
Galectin-3 Is a Natural Binding Ligand of MCAM (CD146, MUC18) in Melanoma Cells and Their Interaction Promotes Melanoma Progression
 , and
, and 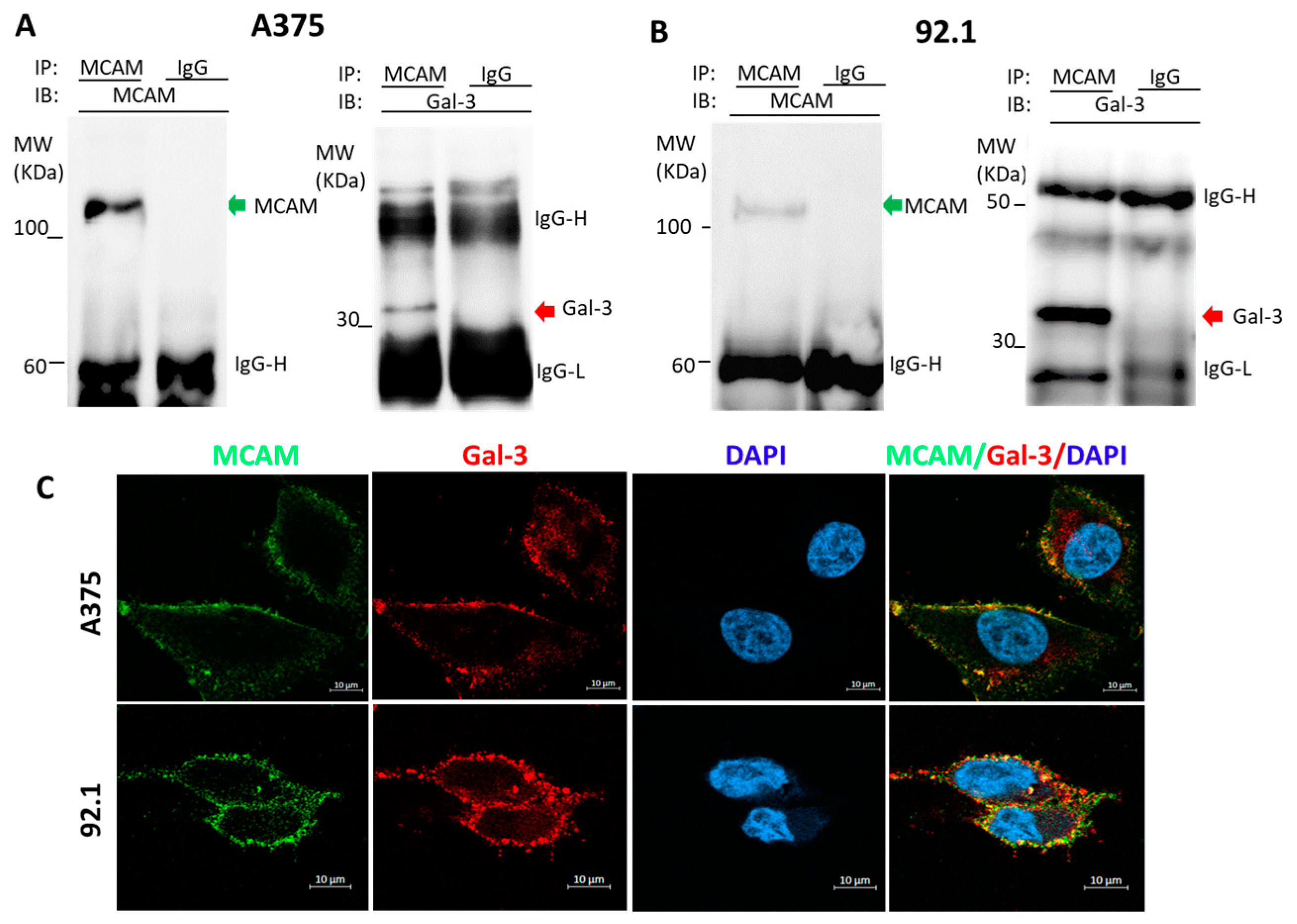{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cells
2.3. Production of Recombinant Galectin-3 and Truncated Galectin-3
2.4. Cell Proliferation
2.5. Cell Migration
2.6. Cell Adhesion
2.7. Cell Invasion
2.8. Colony Formation
2.9. ShRNA MCAM Suppression
2.10. MCAM Dimerization
2.11. Immunofluorescence and Confocal Microscopy
2.12. Co-Immunoprecipitation
2.13. Protein De-Glycosylation
2.14. Statistical Analysis
3. Results
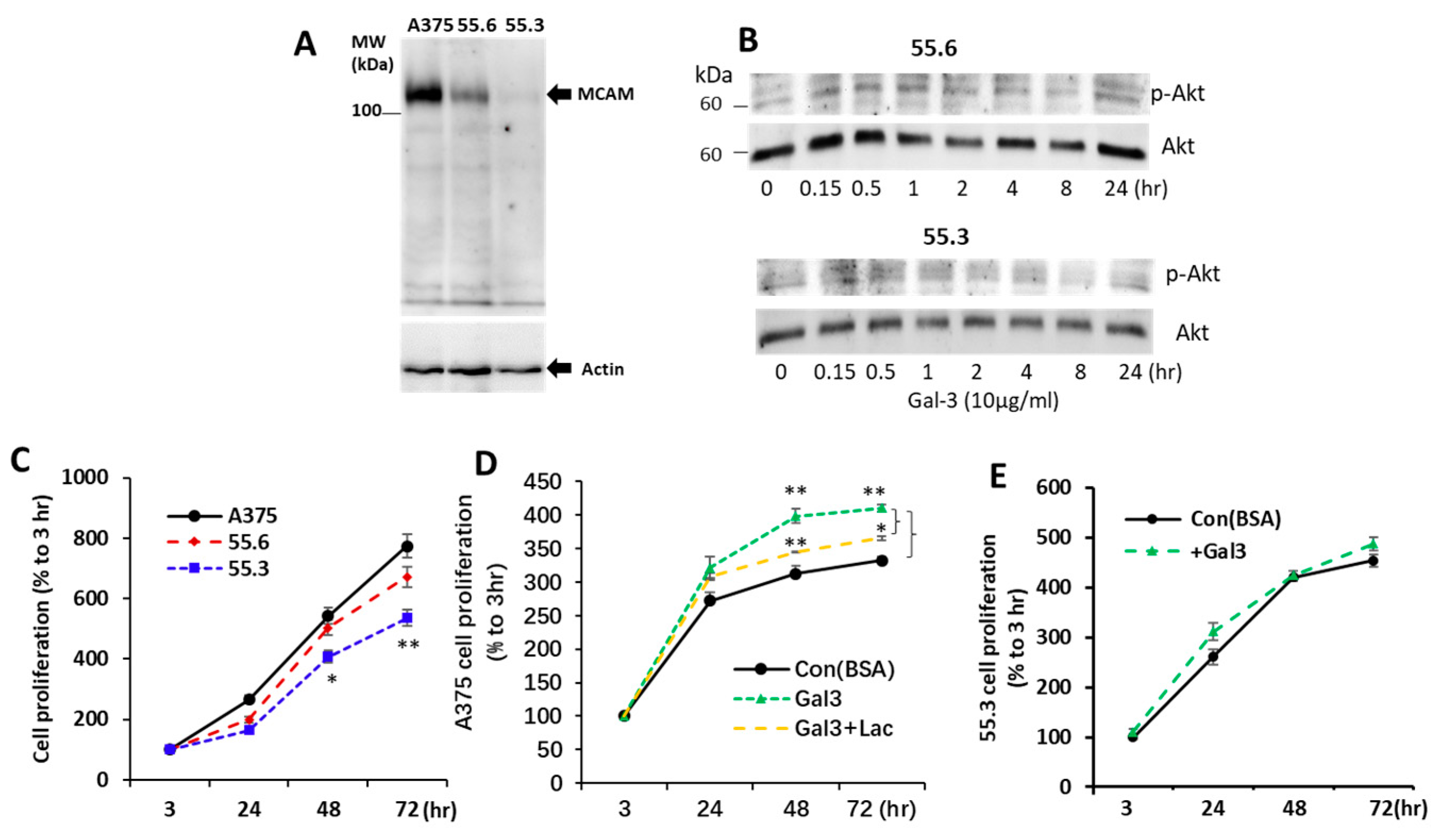
3.1. Galectin-3 Is a Natural Ligand of MCAM in Human Melanoma Cells
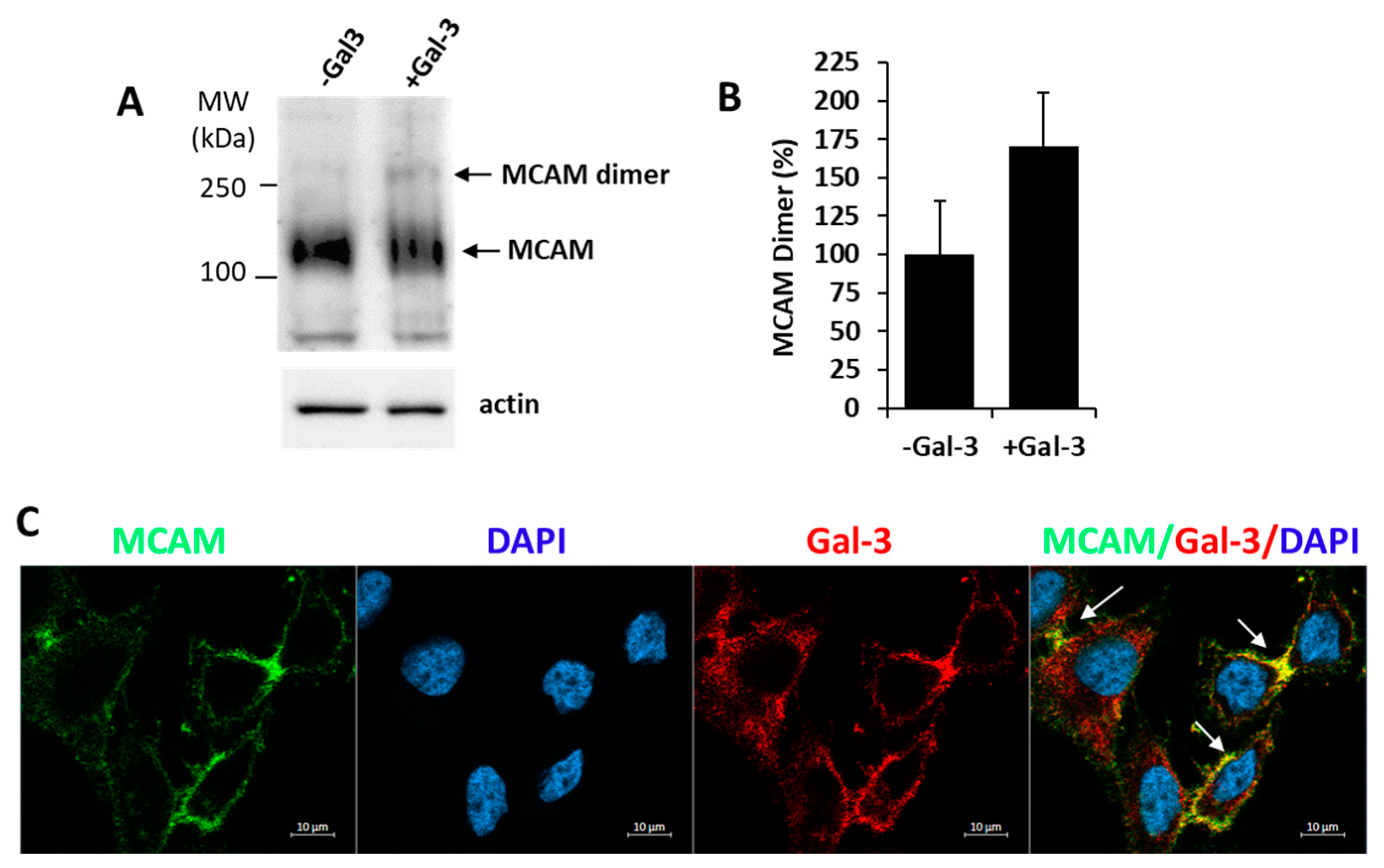
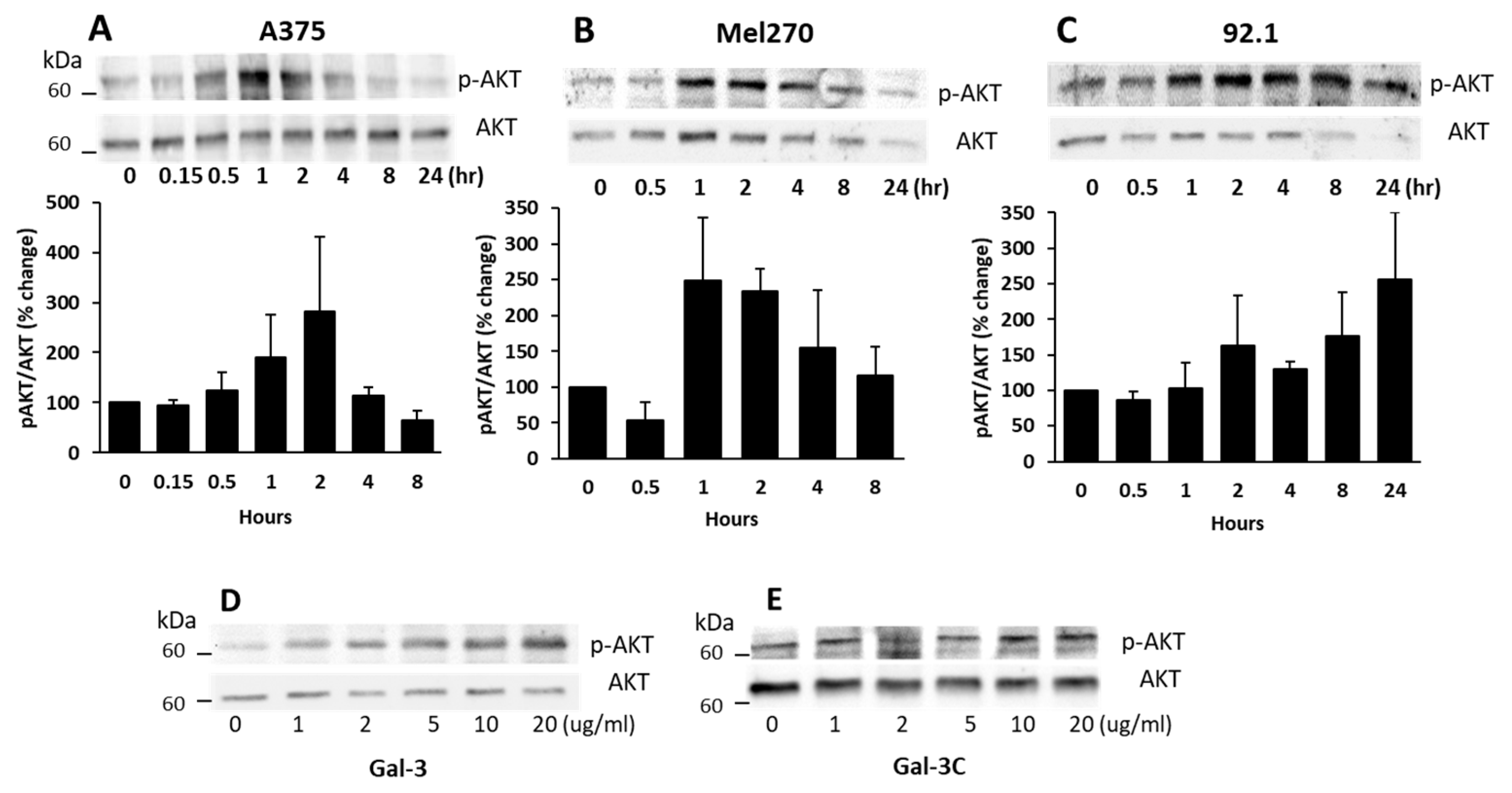
3.2. Galectin-3 Binding to MCAM Induces MCAM Dimerization and Subsequent Downstream AKT Signalling
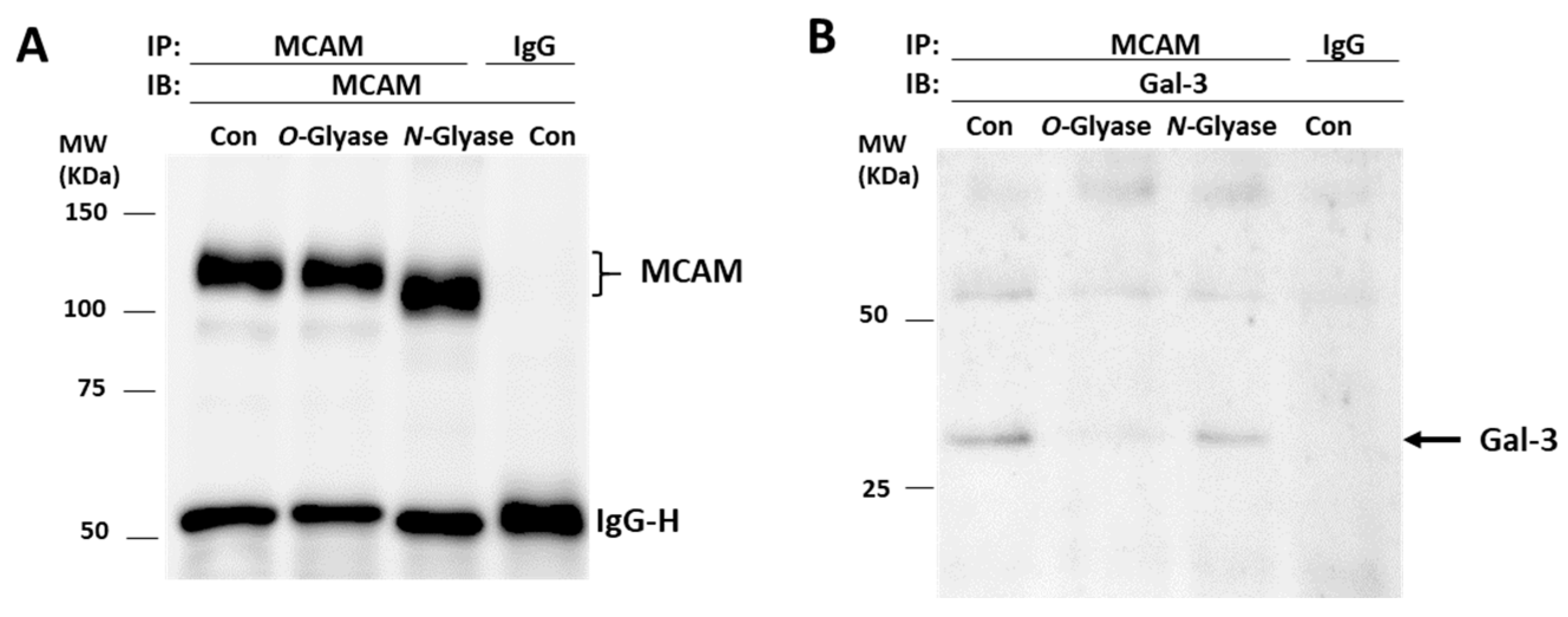
3.3. Galectin-3 Binds to MCAM via O-Linked Glycans
3.4. Suppression of MCAM Expression by shRNA Abolishes Galectin-3-Induced AKT Activation
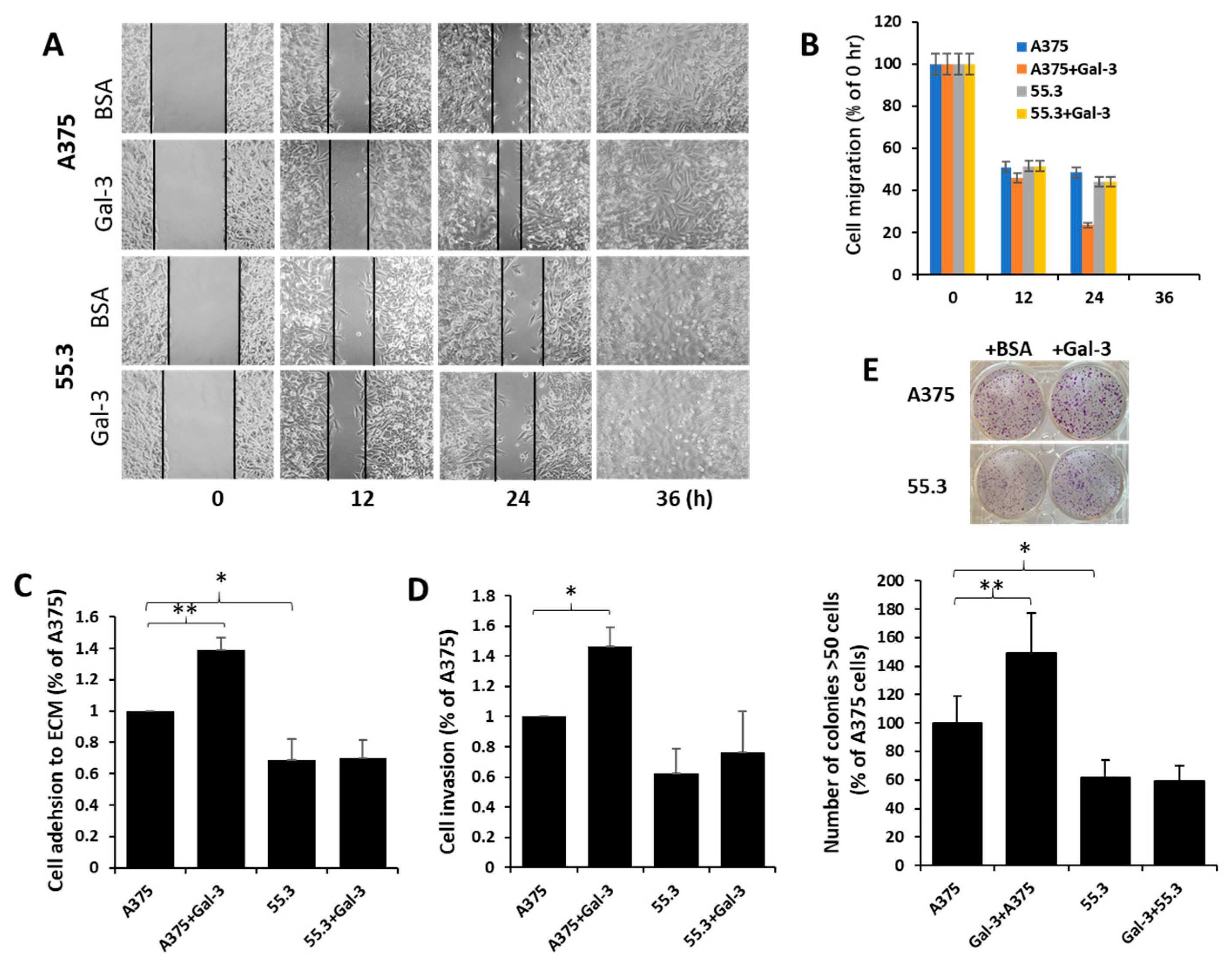
3.5. Galectin-3–MCAM Interaction Promotes Melanoma Cell Proliferation, Adhesion, Migration, and Invasion
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Prim. 2015, 1, 15003. [Google Scholar] [CrossRef] [PubMed]
- Sers, C.; Riethmüller, G.; Johnson, J.P. MUC18, a melanoma-progression associated molecule, and its potential role in tumor vascularization and hematogenous spread. Cancer Res. 1994, 54, 5689–5694. [Google Scholar] [PubMed]
