
Copper-Dependent Kinases and Their Role in Cancer Inception, Progression and Metastasis

Abstract
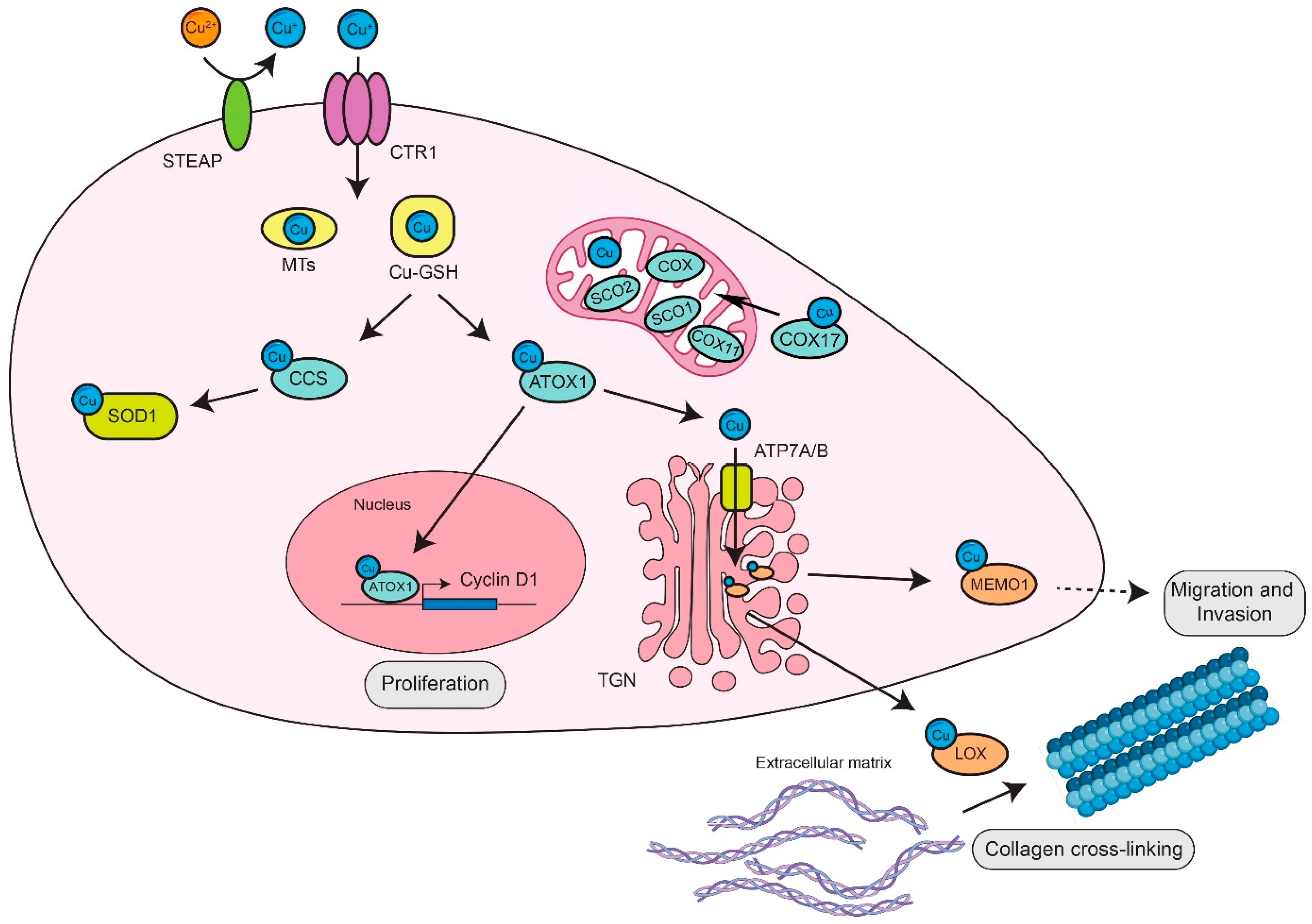
:1. Systemic and Cellular Copper Homeostasis
2. Role of Copper Homeostasis and Copper-Binding Proteins in Cancer
3. Copper-Dependent Kinases and Their Pathological Roles in Cancer
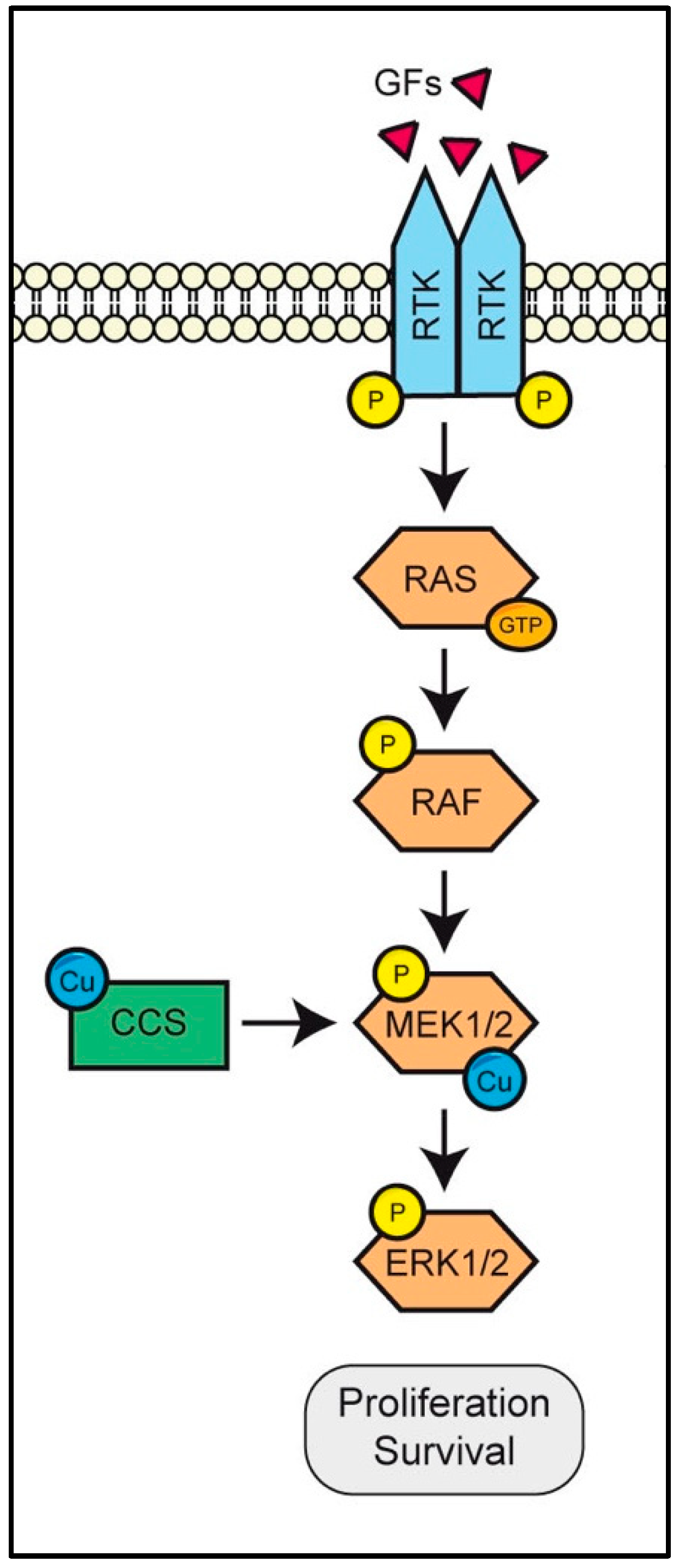
3.1. MEK Is a Copper-Binding Kinase That Triggers the RAS/RAF/MEK/ERK Pathway
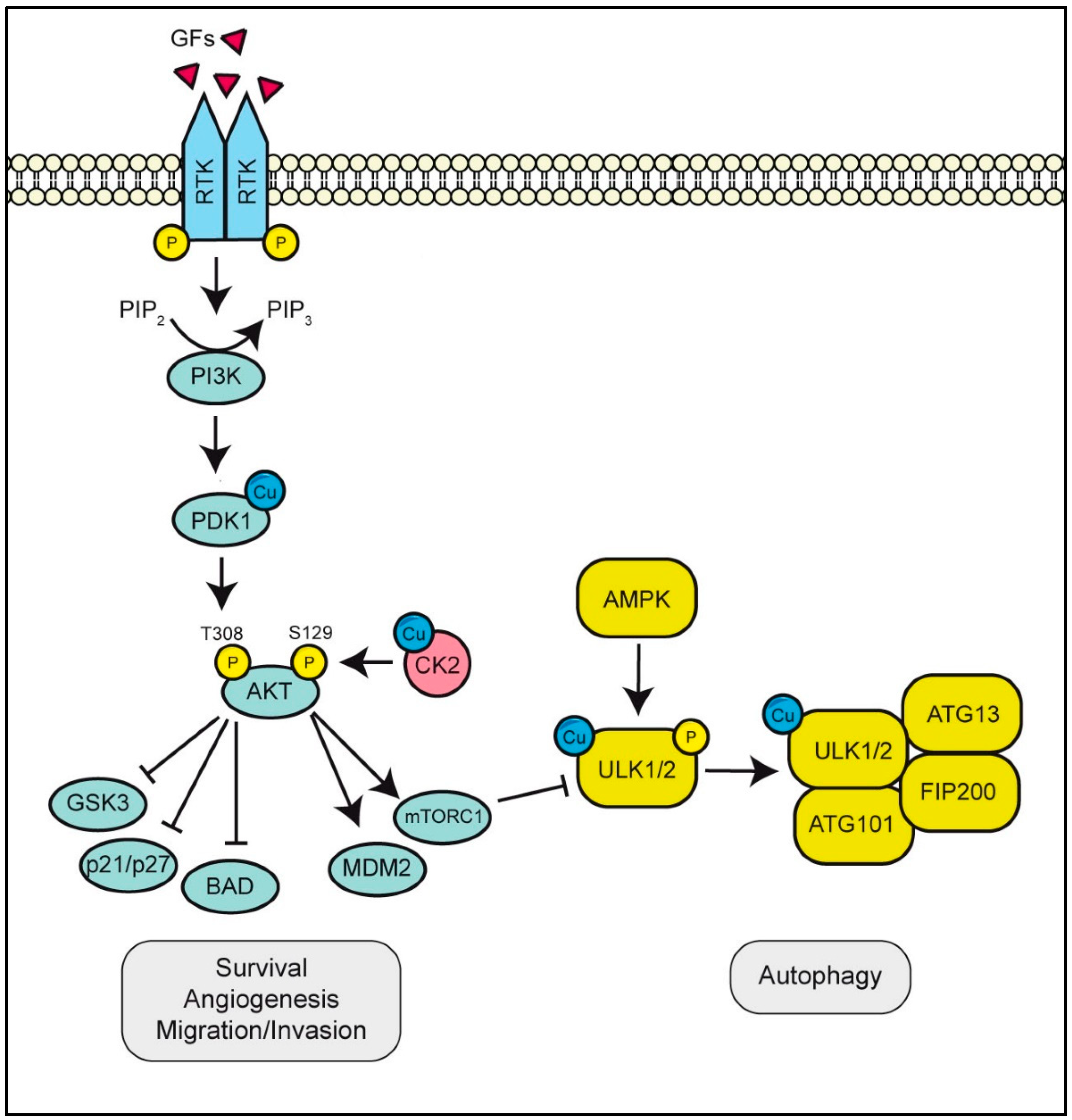
3.2. ULK1/2 Is A Copper-Binding Kinase Essential for Autophagy Initiation
3.3. PDK1 or CK2 Binds Cu Activating the PI3K/AKT Pathway
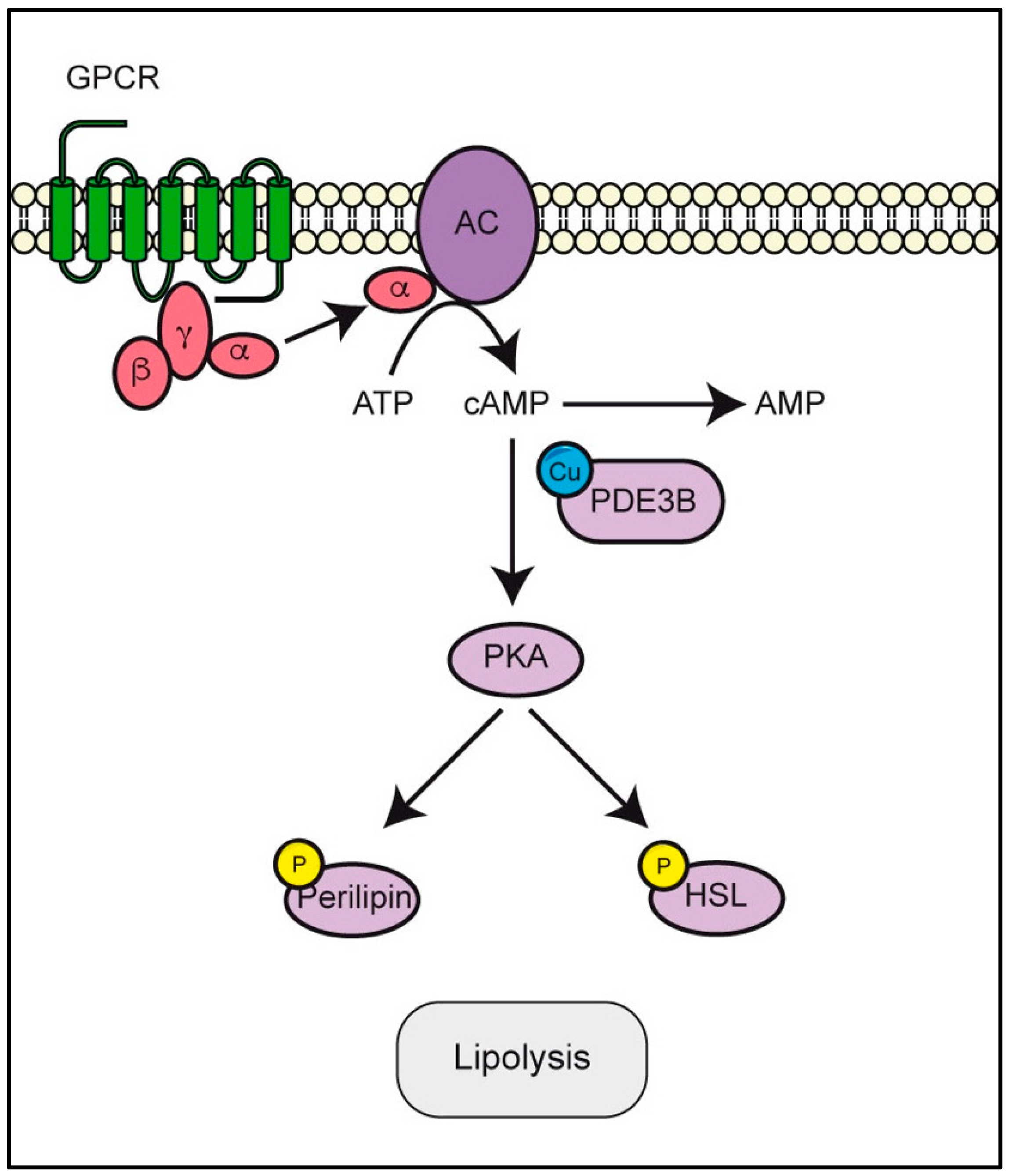
3.4. PDE3B Requires Copper to Modulate cAMP/PKA Pathway
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP-1/2 | adaptor complex |
| ATOX1/HAH1 | Antioxidant-protein 1 |
| ATP7A/B | P-type ATPases |
| BCS | bathocuproine disulfonate |
| cAMP | cyclic adenosine monophosphate |
| CCS | copper chaperone for superoxide dismutase |
| CIP4 | CDC24 interacting protein 4 |
| CK2 | casein kinase 2 |
| COX | cytochrome c oxidase |
| COX17 | cytochrome c oxidase copper chaperone |
| CR | conserved regions |
| CTD | C-terminal interacting domain |
| ECM | extracellular matrix |
| EMT | epithelial to mesenchymal transition |
| FAK | focal adhesion kinase |
| GAP | GTPases-activating proteins |
| GPCRs | G-protein coupled receptors |
| GSH | tripeptide glutathione |
| GSK3 | glycogen synthase kinase 3 |
| HIF-1 | hypoxia inducible factor-1 |
| HREs | hypoxia responsive elements |
| IRS1 | insulin-like receptor substrate 1 |
| KD | kinase domain |
| LOX | lysyl oxidase |
| LOXL | lysyl oxidase-like |
| MEK1/2 | mitogen-activated protein kinase kinase 1/2 |
| MEMO1 | mediator of ErbB2-driven cell motility |
| MT1-4 | metallothionein family |
| NTA | N-terminal acidic motif |
| PDK1 | phosphoinositide-dependent kinase-1 |
| PI3K | phosphoinositide 3-kinases |
| PIP2 | phosphatidylinositol-4,5-bisphosphate |
| PIP3 | phosphatidylinositol-3,4,5-trisphosphate |
| PKA | protein kinase A |
| RAS | rat sarcoma |
| RBD | RAS-binding domain |
| RTK | receptor tyrosine kinases |
| SOD1/3 | Cu/Zn superoxide dismutase 1 and 3 |
| Sp1 | zinc-finger transcription factor Specificity Protein 1 |
| TEPA or TRIEN | triethylene tetramine |
| TGN | trans-Golgi network |
| TTM | tetrathiomolybdate |
| ULK1/2 | autophagic proteins Unc-51-like kinase 1 and 2 |
References
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper Biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.; Barrientos, A. Mitochondrial Copper Metabolism and Delivery to Cytochrome C Oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangold, J.B.; Klinman, J.P. Mechanism-Based Inactivation of Dopamine β-Monooxygenase by β-Chlorophenethylamine. J. Biol. Chem. 1984, 259, 7772–7779. [Google Scholar] [CrossRef]
- Ash, D.E.; Papadopoulos, N.J.; Colombo, G.; Villafranca, J.J. Kinetic and Spectroscopic Studies of the Interaction of Copper with Dopamine β-Hydroxylase. J. Biol. Chem. 1984, 259, 3395–3398. [Google Scholar] [CrossRef]
- Roberts, E.A.; Sarkar, B. Liver as a Key Organ in the Supply, Storage, and Excretion of Copper. Am. J. Clin. Nutr. 2008, 88, 851–854. [Google Scholar] [CrossRef] [Green Version]
- Mehari, T.F.; Greene, L.; Duncan, A.L.; Olawale Fakayode, S. Trace and Macro Elements Concentrations in Selected Fresh Fruits, Vegetables, Herbs, and Processed Foods in North Carolina, USA. J. Environ. Prot. 2015, 6, 573–583. [Google Scholar] [CrossRef]
- Linder, M.; Hazegh-Azam, M. Copper biochemistry and molecular biology. Am. J. Clin. Nutr. 1996, 63, 797S–811S. [Google Scholar] [CrossRef]
