1. Introduction
Alzheimer’s disease (AD) [
1], the most common cause of senile dementia worldwide, is a degenerative disorder of the central nervous system characterized by memory loss and dysfunctions of language and behavior. Although the pathogenesis of AD is not completely clear, several hypotheses about AD pathogenesis, such as the free-radical injury hypothesis [
2], amyloid peptide hypothesis [
3], cholinergic hypothesis [
4] and Tau hyper-phosphorylation hypothesis [
5], are used as the main basis for its treatment. Therefore, there are several potential drug targets such as acetylcholinesterase [
6], amyloid-β protein [
7] and tau protein [
8], which are used for the treatment and amelioration of the disease.
Monoamine oxidases A and B (MAO-A and MAO-B) are flavin adenine dinucleotide (FAD)-dependent enzymes [
9] which play important roles in the metabolism of biogenic amines in the central nervous system and in peripheral tissues. MAO-A inhibitors can be used as anti-depressants, whereas MAO-B inhibitors are mainly used in the treatment of neurodegenerative diseases such as AD. MAO-B is an important mitochondrial enzyme [
10] which metabolizes β-phenethylamine [
11]. Studies suggested that reactive astrocytes in the brain of an AD mice model aberrantly and abundantly produce an inhibitory glio-transmitter, γ-aminobutyric acid (GABA) through MAO-B, and this leads to an abnormal release of GABA through the bestrophin 1 channel [
12]. The released GABA reduces the normal flow of information by acting on presynaptic GABA receptors. Therefore, the researchers proposed that selective inhibition of GABA synthesis and release by MAO-B inhibitors may be an effective therapeutic strategy for treating memory impairment in AD patients [
13]. Subsequently, several effective MAO-B inhibitors, such as selegiline [
14,
15] and rasagiline [
16], have been shown to be effective in retarding the neurodegeneration normally seen in cases of AD. However, the side effects, including anxiety, hallucinations and dyskinesia, are also obvious [
17].