- Lei, X.; Guan, C.W.; Song, Y.; Wang, H. The multifaceted role of CD146/MCAM in the promotion of melanoma progression. Cancer Cell Int. 2015, 15, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapanotti, M.C.; Cugini, E.; Nuccetelli, M.; Terrinoni, A.; Di Raimondo, C.; Lombardo, P.; Costanza, G.; Cosio, T.; Rossi, P.; Orlandi, A.; et al. MCAM/MUC18/CD146 as a multifaceted warning marker of melanoma progression in liquid biopsy. Int. J. Mol. Sci. 2021, 22, 12416. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.L.; Millward, M.; Pearce, R.; Lee, M.; Frank, M.H.; Ireland, A.; Monshizadeh, L.; Rai, T.; Heenan, P.; Medic, S.; et al. Markers of circulating tumour cells in the peripheral blood of patients with melanoma correlate with disease recurrence and progression. Br. J. Dermatol. 2013, 168, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Ruma, I.M.W.; Putranto, E.W.; Kondo, E.; Murata, H.; Watanabe, M.; Huang, P.; Kinoshita, R.; Futami, J.; Inoue, Y.; Yamauchi, A.; et al. MCAM, as a novel receptor for S100A8/A9, mediates progression of malignant melanoma through prominent activation of NF-kappaB and ROS formation upon ligand binding. Clin. Exp. Metastasis 2016, 33, 609–627. [Google Scholar] [CrossRef]
- Jouve, N.; Bachelier, R.; Despoix, N.; Blin, M.G.; Matinzadeh, M.K.; Poitevin, S.; Aurrand-Lions, M.; Fallague, K.; Bardin, N.; Blot-Chabaud, M.; et al. CD146 mediates VEGF-induced melanoma cell extravasation through FAK activation. Int. J. Cancer 2015, 137, 50–60. [Google Scholar] [CrossRef]
- Leslie, M.C.; Zhao, Y.J.; Lachman, L.B.; Hwu, P.; Bar-Eli, M. Immunization against MUC18/MCAM, a novel antigen that drives melanoma invasion and metastasis. Gene Ther. 2007, 14, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Mills, L.; Tellez, C.; Huang, S.; Baker, C.; Mccarty, M.; Green, L.; Gudas, J.M.; Feng, X.; Bar-Eli, M. Fully human antibodies to MCAM/MUC18 inhibit tumor growth and metastasis of human melanoma. Cancer Res. 2002, 62, 5106–5114. [Google Scholar]