- Linder, M.C. Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review. Int. J. Mol. Sci. 2020, 21, 4932. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper Metabolism as a Unique Vulnerability in Cancer. Biochim. Biophys. Acta. Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Puig, S.; Thiele, D.J. Molecular Mechanisms of Copper Uptake and Distribution. Curr. Opin. Chem. Biol. 2002, 6, 171–180. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Zhou, B.; Cosco, D.; Gitschier, J. The Copper Transporter CTR1 Provides an Essential Function in Mammalian Embryonic Development. Proc. Natl. Acad. Sci. USA 2001, 98, 6836–6841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap Proteins Are Metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Scarl, R.T.; Lawrence, C.M.; Gordon, H.M.; Nunemaker, C.S. STEAP4: Its Emerging Role in Metabolism and Homeostasis of Cellular Iron and Copper. J. Endocrinol. 2017, 234, R123–R134. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity Protein 1 (Sp1) Oscillation is Involved in Copper Homeostasis Maintenance by Regulating Human High-Affinity Copper Transporter 1 Expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Petris, M.J.; Smith, K.; Lee, J.; Thiele, D.J. Copper-Stimulated Endocytosis and Degradation of the Human Copper Transporter, HCtr1. J. Biol. Chem. 2003, 278, 9639–9646. [Google Scholar] [CrossRef] [Green Version]
- Curnock, R.; Cullen, P.J. Mammalian Copper Homeostasis Requires Retromer-Dependent Recycling of the High-Affinity Copper Transporter 1. J. Cell Sci. 2020, 133, jcs249201. [Google Scholar] [CrossRef]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. O-Linked Glycosylation at Threonine 27 Protects the Copper Transporter HCTR1 from Proteolytic Cleavage in Mammalian Cells. J. Biol. Chem. 2007, 282, 20376–20387. [Google Scholar] [CrossRef] [Green Version]
- Öhrvik, H.; Nose, Y.; Wood, L.K.; Kim, B.E.; Gleber, S.C.; Ralle, M.; Thiele, D.J. Ctr2 Regulates Biogenesis of a Cleaved Form of Mammalian Ctr1 Metal Transporter Lacking the Copper- and Cisplatin-Binding Ecto-Domain. Proc. Natl. Acad. Sci. USA 2013, 110, E4279–E4288. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. [1] Role of Free Radicals and Catalytic Metal Ions in Human Disease: An Overview. Methods Enzymol. 1990, 186, 1–85. [Google Scholar] [CrossRef]
- Manzl, C.; Enrich, J.; Ebner, H.; Dallinger, R.; Krumschnabel, G. Copper-Induced Formation of Reactive Oxygen Species Causes Cell Death and Disruption of Calcium Homeostasis in Trout Hepatocytes. Toxicology 2004, 196, 57–64. [Google Scholar] [CrossRef]
- Gudekar, N.; Shanbhag, V.; Wang, Y.; Ralle, M.; Weisman, G.A.; Petris, M.J. Metallothioneins Regulate ATP7A Trafficking and Control Cell Viability during Copper Deficiency and Excess. Sci. Rep. 2020, 10, 7856. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular Glutathione Plays a Key Role in Copper Uptake Mediated by Human Copper Transporter 1. Am. J. Physiol. Cell Physiol. 2013, 304, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Calvo, J.; Jung, H.; Meloni, G. Copper Metallothioneins. IUBMB Life 2017, 69, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, S.; Lee, J.; Lau, M.; Thiele, D.J. Biochemical and Genetic Analyses of Yeast and Human High Affinity Copper Transporters Suggest a Conserved Mechanism for Copper Uptake. J. Biol. Chem. 2002, 277, 26021–26030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casareno, R.L.B.; Waggoner, D.; Gitlin, J.D. The Copper Chaperone CCS Directly Interacts with Copper/Zinc Superoxide Dismutase. J. Biol. Chem. 1998, 273, 23625–23628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertinato, J.; L’Abbé, M.R. Copper Modulates the Degradation of Copper Chaperone for Cu, Zn Superoxide Dismutase by the 26 S Proteosome. J. Biol. Chem. 2003, 278, 35071–35078. [Google Scholar] [CrossRef] [Green Version]
- Arciello, M.; Capo, C.R.; Annibale, S.D.; Cozzolino, M.; Ferri, A.; Carri, M.T.; Rossi, L. Copper Depletion Increases the Mitochondrial-Associated SOD1 in Neuronal Cells. BioMetals 2011, 24, 269–278. [Google Scholar] [CrossRef]
- Barry, A.N.; Shinde, U.; Lutsenko, S. Structural Organization of Human Cu-Transporting ATPases: Learning from Building Blocks. J. Biol. Inorg. Chem. 2010, 15, 47–59. [Google Scholar] [CrossRef]
- Gourdon, P.; Sitsel, O.; Karlsen, J.L.; Møller, L.B.; Nissen, P. Structural Models of the Human Copper P-Type ATPases ATP7A and ATP7B. Biol. Chem. 2012, 393, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Kaler, S.G. ATP7A-Related Copper Transport Diseasesg-Emerging Concepts and Future Trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef]
