
Potassium Channels as Therapeutic Targets in Pulmonary Arterial Hypertension

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
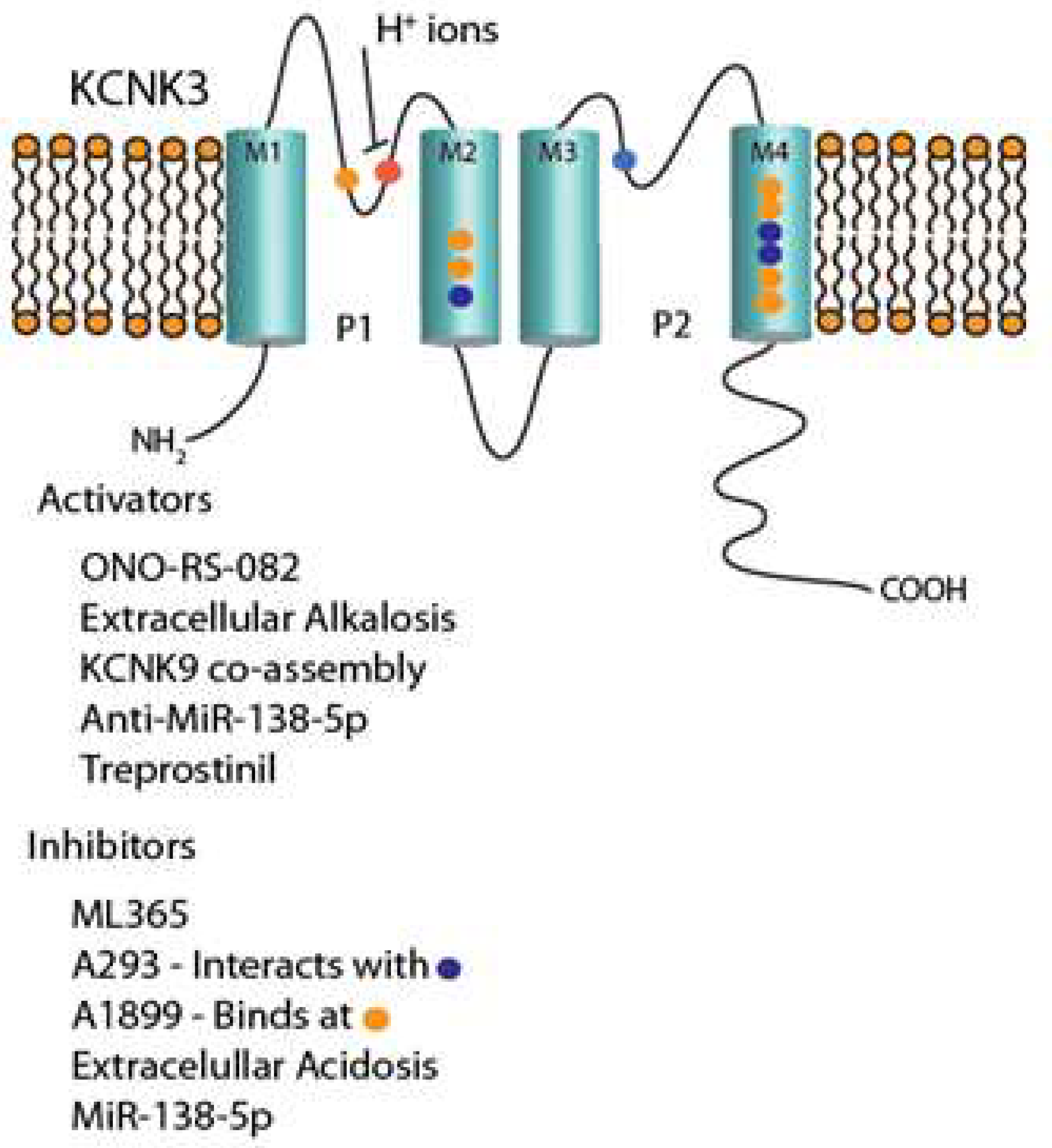
2. KCNK3
2.1. Introduction and Molecular Biology
2.2. KCNK3 Channel in PH
2.3. KCNK3 Mutations in PAH
2.4. KCNK3 Dysfunction in Human and Animal Models of PH
2.5. Cellular Electrophysiology and Pharmacology of KCNK3 PH Rats
2.6. KCNK3 Currents and Pulmonary Arterial Constriction
2.7. KCNK3 Dysfunction in PH Using a CRISPR/Cas9 Rat
2.8. Signaling Pathways Involved in KCNK3 Regulation
2.9. KCNK3 and Immune Cells in PAH
2.10. KCNK3 Inhibitors and Activators
2.11. KCNK3 Inhibitors
2.11.1. A293
2.11.2. A1899
2.11.3. ML365
2.12. KCNK3 Activators
2.12.1. ONO-RS-082
2.12.2. KCNK9
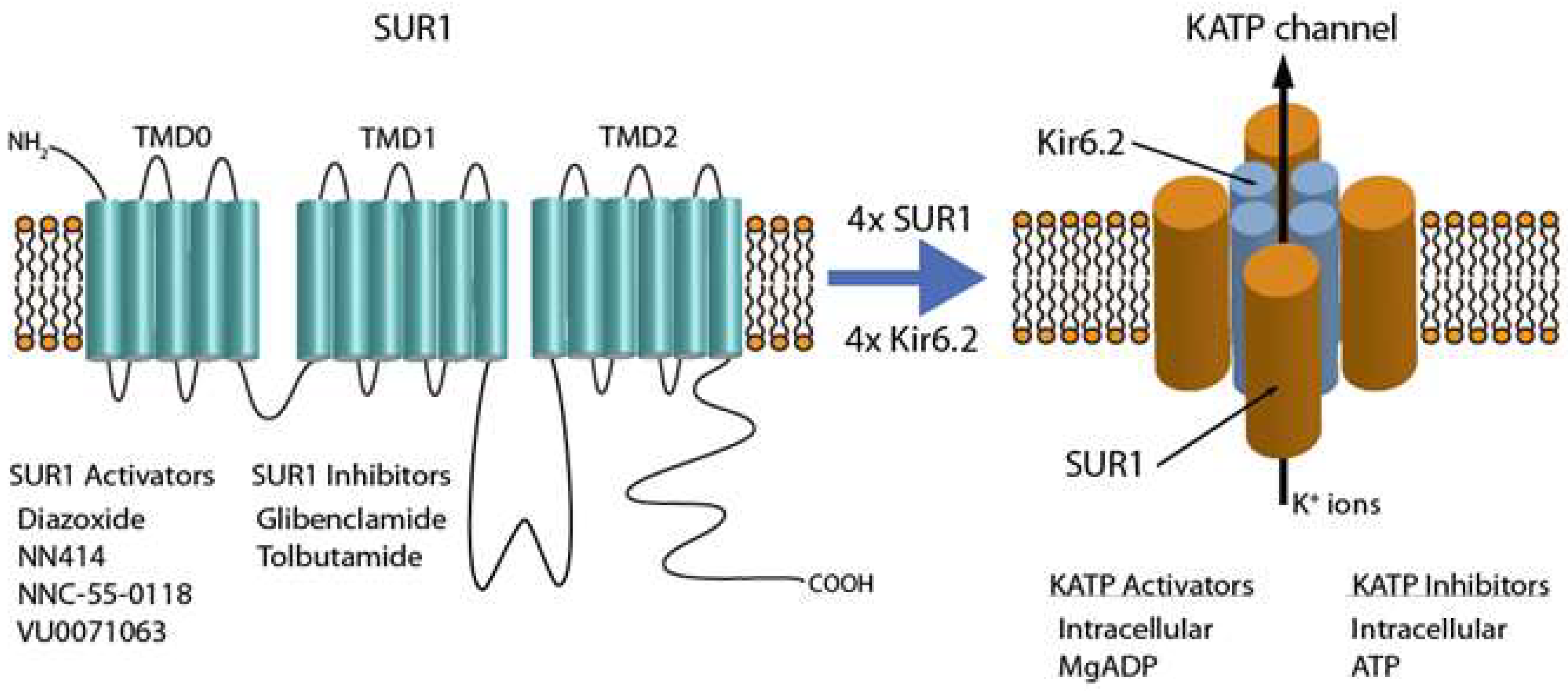
3. ABCC8/SUR1
3.1. Introduction and Molecular Biology
3.2. SUR1/KATP Channels in PAH
3.3. SUR1 Loss-of-Function in PAH
3.4. SUR1/KATP Channels and Vasoreactivity in the Pulmonary Vasculature