- Feng, R.; Wang, Y.; Ramachandran, V.; Ma, Q.; May, M.M.; Li, M.; Zhou, J.X.; Xu, X.; Xu, K.; Fang, S.; et al. Characterization of novel neutralizing mouse monoclonal antibody JM1-24-3 developed against MUC18 in metastatic melanoma. J. Exp. Clin. Cancer Res. 2020, 39, 273. [Google Scholar] [CrossRef]
- Sechler, M.; Parrish, J.K.; Birks, D.K.; Jedlicka, P. The histone demethylase KDM3A, and its downstream target MCAM, promote Ewing Sarcoma cell migration and metastasis. Oncogene 2017, 36, 4150–4160. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Gaur, R.L.; Elmageed, Z.Y.A.; Fernando, A.; Thouta, R.; Trappey, A.K.; Abdraboh, M.E.; El-Sayyad, H.I.; Rao, P.; Raj, M.G. Towards understanding the mode of action of the multifaceted cell adhesion receptor CD146. Biochim. Biophys. Acta 2009, 1795, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yan, X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013, 330, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Kalabis, J.; Xu, X.; Meier, F.; Oka, M.; Bogenrieder, T.; Herlyn, M. Reciprocal regulation of MelCAM and AKT in human melanoma. Oncogene 2003, 22, 6891–6899. [Google Scholar] [CrossRef] [Green Version]
- Colomb, F.; Wang, W.; Simpson, D.; Zafar, M.; Beynon, R.; Rhodes, J.M.; Yun, L.G. Galectin-3 interacts with the cell-surface glycoprotein CD146 (MCAM, MUC18) and induces secretion of metastasis-promoting cytokines from vascular endothelial cells. J. Biol. Chem. 2017, 292, 8381–8389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Duckworth, C.A.; Zhao, Q.; Pritchard, D.M.; Rhodes, J.M.; Yu, L.