With the circumstances that machine learning has developed rapidly, in silico approaches provide vast help in modern drug discovery. This work describes the discovery of new MAO-B inhibitors with computational approaches. First, machine learning approaches, including SVM, KNN, C4.5, random forest, random tree, AdaBoost and bagging were applied to analyze MAO-B inhibitors qualitatively and predict whether a compound had the ability to inhibit MAO-B. Then, Topomer CoMFA was used to predict the activity of the inhibitors and to identify their stereo-electronic requirements, as well as to reveal the structural factors essential to the interactions with their substrate. Additionally, molecular docking technology was also performed to predict the interactions between MAO-B and MAO-B inhibitors. Finally, 1-(4-hydroxyphenyl)-3-(2,4,6-trimethoxyphenyl)propan-1-one (also called LB) was proposed as a potential MAO-B inhibitor based on the MAO-B inhibitor identification model, Topomer CoMFA model and molecule docking. The in silico findings were further validated with multi-spectroscopy approaches, including fluorescence spectra and UV-vis absorption spectra.
4. Discussion
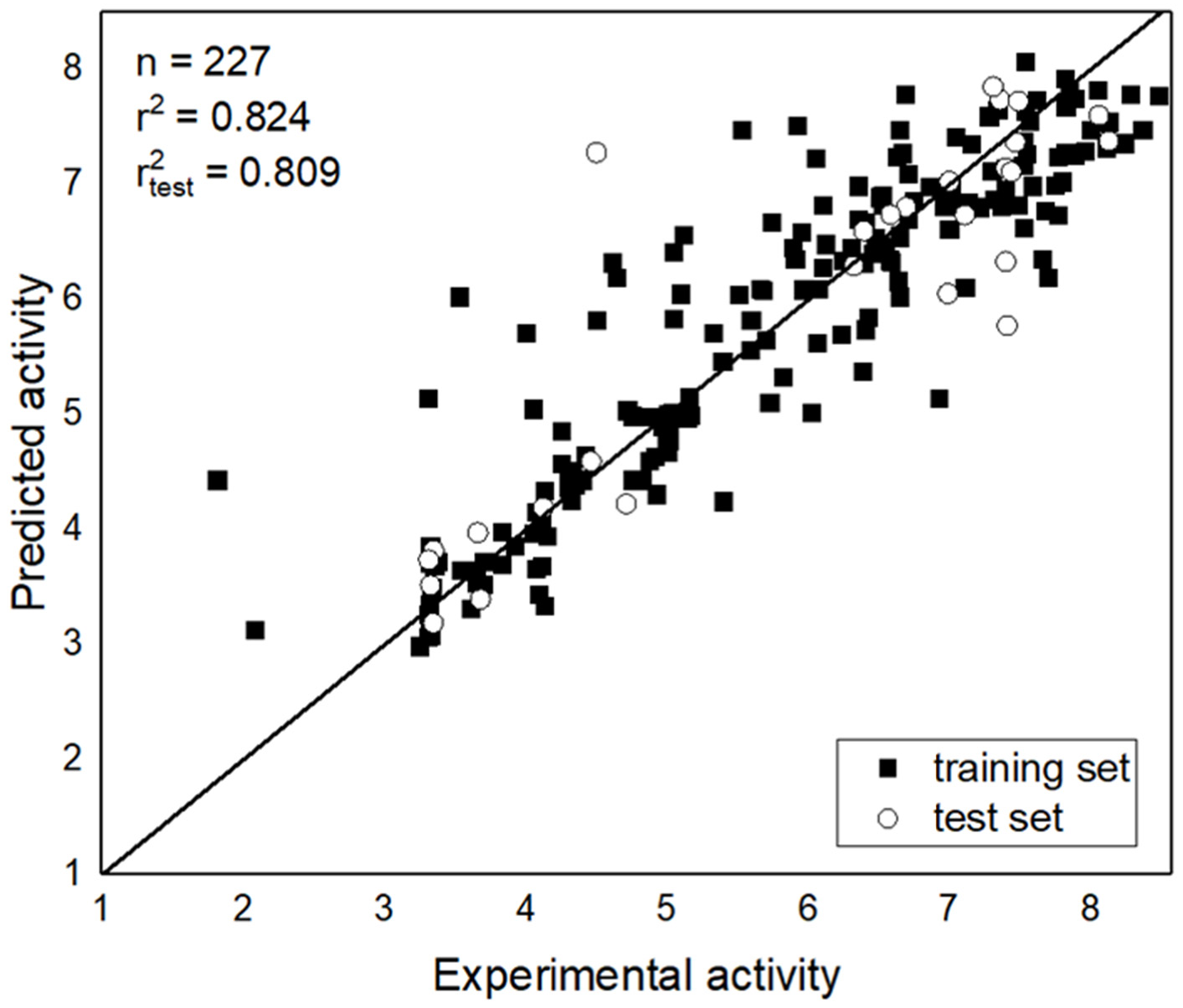
It is necessary for a good prediction model to select appropriate features because of the usual existence of irrelevant features. In this case, a subset containing eight features was used to build the MAO-B inhibitor prediction model. The prediction accuracy of the model was deemed to be very good, as it included both a training set and an independent test set. The results showed that the original data contained some redundant features. Thus, feature selection is a necessary step during the building of a useful prediction model.
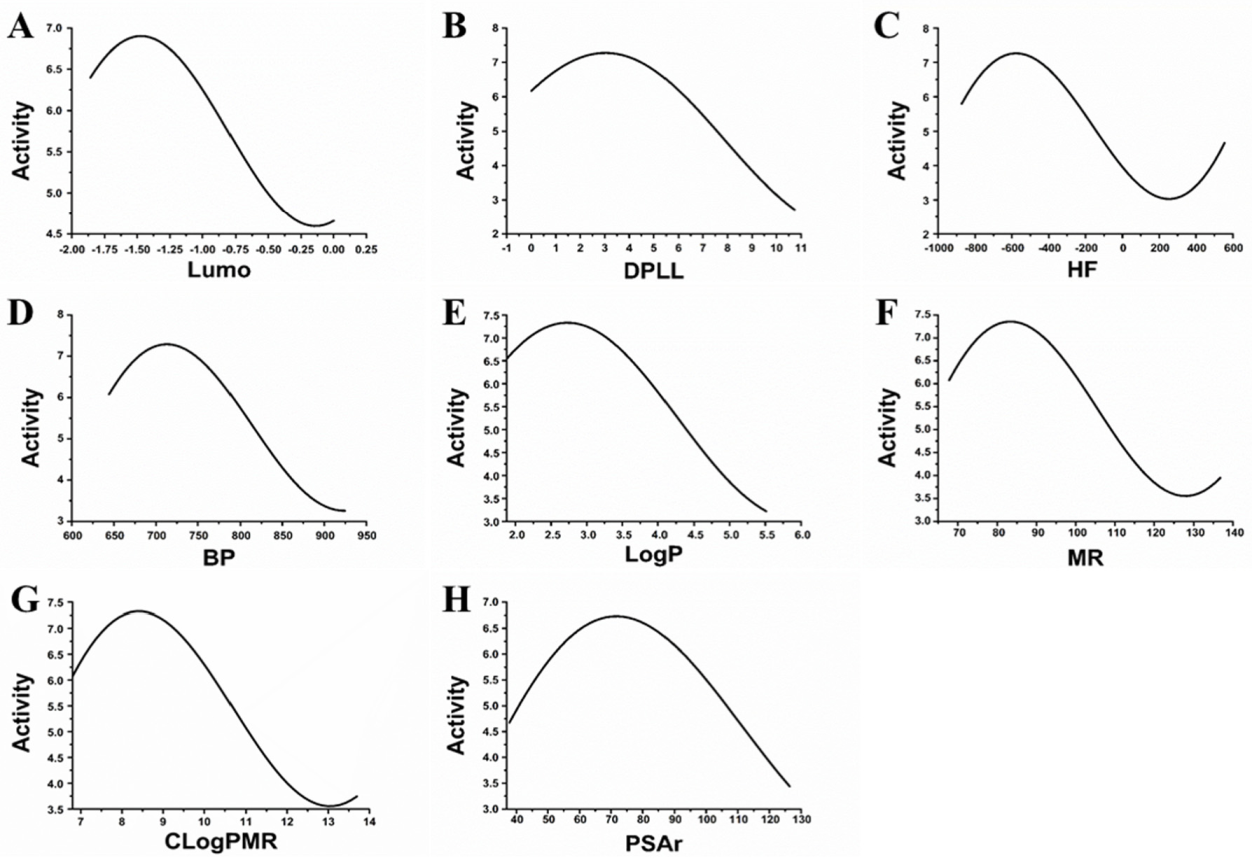
According to the results of the cross-validation, there was a relationship between the eight descriptors used (Lumo, DPLL, HF, BP, LogP, MR, CLogPMR and PSAr) and the inhibitory activities of MAO-B inhibitors. The sensitivity analysis was further applied to reflect on the relationship between these descriptors and the inhibitor activities directly. For example,
Figure 1A shows the relationship between the activity and Lumo. When the Lumo was approximately −0.15 and −1.48, the activity was at the maximum and minimum, respectively.
