Nat. Neurosci. 2021, 24, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.J.; Willis, D.E. To the End of the Line: Axonal mRNA Transport and Local Translation in Health and Neurodegenerative Disease. Dev. Neurobiol. 2018, 78, 209–220. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Espin, J.; Gatius, A.; Armengol, J.Á.; Arumugam, S.; Moradi, M.; Sendtner, M.; Calderó, J.; Tabares, L. SMN Is Physiologically Downregulated at Wild-Type Motor Nerve Terminals but Aggregates Together with Neurofilaments in SMA Mouse Models. Biomolecules 2022, 12, 1524. https://doi.org/10.3390/biom12101524
Franco-Espin J, Gatius A, Armengol JÁ, Arumugam S, Moradi M, Sendtner M, Calderó J, Tabares L. SMN Is Physiologically Downregulated at Wild-Type Motor Nerve Terminals but Aggregates Together with Neurofilaments in SMA Mouse Models. Biomolecules. 2022; 12(10):1524. https://doi.org/10.3390/biom12101524
Chicago/Turabian StyleFranco-Espin, Julio, Alaó Gatius, José Ángel Armengol, Saravanan Arumugam, Mehri Moradi, Michael Sendtner, Jordi Calderó, and Lucia Tabares. 2022. "SMN Is Physiologically Downregulated at Wild-Type Motor Nerve Terminals but Aggregates Together with Neurofilaments in SMA Mouse Models" Biomolecules 12, no. 10: 1524. https://doi.org/10.3390/biom12101524