-G. Increased circulation of galectin-3 in cancer induces secretion of metastasis-promoting cytokines from blood vascular endothelium. Clin. Cancer Res. 2013, 19, 1693–1704. [Google Scholar] [CrossRef] [Green Version]
- Newlaczyl, U.A.; Yu, L.G. Galectin-3—A jack-of-all-trades in cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef]
- Vereecken, P.; Awada, A.; Suciu, S.; Castro, G.; Morandini, R.; Litynska, A.; Lienard, D.; Ezzedine, K.; Ghanem, G.; Heenen, M. Evaluation of the prognostic significance of serum galectin-3 in American Joint Committee on Cancer stage III and stage IV melanoma patients. Melanoma Res. 2009, 19, 316–320. [Google Scholar] [CrossRef]
- Prieto, V.G.; Mourad-Zeidan, A.A.; Melnikova, V.; Johnson, M.M.; Lopez, A.; Diwan, A.H.; Lazar, A.J.; Shen, S.S.; Zhang, P.S.; Reed, J.A.; et al. Galectin-3 expression is associated with tumor progression and pattern of sun exposure in melanoma. Clin. Cancer Res. 2006, 12, 6709–6715. [Google Scholar] [CrossRef] [Green Version]
- Dye, D.E.; Emedic, S.; Eziman, M.; Coombe, D.R. Melanoma biomolecules: Independently identified but functionally intertwined. Front. Oncol. 2013, 3, 252. [Google Scholar] [CrossRef] [Green Version]
- Souri, Z.; Wierenga, A.P.A.; Kroes, W.G.M.; van der Velden, P.A.; Verdijk, R.M.; Eikmans, M.; Luyten, G.P.M.; Jager, M.J. LAG3 and its ligands show increased expression in high-risk uveal melanoma. Cancers 2021, 13, 4445. [Google Scholar] [CrossRef]