Figure 1B shows the relationship between the activity and DPLL. When the DPLL was approximately 3.07, the activity value was at its peak.
Figure 1C shows the relationship between the activity and HF. When the HF was approximately −581.08 and 263.38, the activity values were at the maximum and minimum, respectively.
Figure 1D shows the relationship between the activity and BP. When the BP was approximately 712.82, the activity value was at its peak.
Figure 1E shows the relationship between the activity and LogP. When the LogP was approximately 2.70, the activity value was at its peak.
Figure 1F shows the relationship between the activity and MR. When the MR was approximately 83.27 and 128.32, the activity values were at the maximum and minimum, respectively.
Figure 1G shows the relationship between the activity and CLogPMR. When the CLogPMR was approximately 8.35 and 12.99, the activity values were at the maximum and minimum, respectively.
Figure 1H shows the relationship between the activity and PSAr. When the PSAr was approximately 72.16, the activity value was at its peak.
The use of a MAO-B inhibitor prediction model indicated that these eight molecular descriptors are able to classify whether a compound is capable of acting as a MAO-B inhibitor. Moreover, these eight molecular descriptors may be related to the activity of the MAO-B.
The activities of MAO-B inhibitors are related to the integrity of the pharmacophore [
36]. In the Topomer CoMFA model, the pharmacophore is related to cutting style, which is important for the model’s predictive performance [
17,
37,