- Bartee, M.Y.; Lutsenko, S. Hepatic Copper-Transporting ATPase ATP7B: Function and Inactivation at the Molecular and Cellular Level. BioMetals 2007, 20, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, C.; Zlatic, S.A.; Wallin, M.; Vrailas-Mortimer, A.; Fahrni, C.J.; Faundez, V. Trafficking Mechanisms of P-Type ATPase Copper Transporters. Curr. Opin. Cell Biol. 2019, 59, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Ackerman, C.M.; Carroll, E.C.; Jia, S.; Hoagland, A.; Chan, J.; Thai, B.; Liu, C.S.; Isacoff, E.Y.; Chang, C.J. Copper Regulates Rest-Activity Cycles through the Locus Coeruleus-Norepinephrine System. Nat. Chem. Biol. 2018, 14, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, S.; Kruse, R.L.; Hamilton, J.P.; Lutsenko, S. Wilson Disease: Update on Pathophysiology and Treatment. Front. Cell Dev. Biol. 2022, 10, 871877. [Google Scholar] [CrossRef] [PubMed]
- Horn, N.; Møller, L.B.; Nurchi, V.M.; Aaseth, J. Chelating Principles in Menkes and Wilson Diseases: Choosing the Right Compounds in the Right Combinations at the Right Time. J. Inorg. Biochem. 2019, 190, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, R.; Spinelli, F.; Contino, M.; Colabufo, N.A. The Pivotal Role of Copper in Neurodegeneration: A New Strategy for the Therapy of Neurodegenerative Disorders. Mol. Pharm. 2018, 15, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.J.; Bartnikas, T.B.; Gitlin, J.D. The Role of Copper in Neurodegenerative Disease. Neurobiol. Dis. 1999, 6, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Siotto, M.; Arciello, M.; Rossi, L. Non-Ceruloplasmin Bound Copper and: ATP7B Gene Variants in Alzheimer’s Disease. Metallomics 2016, 8, 863–873. [Google Scholar] [CrossRef]
- Zuily, L.; Lahrach, N.; Fassler, R.; Genest, O.; Faller, P.; Sénèque, O.; Denis, Y.; Castanié-Cornet, M.P.; Genevaux, P.; Jakob, U.; et al. Copper Induces Protein Aggregation, a Toxic Process Compensated by Molecular Chaperones. MBio 2022, 13, e03251-21. [Google Scholar] [CrossRef]
- Raju, K.S.; Alessandri, G.; Ziche, M.; Gullino, P.M. Ceruloplasmin, Copper Ions, and Angiogenesis. J. Natl. Cancer Inst. 1982, 69, 1183–1188. [Google Scholar]
- Lane, T.F.; Luisa Iruela-Arispe, M.; John, R.S.; Sage, E.H. SPARC is a source of copper-binding peptides that stimulate angiogenesis. J. Cell Biol. 1994, 125, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Soncin, F.; Guitton, J.D.; Cartwright, T.; Badet, J. Interaction of Human Angiogenin with Copper Modulates Angiogenin Binding to Endothelial Cells. Biochem. Biophys. Res. Commun. 1997, 236, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landriscina, M.; Bagalá, C.; Mandinova, A.; Soldi, R.; Micucci, I.; Bellum, S.; Prudovsky, I.; Maciag, T. Copper Induces the Assembly of a Multiprotein Aggregate Implicated in the Release of Fibroblast Growth Factor 1 in Response to Stress. J. Biol. Chem. 2001, 276, 25549–25557. [Google Scholar] [CrossRef] [Green Version]
- Juarez, J.C.; Betancourt, O., Jr.; Pirie-Shepherd, S.R.; Guan, X.; Price, M.L.; Shaw, D.E.; Mazar, A.P.; Doñate, F. Copper Binding by Tetrathiomolybdate Attenuates Angiogenesis and Tumor Cell Proliferation through the Inhibition of Superoxide Dismutase 1. Clin. Cancer Res. 2006, 12, 4974–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margalioth, E.J.; Schenker, J.G.; Chevion, M. Copper and Zinc Levels in Normal and Malignant Tissues. Cancer 1983, 52, 868–872. [Google Scholar] [CrossRef]
- Coates, R.J.; Weiss, N.S.; Daling, J.R.; Rettmer, R.L.; Warnick, G.R. Cancer Risk in Relation to Serum Copper Levels. Cancer Res. 1989, 49, 4353–4356. [Google Scholar] [PubMed]
- Díez, M.; Cerdà, F.J.; Arroyo, M.; Balibrea, J.L. Use of the copper/zinc ratio in the diagnosis of lung cancer. Cancer 1989, 63, 726–730. [Google Scholar] [CrossRef]
- Oyama, T.; Matsuno, K.; Kawamoto, T.; Mitsudomi, T.; Shirakusa, T.; Kodama, Y. Efficiency of Serum Copper/Zinc Ratio for Differential Diagnosis of Patients with and without Lung Cancer. Biol. Trace Elem. Res. 1994, 42, 115–127. [Google Scholar] [CrossRef]
- Jin, Y.; Zhang, C.; Xu, H.; Xue, S.; Wang, Y.; Hou, Y.; Kong, Y.; Xu, Y. Combined Effects of Serum Trace Metals and Polymorphisms of CYP1A1 or GSTM1 on Non-Small Cell Lung Cancer: A Hospital Based Case-Control Study in China. Cancer Epidemiol. 2011, 35, 182–187. [Google Scholar] [CrossRef]