- Liu, T.F.; Rabinovich, G.A. Galectins as modulators of tumour progression. Nat. Rev. Cancer 2005, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Barrow, H.; Rhodes, J.M.; Yu, L.-G. The role of galectins in colorectal cancer progression. Int. J. Cancer 2011, 129, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dange, M.C.; Srinivasan, N.; More, S.K.; Bane, S.M.; Upadhya, A.; Ingle, A.D.; Gude, R.P.; Mukhopadhyaya, R.; Kalraiya, R.D. Galectin-3 expressed on different lung compartments promotes organ specific metastasis by facilitating arrest, extravasation and organ colonization via high affinity ligands on melanoma cells. Clin. Exp. Metastasis 2014, 31, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Sindrewicz, P.; Li, X.; Yates, E.A.; Turnbull, J.E.; Lian, L.-Y.; Yu, L.-G. Intrinsic tryptophan fluorescence spectroscopy reliably determines galectin-ligand interactions. Sci. Rep. 2019, 9, 11851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, C.A.; Guimond, S.E.; Sindrewicz, P.; Hughes, A.J.; French, N.S.; Lian, L.-Y.; Yates, E.A.; Pritchard, D.M.; Rhodes, J.M.; Turnbull, J.E.; et al. Chemically modified, non-anticoagulant heparin derivatives are potent galectin-3 binding inhibitors and inhibit circulating galectin-3-promoted metastasis. Oncotarget 2015, 6, 23671–23687. [Google Scholar] [CrossRef] [Green Version]
- Barrow, H.; Guo, X.; Wandall, H.H.; Pedersen, J.W.; Fu, B.; Zhao, Q.; Chen, C.; Rhodes, J.M.; Yu, L.-G. Serum galectin-2,-4, and -8 are greatly increased in colon and breast cancer patients and promote cancer cell adhesion to blood vascular endothelium. Clin. Cancer Res. 2011, 17, 7035–7046. [Google Scholar] [CrossRef] [Green Version]