38]. In the Topomer CoMFA analysis, the training set is split into two fragments. If the fragmentation is complete, the input structures are standardized and the topomers are generated. All of the topomers share the same identical substructure. If the same identical substructures in the test set are recognized, the model’s predictive ability is promising. The presence of identical substructures is considered as the pharmacophore. In this study, the compounds were fragmented into R1 and R2 groups, with the two models being obtained based on different fragmentation procedures. Model 2, with the highest q
2 and r
2 values, was selected as the final model for further subsequent evaluation.
The active site in Model 2 was modified based on the active site in R1 of Model 1, which contributed to the model’s predictive ability (
Table 2). Thus, we can speculate that R1 and R2 groups in Model 1 have identical substructures. Generally, a substrate interacts with a receptor through its active pharmacophore. Active pharmacophores have a particular conformation which interacts with key amino acid residues that are the active residues in the active pocket of MAO-B. The molecule is often divided into the active pharmacophore and other structures which are regarded as having the same identical substructures.
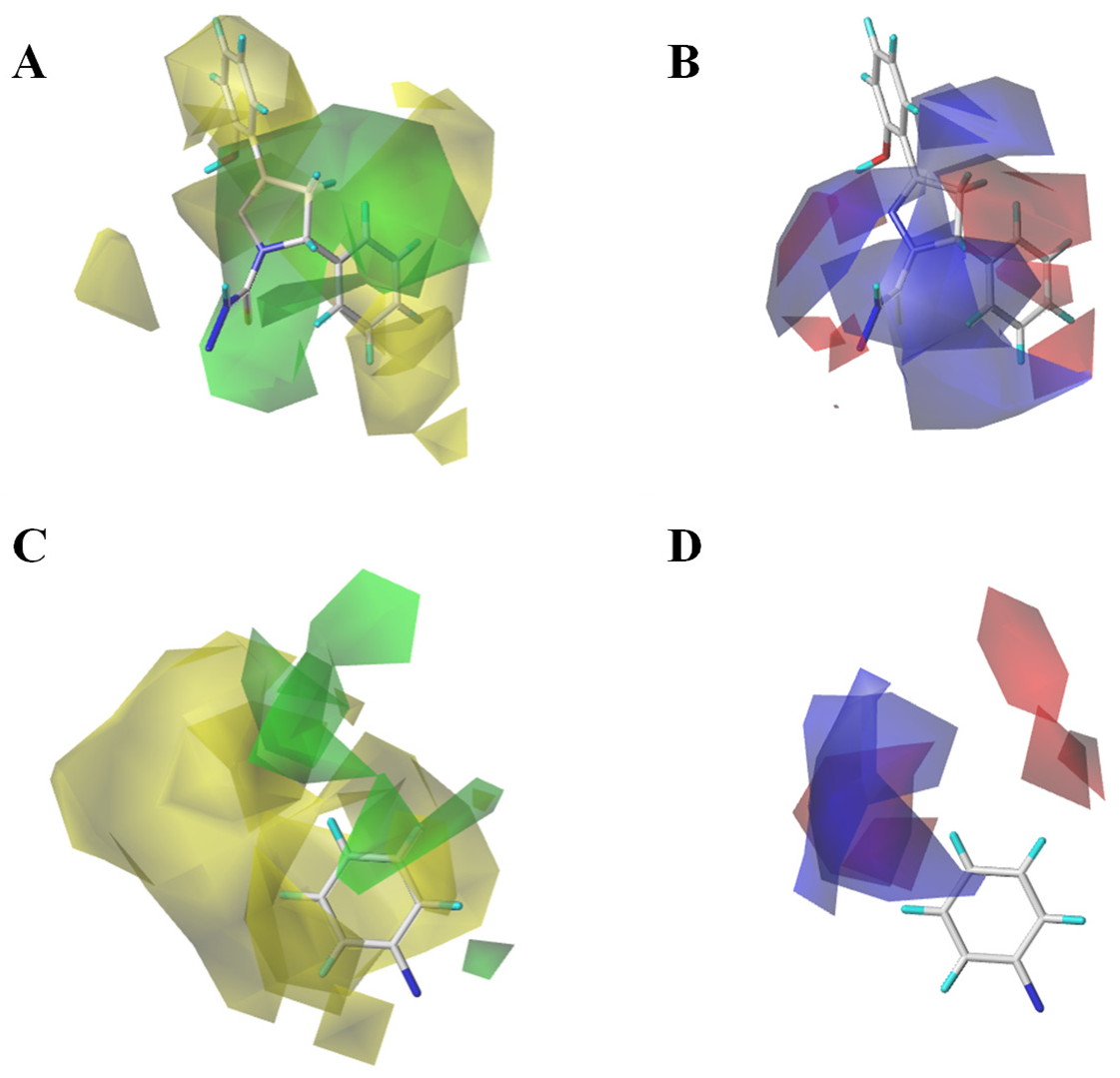
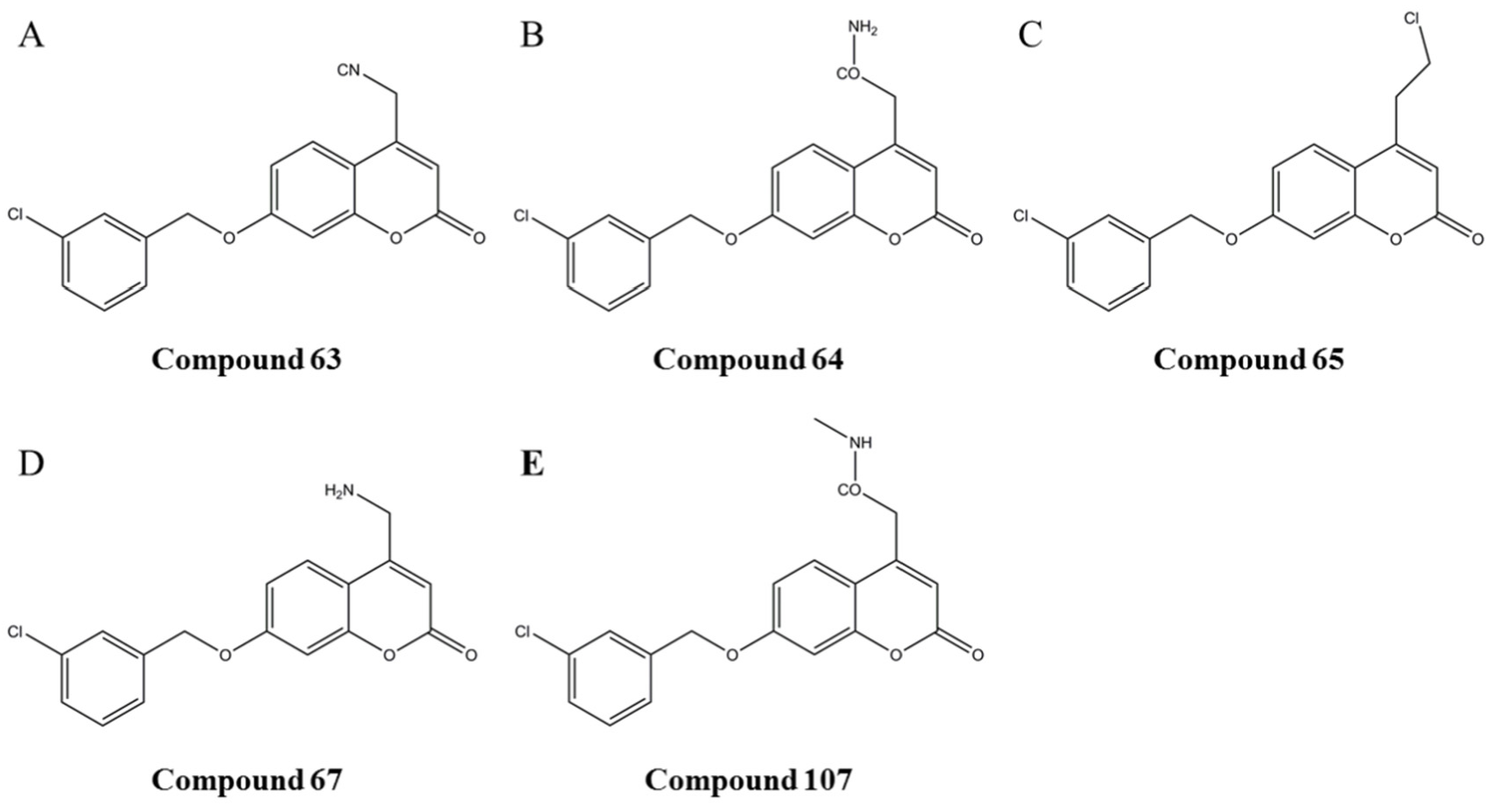
In order to further enhance the biological activities of MAO-B inhibitors, the Topomer CoMFA model provides alternative possibilities for modifying MAO-B inhibitors. Compound
63 (
Figure 3A) was chosen to study the effects of R1 and R2 groups on the activity. In the R1 group, large and negatively charged groups in the chlorobenzene ring may increase the compound’s biological activity (
Figure 8). In the R2 group, small groups with a positive charge on the propiononitrile may also increase the compound’s biological activity (
Figure 3). Similar studies have reported that Topomer CoMFA was used in designing xanthine oxido-reductase inhibitors [
23]. Additionally, it can also be used in virtual screening for identification of novel antagonists [
39].