- Brem, S.S.; Zagzag, D.; Tsanaclis, A.M.; Gately, S.; Elkouby, M.P.; Brien, S.E. Inhibition of Angiogenesis and Tumor Growth in the Brain. Suppression of Endothelial Cell Turnover by Penicillamine and the Depletion of Copper, an Angiogenic Cofactor. Am. J. Pathol. 1990, 137, 1121–1142. [Google Scholar]
- Brem, S.; Tsanaclis, A.M.; Zagzag, D. Anticopper Treatment Inhibits Pseudopodial Protrusion and the Invasive Spread of 9L Gliosarcoma Cells in the Rat Brain. Neurosurgery 1990, 26, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of Metastatic Cancer with Tetrathiomolybdate, an Anticopper, Antiangiogenic Agent: Phase I Study1. Clin. Cancer Res. 2000, 6, 1–10. [Google Scholar] [PubMed]
- Cox, C.; Teknos, T.N.; Barrios, M.; Brewer, G.J.; Dick, R.D.; Merajver, S.D. The Role of Copper Suppression as an Antiangiogenic Strategy in Head and Neck Squamous Cell Carcinoma. Laryngoscope 2001, 111, 696–701. [Google Scholar] [CrossRef]
- Wen Kuo, H.; Fan Chen, S.; Ching, C.W.; Ren Chen, D.; Hung Lee, J. Serum and Tissue Trace Elements in Patients with Breast Cancer in Taiwan. Biol. Trace Elem. Res. 2002, 89, 1–11. [Google Scholar]
- Feng, Y.; Zeng, J.-W.; Ma, Q.; Zhang, S.; Tang, J.; Feng, J.-F. Serum Copper and Zinc Levels in Breast Cancer: A Meta-Analysis. J. Trace Elem. Med. Biol. 2020, 62, 126629. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jiang, M.; Jing, H.; Sheng, W.; Wang, X.; Han, J.; Wang, L. Analysis of Serum Levels of 15 Trace Elements in Breast Cancer Patients in Shandong, China. Environ. Sci. Pollut. Res. 2014, 22, 7930–7935. [Google Scholar] [CrossRef]
- Adeoti, M.L.; Oguntola, A.S.; Akanni, E.O.; Agodirin, O.S.; Oyeyemi, G.M. Trace Elements; Copper, Zinc and Selenium, in Breast Cancer Afflicted Female Patients in LAUTECH Osogbo, Nigeria. Indian J. Cancer 2015, 52, 106–109. [Google Scholar] [CrossRef]
- Pavithra, V.; Sathisha, T.G.; Kasturi, K.; Mallika, D.S.; Amos, S.J.; Ragunatha, S. Serum Levels of Metal Ions in Female Patients with Breast Cancer. J. Clin. Diagn. Res. 2015, 9, BC25. [Google Scholar] [CrossRef]
- Nayak, S.B.; Bhat, V.R.; Upadhyay, D.; Udupa, S.L. Copper and Ceruloplasmin Status in Serum of Prostate and Colon Cancer Patients. Indian, J. Physiol. Pharmacol. 2003, 47, 108–110. [Google Scholar]
- Stepien, M.; Jenab, M.; Freisling, H.; Becker, N.-P.; Czuban, M.; Tjønneland, A.; Olsen, A.; Overvad, K.; Boutron-Ruault, M.-C.; Mancini, F.R.; et al. Bueno-de-Mesquita 15. José María Huerta 2017, 38, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Sohrabi, M.; Gholami, A.; Azar, M.H.; Yaghoobi, M.; Shahi, M.M.; Shirmardi, S.; Nikkhah, M.; Kohi, Z.; Salehpour, D.; Khoonsari, M.R.; et al. Trace Element and Heavy Metal Levels in Colorectal Cancer: Comparison Between Cancerous and Non-cancerous Tissues. Biol. Trace Element Res. 2017, 183, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Celauro, E.; Hildesjö, C.; Feizi, A.; Stål, O.; Fierro-González, J.C.; Wittung-Stafshede, P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 2017, 9, 112–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, S.S.; Karjodkar, F.R. Circulating Immune Complexes and Trace Elements (Copper, Iron and Selenium) as Markers in Oral Precancer and Cancer: A Randomised, Controlled Clinical Trial. Head Face Med. 2006, 2, 33. [Google Scholar] [CrossRef] [Green Version]
- Baltaci, A.K.; Dundar, T.K.; Aksoy, F.; Mogulkoc, R. Changes in the Serum Levels of Trace Elements Before and After the Operation in Thyroid Cancer Patients. Biol. Trace Elem. Res. 2017, 175, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yaman, M.; Kaya, G.; Yekeler, H. Distribution of Trace Metal Concentrations in Paired Cancerous and Non-Cancerous Human Stomach Tissues. World J. Gastroenterol. 2007, 13, 612–618. [Google Scholar] [CrossRef]