- Erdbruegger, U.; Dhaygude, A.; Haubitz, M.; Woywodt, A. Circulating endothelial cells: Markers and mediators of vascular damage. Curr. Stem Cell Res. Ther. 2010, 5, 294–302. [Google Scholar] [CrossRef]
- Jiang, T.; Zhuang, J.; Duan, H.; Luo, Y.; Zeng, Q.; Fan, K.; Yan, H.; Lu, D.; Ye, Z.; Hao, J.; et al. CD146 is a coreceptor for VEGFR-2 in tumor angiogenesis. Blood 2012, 120, 2330–2339. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Zhang, C.; Yan, H.; Luo, Y.; Kong, R.; Wen, P.; Ye, Z.; Chen, J.; Feng, J.; Liu, F.; et al. CD146 acts as a novel receptor for netrin-1 in promoting angiogenesis and vascular development. Cell Res. 2015, 25, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Jouve, N.; Despoix, N.; Espeli, M.; Gauthier, L.; Cypowyj, S.; Fallague, K.; Schiff, C.; Dignat-George, F.; Vély, F.; Leroyer, A.S. The involvement of CD146 and its novel ligand Galectin-1 in apoptotic regulation of endothelial cells. J. Biol. Chem. 2013, 288, 2571–2579. [Google Scholar] [CrossRef]
- Flanagan, K.; Fitzgerald, K.; Baker, J.; Regnstrom, K.; Gardai, S.; Bard, F.; Mocci, S.; Seto, P.; You, M.; LaRochelle, C.; et al. Laminin-411 is a vascular ligand for MCAM and facilitates TH17 cell entry into the CNS. PLoS ONE 2012, 7, e40443. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, E.M.; Geddes-Sweeney, J.E.; Cedeno-Laurent, F.; Walley, K.C.; Barthel, S.R.; Opperman, M.J.; Liang, J.; Lin, J.Y.; Schatton, T.; Laga, A.C.; et al. Melanoma cell galectin-1 ligands functionally correlate with malignant potential. J. Investig. Dermatol. 2015, 135, 1849–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Zheng, C.; Zhang, J.; Lu, D.; Xing, S.; Feng, J.; Yang, D.; Yan, X. Recognition of CD146 as an ERM-binding protein offers novel mechanisms for melanoma cell migration. Oncogene 2012, 31, 306–321. [Google Scholar] [CrossRef] [Green Version]