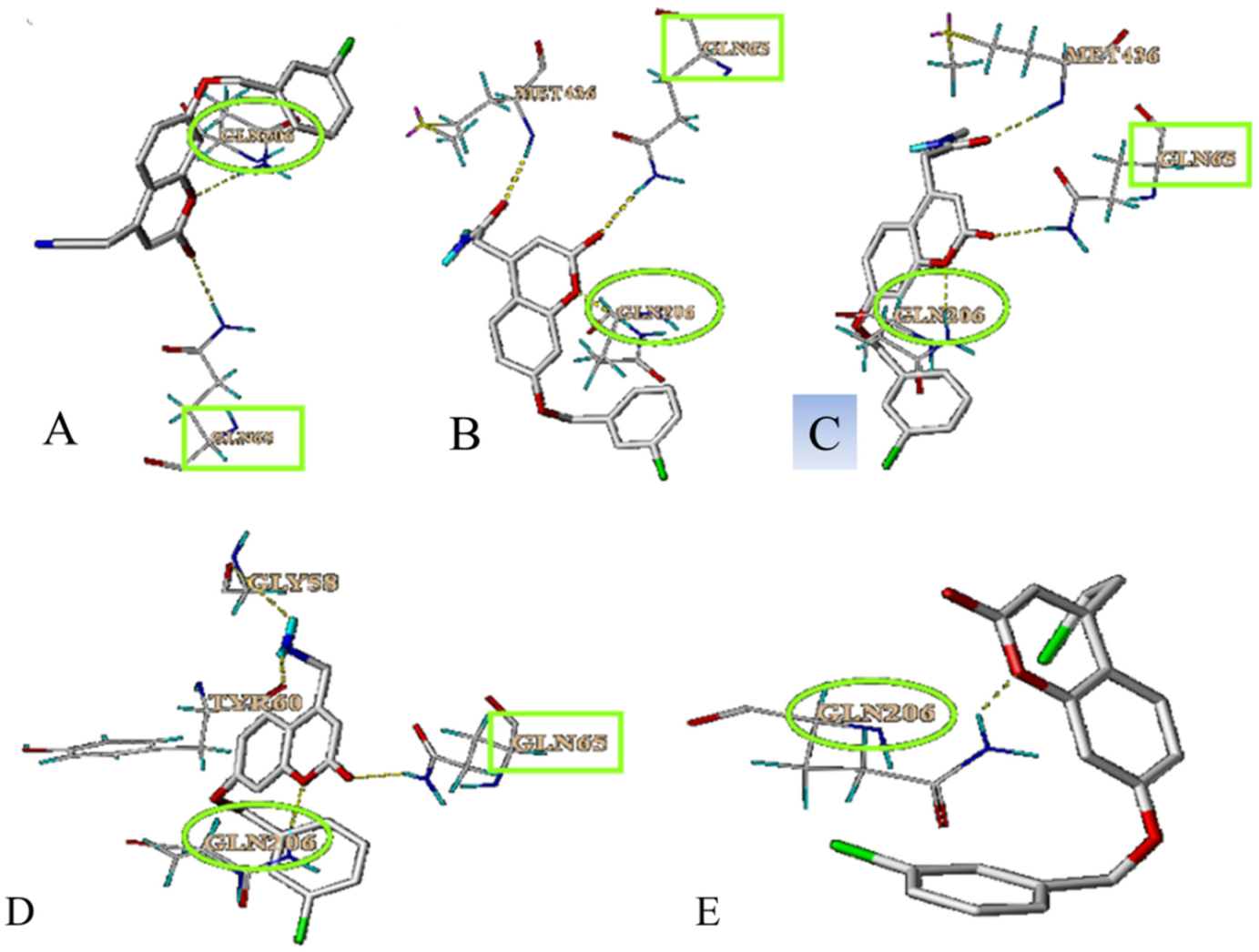
Molecular docking is used as a method of predicting the interaction sites between the compounds and MAO-B. As shown in
Figure 5, there were binding sites between these five compounds and MAO-B. Five compounds can interact with GLN206, which is one of the active sites of MAO-B. Compounds
64,
65 and
67 also can interact with GLN206. Therefore, we hypothesized that GLN206 might be a new active site of MAO-B.