- Itoh, S.; Ha, W.K.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel Role of Antioxidant-1 (Atox1) as a Copper-Dependent Transcription Factor Involved in Cell Proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.-E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A Novel Role for Copper in Ras/Mitogen-Activated Protein Kinase Signaling. Mol. Cell Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Aubert, L.; Nandagopal, N.; Steinhart, Z.; Lavoie, G.; Nourreddine, S.; Berman, J.; Saba-El-Leil, M.K.; Papadopoli, D.; Lin, S.; Hart, T.; et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat. Commun. 2020, 11, 3701. [Google Scholar] [CrossRef]
- Grasso, M.; Bond, G.J.; Kim, Y.J.; Boyd, S.; Dzebo, M.M.; Valenzuela, S.; Tsang, T.; Schibrowsky, N.A.; Alwan, K.B.; Blackburn, N.J.; et al. The Copper Chaperone CCS Facilitates Copper Binding to MEK1/2 to Promote Kinase Activation. J. Biol. Chem. 2021, 297, 101314. [Google Scholar] [CrossRef]
- Millanes-Romero, A.; Herranz, N.; Perrera, V.; Iturbide, A.; Loubat-Casanovas, J.; Gil, J.; Jenuwein, T.; García de Herreros, A.; Peiró, S. Regulation of Heterochromatin Transcription by Snail1/LOXL2 during Epithelial-to-Mesenchymal Transition. Mol. Cell 2013, 52, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wu, J.-S.; Muzik, O.; Hsieh, J.-T.; Lee, R.J.; Peng, F. Reduced 64 Cu Uptake and Tumor Growth Inhibition by Knockdown of Human Copper Transporter 1 in Xenograft Mouse Model of Prostate Cancer. J. Nucl. Med. 2014, 55, 622–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Singh, M.K.; Singh, T.B.; Bhartiya, S.K.; Singh, S.P.; Shukla, V.K. Heavy and Trace Metals in Carcinoma of the Gallbladder. World J. Surg. 2013, 37, 2641–2646. [Google Scholar] [CrossRef]
- Saleh, S.A.K.; Adly, H.M.; Abdelkhaliq, A.A.; Nassir, A.M. Serum Levels of Selenium, Zinc, Copper, Manganese, and Iron in Prostate Cancer Patients. Curr. Urol. 2020, 14, 44–49. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper Is Required for Oncogenic BRAF Signalling and Tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.; Nalvarte, I.; Smirnova, T.; Vecchi, M.; Aceto, N.; Dolemeyer, A.; Frei, A.; Lienhard, S.; Wyckoff, J.; Hess, D.; et al. Memo Is a Copper-Dependent Redox Protein with an Essential Role in Migration and Metastasis. Sci. Signal. 2014, 7, ra56. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 6836–6841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Chang, C.; Liu, B.; Li, Z.; Li, H.; Cai, N.; Wang, H.H. Copper (II) Ions Activate Ligand-Independent Receptor Tyrosine Kinase (RTK) Signaling Pathway. Biomed. Res. Int. 2019, 2019, 4158415. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bond, G.J.; Tsang, T.; Posimo, J.M.; Busino, L.; Brady, D.C. Copper Chaperone ATOX1 Is Required for MAPK Signaling and Growth in: BRAF Mutation-Positive Melanoma. Metallomics 2019, 11, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Tsang, T.; Posimo, J.M.; Gudiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper Is an Essential Regulator of the Autophagic Kinases ULK1/2 to Drive Lung Adenocarcinoma. Nat. Cell Biol. 2020, 22, 412–424. [Google Scholar] [CrossRef]
- Voli, F.; Valli, E.; Lerra, L.; Kimpton, K.; Saletta, F.; Giorgi, F.M.; Mercatelli, D.; Rouaen, J.R.C.; Shen, S.; Murray, J.E.; et al. Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion. Cancer Res. 2020, 80, 4129–4144. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Gouw, A.M.; LaGory, E.L.; Guo, S.; Attarwala, N.; Tang, Y.; Qi, J.; Chen, Y.-S.; Gao, Z.; Casey, K.M.; et al. Mitochondrial Copper Depletion Suppresses Triple-Negative Breast Cancer in Mice. Nat. Biotechnol. 2021, 39, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jian, Z.; Liu, H.; Cui, H.; Deng, H.; Fang, J.; Zuo, Z.; Wang, X.; Zhao, L.; Geng, Y.; et al. TGF-β1-Induced EMT Activation via Both Smad-Dependent and MAPK Signaling Pathways in Cu-Induced Pulmonary Fibrosis. Toxicol. Appl. Pharmacol. 2021, 418, 115500. [Google Scholar] [CrossRef] [PubMed]
- Chojnowski, J.E.; Li, R.; Tsang, T.; Alfaran, F.H.; Dick, A.; Cocklin, S.; Brady, D.C.; Strochlic, T.I. Copper Modulates the Catalytic Activity of Protein Kinase CK2. Front. Mol. Biosci. 2022, 9, 878652. [Google Scholar] [CrossRef]