- Sauer, N.; Szlasa, W.; Jonderko, L.; Oślizło, M.; Kunachowicz, D.; Kulbacka, J.; Karłowicz-Bodalska, K. LAG-3 as a potent target for novel anticancer therapies of a wide range of tumors. Int. J. Mol. Sci. 2022, 23, 9958. [Google Scholar] [CrossRef]
- Yu, L.G. The oncofetal Thomsen-Friedenreich carbohydrate antigen in cancer progression. Glycoconj. J. 2007, 24, 411–420. [Google Scholar] [CrossRef]
- Sindrewicz, P.; Lian, L.Y.; Yu, L.G. Interaction of the oncofetal Thomsen-Friedenreich antigen with galectins in cancer progression and metastasis. Front. Oncol. 2016, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.-G.; Andrews, N.; Zhao, Q.; McKean, D.; Williams, J.F.; Connor, L.J.; Gerasimenko, O.V.; Hilkens, J.; Hirabayashi, J.; Kasai, K.; et al. Galectin-3 interaction with Thomsen-Friedenreich disaccharide on cancer-associated MUC1 causes increased cancer cell endothelial adhesion. J. Biol. Chem. 2007, 282, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Shih, I.M.; Elder, D.E.; Speicher, D.; Johnson, J.P.; Herlyn, M. Isolation and functional characterization of the A32 melanoma-associated antigen. Cancer Res. 1994, 54, 2514–2520. [Google Scholar]
- Vellis, D.C.; Pietrobono, S.; Stecca, B. The role of glycosylation in melanoma progression. Cells 2021, 10, 2136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Miller, M.C.; Xu, X.; Song, C.; Zhang, F.; Zheng, Y.; Zhou, Y.; Tai, G.; Mayo, K.H. NMR-based insight into galectin-3 binding to endothelial cell adhesion molecule CD146: Evidence for noncanonical interactions with the lectin’s CRD beta-sandwich F-face. Glycobiology 2019, 29, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Nabi, R.I.