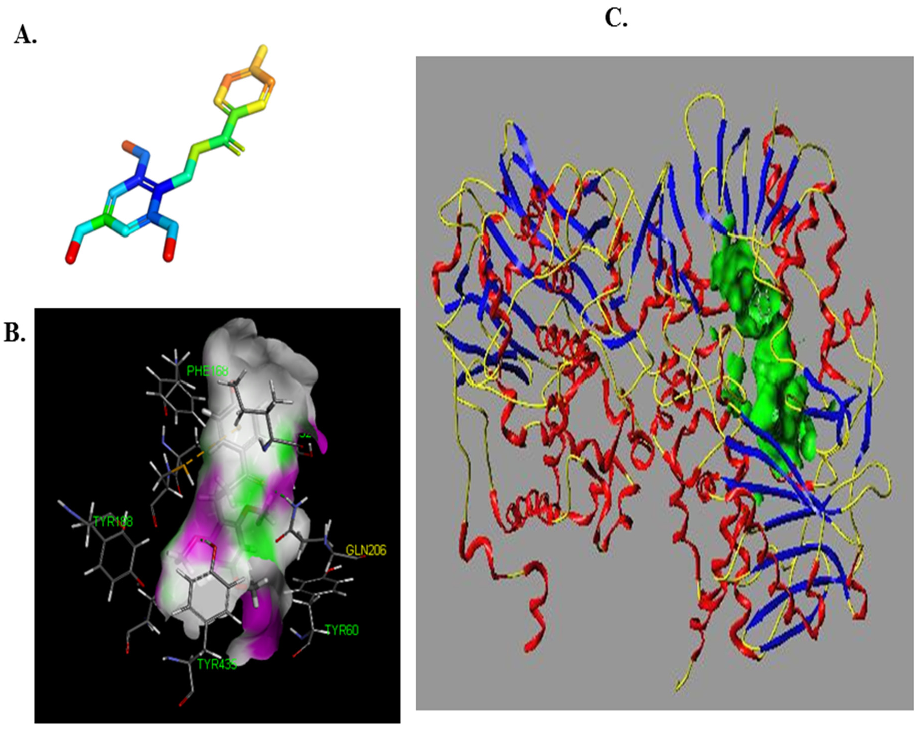
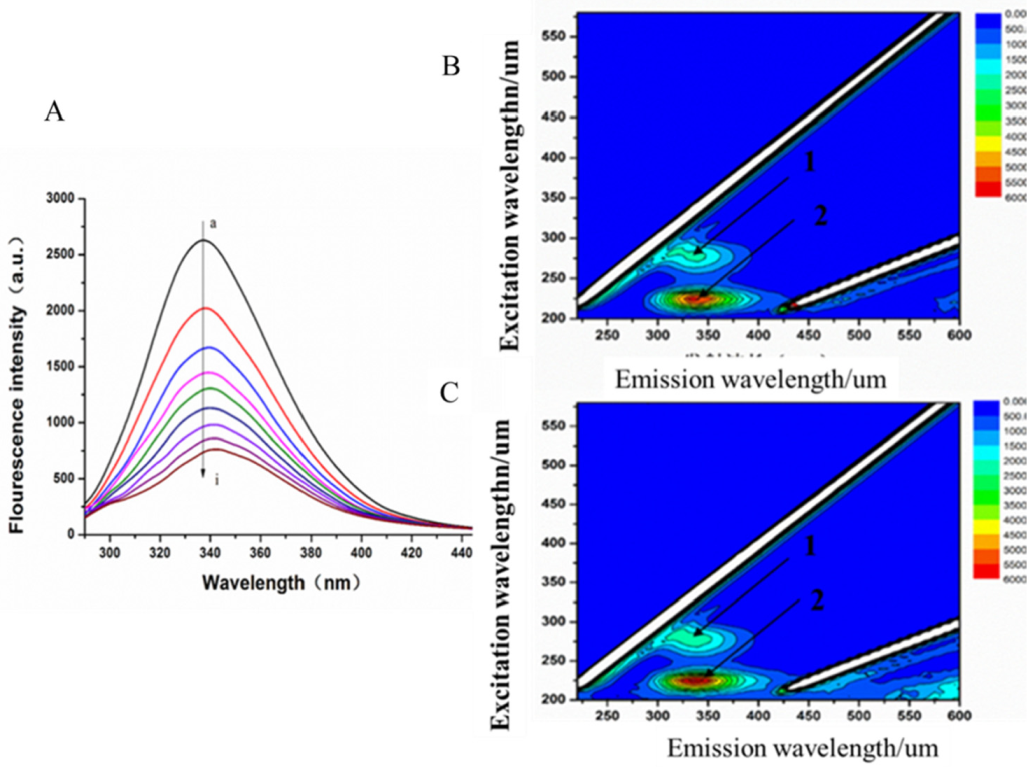
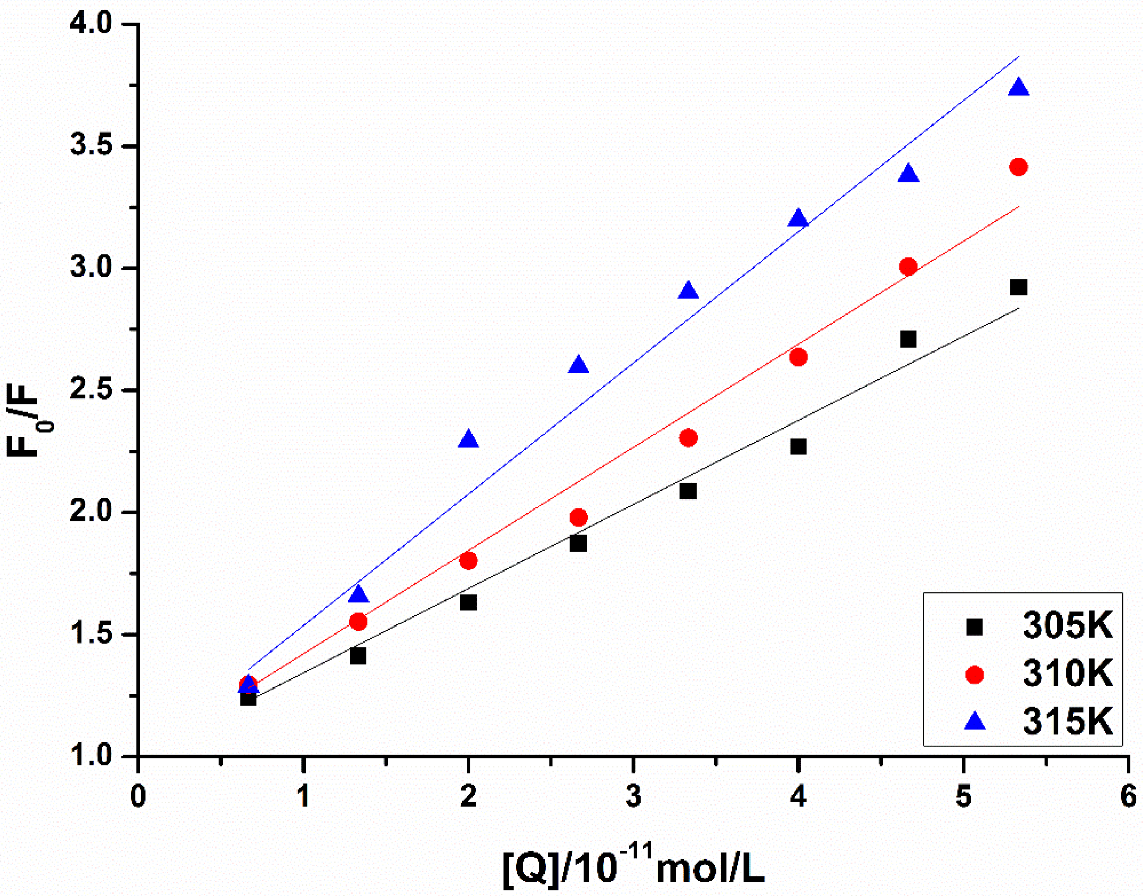
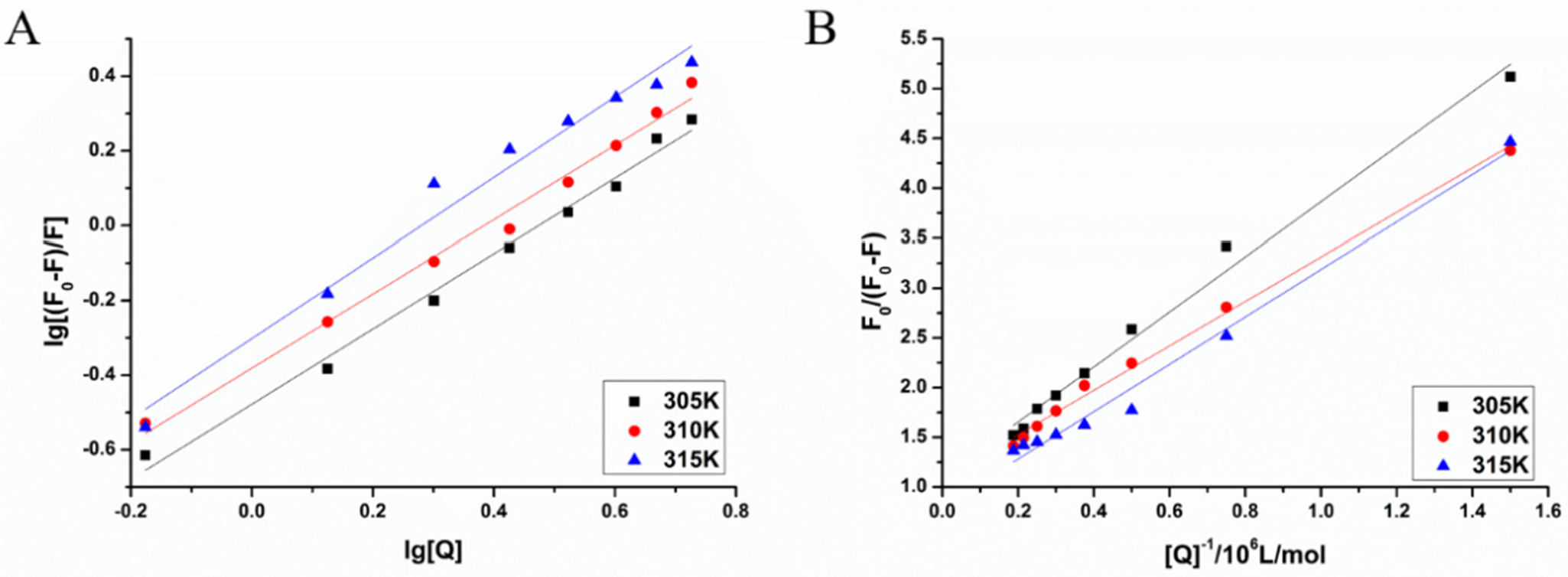
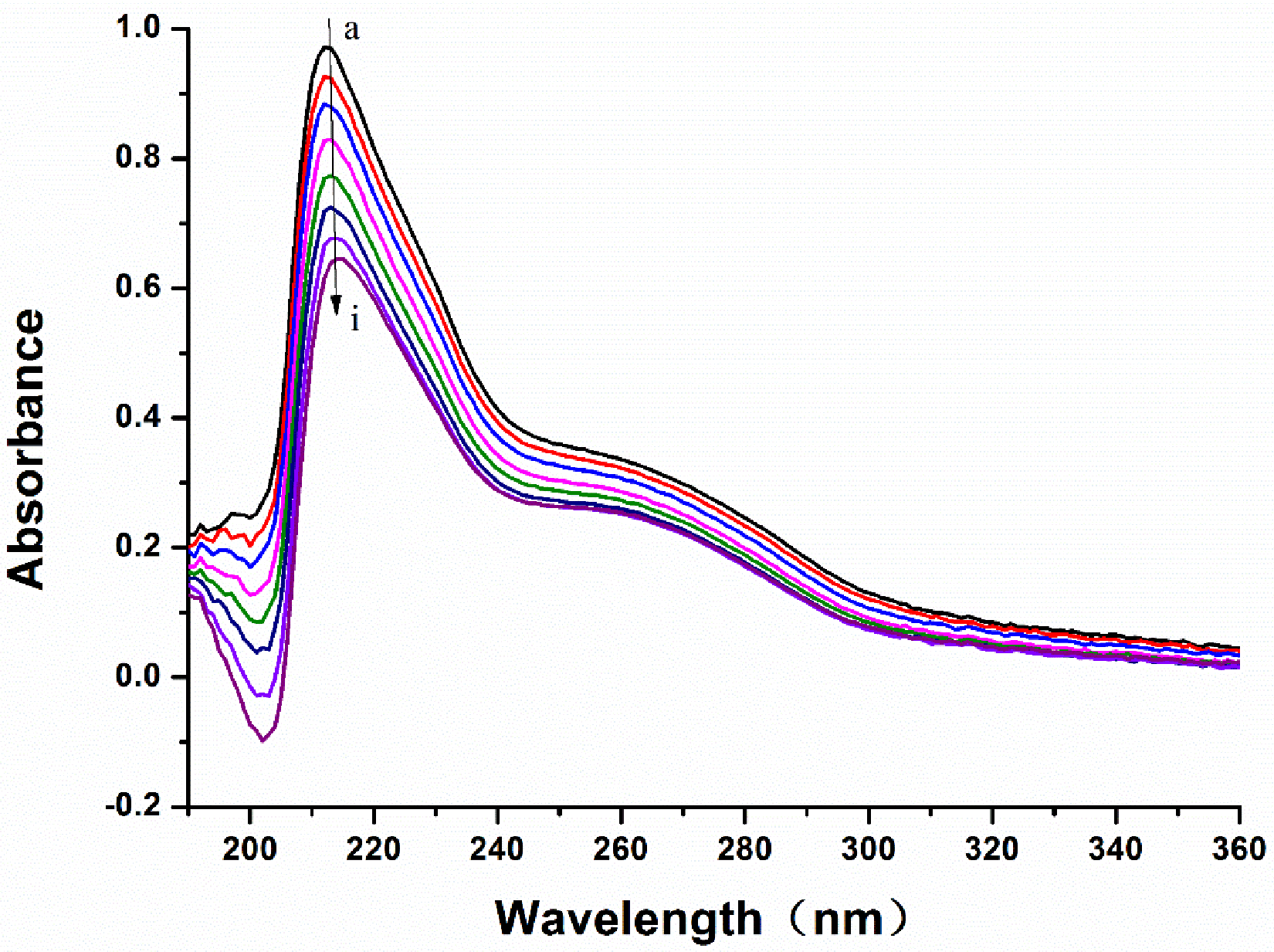
Reliable prediction model has the capacity to correctly predict potential candidate drugs. In this study, a virtual screening was applied based on our MAO-B inhibitor identification and activity models. As a result, LB was predicted as a potential MAO-B inhibitor. Our experiment also showed that LB inhibits MAO-B well. According to the experimental results of the fluorescence spectrum, compound LB can also cause good fluorescence quenching of MAO-B. Molecular structure and chemical environment are important factors affecting the emission fluorescence and fluorescence intensity of substances. In biological macromolecules, higher numbers of aromatic hydrocarbons or conjugated double bonds lead to strong fluorescence produced by organic compounds. Meanwhile, the nature of substituents also has a great impact on the fluorescence intensity of phosphors. Substituents on a benzene ring will lead to the