- Chen, G.-F.; Sudhahar, V.; Youn, S.-W.; Das, A.; Cho, J.; Kamiya, T.; Urao, N.; McKinney, R.D.; Surenkhuu, B.; Hamakubo, T.; et al. Copper Transport Protein Antioxidant-1 Promotes Inflammatory Neovascularization via Chaperone and Transcription Factor Function. Sci. Rep. 2015, 5, 14780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Ge, G. Lysyl Oxidase, Extracellular Matrix Remodeling and Cancer Metastasis. Cancer Microenviron. 2012, 5, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, A.V.; Chen, J. MEMO1, a new IRS1-interacting protein, induces epithelial–mesenchymal transition in mammary epithelial cells. Oncogene 2012, 32, 3130–3138. [Google Scholar] [CrossRef]
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper Regulation of Hypoxia-Inducible Factor-1 Activity. Mol. Pharmacol. 2009, 75, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J.A. Signal Transduction Mediated by the Ras/Raf/MEK/ERK Pathway from Cytokine Receptors to Transcription Factors: Potential Targeting for Therapeutic Intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Montagut, C.; Settleman, J. Targeting the RAF-MEK-ERK Pathway in Cancer Therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, F.; Nucera, C.; Pellegriti, G.; Gangemi, P.; Attard, M.; Stella, M.; Loda, M.; Vella, V.; Giordano, C.; Trimarchi, F.; et al. BRAF(V600E) Mutation and the Biology of Papillary Thyroid Cancer. Endocr. Relat. Cancer 2008, 15, 191–205. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radzlejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A Family of Protein-Serine/Threonine Kinases That Are Activated and Tyrosine Phosphorylated in Response to Insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Zehorai, E.; Yao, Z.; Plotnikov, A.; Seger, R. The Subcellular Localization of MEK and ERK-A Novel Nuclear Translocation Signal (NTS) Paves a Way to the Nucleus. Mol. Cell Endocrinol. 2010, 314, 213–220. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK Cascades: Signaling Components, Nuclear Roles and Mechanisms of Nuclear Translocation. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular, Interpretation of ERK Signal Duration by Immediate Early Gene Products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A Reinvestigation of the Multisite Phosphorylation of the Transcription Factor C-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-Dependent Phosphorylation Pathways Regulate Myc Protein Stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Zhang, W.; Ballif, B.A.; Elliott, H.L.; Danuser, G.; Blenis, J. ERK-MAPK Drives Lamellipodia Protrusion by Activating the WAVE2 Regulatory Complex. Mol. Cell 2011, 41, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; De Lanerolle, P.; Cheresh, D.A. Regulation of Cell Motility by Mitogen-Activated Protein Kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Lake, D.; Corrêa, S.A.L.; Müller, J. Negative Feedback Regulation of the ERK1/2 MAPK Pathway. Cell Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barret, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP Kinase Pathway Suppresses Growth of Colon Tumors in Vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J. Searching for Harmony in Transition-Metal Signaling. Nat. Chem. Biol. 2015, 11, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated Copper and Oxidative Stress in Cancer Cells as a Target for Cancer Treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable Copper Modulates Oxidative Phosphorylation and Growth of Tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J.; et al. Inhibition of Human Copper Trafficking by a Small Molecule Significantly Attenuates Cancer Cell Proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Deng, R.; Luo, R.Z.; Shen, G.P.; Cai, M.Y.; Du, Z.M.; Jiang, S.; Yang, M.T.; Fu, J.H.; Zhu, X.F. Low Expression of ULK1 Is Associated with Operable Breast Cancer Progression and Is an Adverse Prognostic Marker of Survival for Patients. Breast Cancer Res. Treat. 2012, 134, 549–560. [Google Scholar] [CrossRef]