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell Sci. 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Guo, X.; Nash, G.B.; Stone, P.C.; Hilkens, J.; Rhodes, J.M.; Yu, L.-G. Circulating galectin-3 promotes metastasis by modifying MUC1 localization on cancer cell surface. Cancer Res. 2009, 69, 6799–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piyush, T.; Chacko, A.R.; Sindrewicz, P.; Hilkens, J.; Rhodes, J.M.; Yu, L.-G. Interaction of galectin-3 with MUC1 on cell surface promotes EGFR dimerization and activation in human epithelial cancer cells. Cell Death Differ. 2017, 24, 1937–1947. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Y.; Maxwell, E.; Sindrewicz-Goral, P.; Shapanis, A.; Li, S.; Morgan, M.; Yu, L.-G. Galectin-3 Is a Natural Binding Ligand of MCAM (CD146, MUC18) in Melanoma Cells and Their Interaction Promotes Melanoma Progression. Biomolecules 2022, 12, 1451. https://doi.org/10.3390/biom12101451
Pang Y, Maxwell E, Sindrewicz-Goral P, Shapanis A, Li S, Morgan M, Yu L-G. Galectin-3 Is a Natural Binding Ligand of MCAM (CD146, MUC18) in Melanoma Cells and Their Interaction Promotes Melanoma Progression. Biomolecules. 2022; 12(10):1451. https://doi.org/10.3390/biom12101451
Chicago/Turabian StylePang, Yaoyu, Ellen Maxwell, Paulina Sindrewicz-Goral, Andrew Shapanis, Shun Li, Mark Morgan, and Lu-Gang Yu. 2022. "Galectin-3 Is a Natural Binding Ligand of MCAM (CD146, MUC18) in Melanoma Cells and Their Interaction Promotes Melanoma Progression" Biomolecules 12, no. 10: 1451. https://doi.org/10.3390/biom12101451