displacement of maximum absorption wavelength and the change of fluorescence peak. Generally, electronic groups, including—NH2, -Oh, -OCH3, -nhch3 and -n (CH3) 2, can enhance the fluorescence, while electron-absorbing groups, including—Cl, -Br, -I, -nhcoch3, -NO2 and -COOH, will weaken the fluorescence. Compound LB has several -OCH3, and when -OCH3 is connected with the benzene ring, it can be used as an electron donor group only if it forms a para-position effect. In the molecular structure of compound LB, the methoxy group is an electron-absorbing group, so compound LB has stronger fluorescence quenching of MAO-B, which also shows that compound LB has a greater impact on the aromatic amino acids in MAO-B molecule. In addition, as the structure of compound LB is symmetrical, the charge distribution is also uniform and symmetrical, which cause smaller dipole moment. Therefore, the polarity of compound LB is small, while MAO-B focuses on the decomposition and deamination of non-polar aromatic amine phenylethylamine, which may be the reason for the better inhibition effect of L. H-bond force is an important force for the formation of stable protein ground complex, and compound LB has a better inhibitory effect on MAO-B.
The results of molecule docking showed that LB binds to MAO-B at Phe168, CYS 172, ILE198, GLN206 and TYR435 through H-bond.
Figure 5 showed that GLN206 is a main amino acid residue which could interact with high activity inhibitors. Hence, GLN206 could be regarded as an important site for inhibitors. Interestingly, our experiments of the fluorescence quenching of MAO-B showed that there is only one binding site for LB to interact with MAO-B, which is different from the docking results. The possible explanation may be that tryptophan (try), tyrosine (Tyr) and phenylalanine (PHE) are the only components of natural amino acids that can emit fluorescence, which can be determined by the fluorescence method. In our molecule docking study, Phe168 was one of the acting sites among the five amino acid residues. Hence, in the fluorescence quenching experiments, the fluorescence intensity decreased, and only one site was concluded.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