- Ouyang, L.; Zhang, L.; Fu, L.; Liu, B. A Small-Molecule Activator Induces ULK1-Modulating Autophagy-Associated Cell Death in Triple Negative Breast Cancer. Autophagy 2017, 13, 777–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xu, H.; Chen, X.; Chen, J.; Li, X.; Qiao, G.; Tian, Y.; Yuan, R.; Su, S.; Liu, X.; et al. Aqueous Extract of Clove Inhibits Tumor Growth by Inducing Autophagy through AMPK/ULK Pathway. Phyther. Res. 2019, 33, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, T.; Gu, X.; Davis, C.I.; Posimo, J.M.; Miller, Z.A.; Brady, D.C. BRAFV600E-Driven Lung Adenocarcinoma Requires Copper to Sustain Autophagic Signaling and Processing. Mol. Cancer Res. 2022, 20, 1096–1107. [Google Scholar] [CrossRef]
- Guo, J.; Cheng, J.; Zheng, N.; Zhang, X.; Dai, X.; Zhang, L.; Hu, C.; Wu, X.; Jiang, Q.; Wu, D.; et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv. Sci. 2021, 8, e2004303. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt Signal Transduction for Cancer Therapy. Signal. Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist Transcriptionally Up-Regulates AKT2 in Breast Cancer Cells Leading to Increased Migration, Invasion, and Resistance to Paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef] [Green Version]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar] [CrossRef]
- Shin, I.; Yakes, F.M.; Rojo, F.; Shin, N.-Y.; Bakin, A.V.; Baselga, J.; Arteaga, C.L. The Sequential Activation of Cyclin-Dependent Kinases (CDKs) Reg-Ulates the Eukaryotic Cell Cycle. CDK Inhibitors (CKIs) Negatively Regulate CDKs, and CDK-Bound G1 Cyclins Can Activate CDKs. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef]
- Leroy, C.; Ramos, P.; Cornille, K.; Bonenfant, D.; Fritsch, C.; Voshol, H.; Bentires-Alj, M. Activation of IGF1R/P110β/AKT/MTOR Confers Resistance to α-Specific PI3K Inhibition. Breast Cancer Res. 2016, 18, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Yang, Z.; Lu, N. A New Role for the PI3K/Akt Signaling Pathway in the Epithelial-Mesenchymal Transition. Cell Adhes. Migr. 2015, 9, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Chua, M.M.J.; Ortega, C.E.; Sheikh, A.; Lee, M.; Abdul-Rassoul, H.; Hartshorn, K.L.; Dominguez, I. CK2 in Cancer: Cellular and Biochemical Mechanisms and Potential Therapeutic Target. Pharmaceuticals 2017, 10, 18. [Google Scholar] [CrossRef]
- Eddy, S.F.; Guo, S.; Demicco, E.G.; Romieu-Mourez, R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. Inducible IκB Kinase/IκB Kinase ε Expression is Induced by CK2 and Promotes Aberrant Nuclear Factor-ΚB Activation in Breast Cancer Cells. Cancer Res. 2005, 65, 11375–11383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. ACK2-Dependent Mechanism for Activation of the JAK-STAT Signaling Pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, L.; Cotruvo, J.A.; Chan, J.; Kaluarachchi, H.; Muchenditsi, A.; Pendyala, V.S.; Jia, S.; Aron, A.T.; Ackerman, C.M.; Wal, M.N.V.; et al. Copper Regulates Cyclic-AMP-Dependent Lipolysis. Nat. Chem. Biol. 2016, 12, 586–592. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Gao, L.N.; Cui, Y.L.; Zhang, Y.; Zhou, X. The Cyclic AMP Signaling Pathway: Exploring Targets for Successful Drug Discovery (Review). Mol. Med. Rep. 2016, 13, 3715–3723. [Google Scholar] [CrossRef] [Green Version]
- Tonucci, F.M.; Almada, E.; Borini-Etichetti, C.; Pariani, A.; Hidalgo, F.; Rico, M.J.; Girardini, J.; Favre, C.; Goldenring, J.R.; Menacho-Marquez, M.; et al. Identification of a CIP4 PKA Phosphorylation Site Involved in the Regulation of Cancer Cell Invasiveness and Metastasis. Cancer Lett. 2019, 461, 65–77. [Google Scholar] [CrossRef]
- Jiang, P.; Enomoto, A.; Takahashi, M. Cell Biology of the Movement of Breast Cancer Cells: Intracellular Signalling and the Actin Cytoskeleton. Cancer Lett. 2009, 284, 122–130. [Google Scholar] [CrossRef]
- Howe, A.K. Regulation of Actin-Based Cell Migration by CAMP/PKA. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1692, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Shen, W.J.; Muliro, K.; Patel, S.; Souza, S.C.; Roth, R.A.; Kraemer, F.B. Stimulation of Lipolysis and Hormone-Sensitive Lipase via the Extracellular Signal-Regulated Kinase Pathway. J. Biol. Chem. 2001, 276, 45456–45461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin Promotes Hormone-Sensitive Lipase-Mediated Adipocyte Lipolysis via Phosphorylation-Dependent and -Independent Mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting Copper and Cancer: From Transition Metal Signalling to Metalloplasia. Nat. Rev. Cancer 2021, 22, 102–113. [Google Scholar] [CrossRef]
- Qu, C.; Peng, Y.; Liu, S. Ferroptosis Biology and Implication in Cancers. Front. Mol. Biosci. 2022, 9, 892957. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Evidence/Cuproproteins Involved | Year | Authors | Ref. |
|---|---|---|---|
| Copper is required for angiogenesis | 1982; 1994; 1997; 2001; 2006 | Raju, K.S. et al.; Lane, T.F. et al.; Soncin, F. et al.; Landriscina, M et al.; Juarez, J.G. et al. | [40]; [41]; [42]; [43]; [44] |
| Higher copper serum levels in cancer patients | 1983, 1989 | Margalioth, E.J., et al.; Coates, R.J. et al. | [45]; [46] |
| Elevated copper serum levels in lung cancer patients | 1989, 1994, 2011 | Diez, M. et al.; Oyama, T., et al. Jin, Y. et al. | [47]; [48]; [49] |
| Copper depletion reduced gliosarcoma growth in rat brains | 1990 | Brem, S.S. et al. | [50]; [51] |
| Phase I study: 90 days copper deficiency reduces cancer progression in 5 out of 6 patients with metastatic cancer | 2000 | Brewer, G.J. et al. | [52] |
| TTM treatment of orthotopic mouse model of head and neck squamous cell carcinoma reduces cancer growth and angiogenesis | 2001 | Cox, C. et al. | [53] |
| Elevated copper serum levels in breast cancer patients | 2002, 2012, 2015, 2015, 2015 | Kuo, H.W., et al.; Feng, J.F. et al.; Ding, X. et al.; Adeoti, M. et al.; Pavithra, V. et al. | [54]; [55]; [56]; [57]; [58] |
| Elevated copper serum levels in colorectal cancer patients | 2003, 2017, 2018 | Nayak, S.B., et al.; Stepien, M., et al., Sohrabi, M. et al. | [59]; [60]; [61] |
| Cuproproteins expression is elevated in human cancers | 2017 | Blockhyus, S. et al. | [62] |
| Elevated copper serum levels in oral and thyroid cancer patients | 2006, 2017 | Khanna, S.S. and Karjodkar, F.R.; Baltaci, A.K., et al.; | [63]; [64] |
| Elevated copper serum levels in stomach cancer patients | 2007 | Yaman, M., et al. | [65] |
| The copper chaperone ATOX1 modulates cell proliferation | 2008 | Itoh, S. et al. | [66] |
| Copper regulates the RAS/RAF/MEK/ERK signaling pathway | 2012; 2020, 2021 | Turski, M.L., et al.; Aubert, L. et al.; Grasso, M. et al. | [67]; [68]; [69] |
| The copper-dependent aminoxidase LOXL2 is required for epithelial to mesenchymal transition (EMT) | 2013 | Millanes-Romero, A. et al. | [70] |
| Elevated copper serum levels in gallbladder and prostate cancer patients | 2014; 2013; 2020 | Cai, H., et al.; Basu, S. et al.; Saleh, S.A.H. et al. | [71]; [72]; [73] |
| Copper modulate BRAF signaling | 2014 | Donita, B.C. et al. | [74] |
| MEMO1 is copper-dependent protein exerting a crucial role during EMT and metastatization | 2014 | MacDonald, G., et al. | [75] |
| Copper chelation blocks the growth of BRAFV600E melanoma | 2017 | Brady, D.C., et al. | [76] |
| ATP7A transfers copper to the cuproenzyme LOXL2 and it is involved in tumorigenesis | 2019 | Shanbagh, V. et al. | [77] |
| Copper activates RTK signaling | 2019 | He, F. et al. | [78] |
| ATOX1 modulates MAPK signaling In BRAFV600E | 2019 | Kim, Y.J., et al. | [79] |
| Copper modulates ULK1/2 mediated autophagy in lung carcinoma | 2020 | Tsang, T., et al. | [80] |
| Copper affects PDL-1 expression | 2020 | Voli, F., et al. | [81] |
| Mitochondrial copper depletion impairs triple negative breast cancer growth in a mouse model. | 2020 | Cui, L., et al. | [82] |
| Copper modulates AKT cascade | 2021, 2022 | Guo, J. et al.; Chojnowski, J.E., et al. | [83]; [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitaliti, A.; De Luca, A.; Rossi, L. Copper-Dependent Kinases and Their Role in Cancer Inception, Progression and Metastasis. Biomolecules 2022, 12, 1520. https://doi.org/10.3390/biom12101520
Vitaliti A, De Luca A, Rossi L. Copper-Dependent Kinases and Their Role in Cancer Inception, Progression and Metastasis. Biomolecules. 2022; 12(10):1520. https://doi.org/10.3390/biom12101520
Chicago/Turabian StyleVitaliti, Alessandra, Anastasia De Luca, and Luisa Rossi. 2022. "Copper-Dependent Kinases and Their Role in Cancer Inception, Progression and Metastasis" Biomolecules 12, no. 10: 1520. https://doi.org/10.3390/biom12101520