3.5. Inhibitors of SUR1/KATP Channel Function
3.6. Activators of SUR1/KATP Channel Function
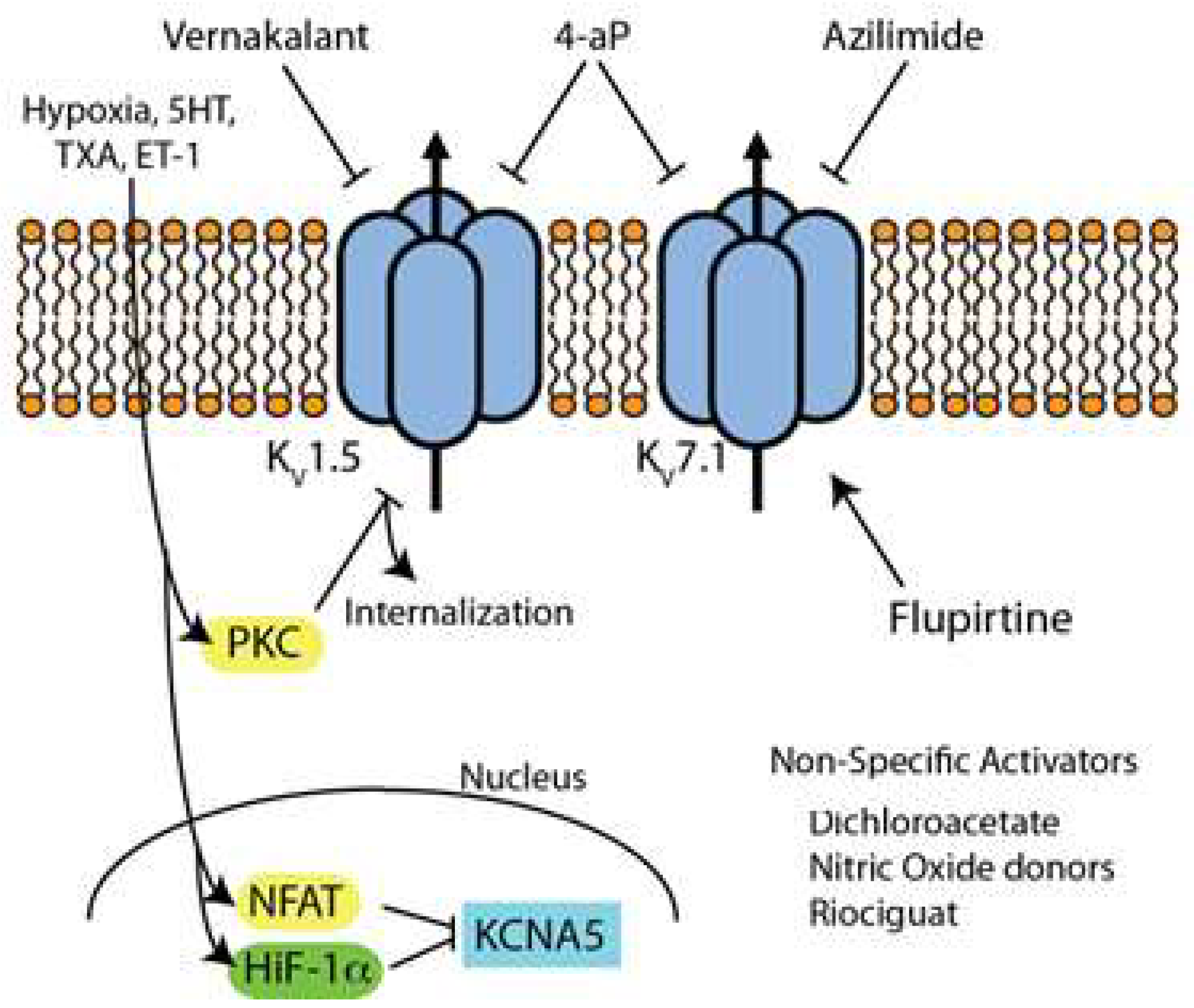
4. Kv Channels
4.1. Introduction and Molecular Biology
4.2. Kv Channels in PH
4.3. PAH-Disposing Mutations and Kv Channels
4.4. Inhibitors/Activators of Kv Channels (Figure 3)
4.5. Kv Channel Blockers
4.6. Kv Channel Activators/Up-Regulators
4.7. Kv Channel Future Studies
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.M.F.; Brida, M.; Carlsen, J.; Coats, A.J.S.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J. 2022, 2200879. [Google Scholar] [CrossRef] [PubMed]
- Boucherat, O.; Chabot, S.; Antigny, F.; Perros, F.; Provencher, S.; Bonnet, S. Potassium channels in pulmonary arterial hypertension. Eur. Respir. J. 2015, 46, 1167–1177. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Capuano, V.; Girerd, B.; Humbert, M.; Montani, D.; Antigny, F. Implication of Potassium Channels in the Pathophysiology of Pulmonary Arterial Hypertension. Biomolecules 2020, 10, 1261. [Google Scholar] [CrossRef]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am.Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Huertas, A.; Guignabert, C.; Barberà, J.A.; Bärtsch, P.; Bhattacharya, J.; Bhattacharya, S.; Bonsignore, M.R.; Dewachter, L.; Dinh-Xuan, A.T.; Dorfmüller, P.; et al. Pulmonary vascular endothelium: The orchestra conductor in respiratory diseases: Highlights from basic research to therapy. Eur. Respir. J. 2018, 51, 1700745. [Google Scholar] [CrossRef]
- Humbert, M.; Lau, E.M.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Yan, Y.; He, Y.-Y.; Jiang, X.; Wang, Y.; Chen, J.-W.; Zhao, J.-H.; Ye, J.; Lian, T.-Y.; Zhang, X.; Zhang, R.-J.; et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci. Adv. 2020, 6, eaba2470. [Google Scholar] [CrossRef]
- Varghese, M.V.; James, J.; Rafikova, O.; Rafikov, R. Glucose-6-phosphate dehydrogenase deficiency contributes to metabolic abnormality and pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L508–L521. [Google Scholar] [CrossRef]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Tregouet, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef]
- Bohnen, M.S.; Ma, L.; Zhu, N.; Qi, H.; McClenaghan, C.; Gonzaga-Jauregui, C.; Dewey, F.E.; Overton, J.D.; Reid, J.G.; Shuldiner, A.R.; et al. Loss-of-Function ABCC8 Mutations in Pulmonary Arterial Hypertension. Circ. Genom. Precis. Med. 2018, 11, e002087. [Google Scholar] [CrossRef]
- Ketchum, K.A.; Joiner, W.J.; Sellers, A.J.; Kaczmarek, L.K.; Goldstein, S.A. A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 1995, 376, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Lesage, F.; Guillemare, E.; Fink, M.; Duprat, F.; Lazdunski, M.; Romey, G.; Barhanin, J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 1996, 15, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.M.; Zilberberg, N.; Goldstein, S.A. Block of Kcnk3 by protons. Evidence that 2-P-domain potassium channel subunits function as homodimers. J. Biol. Chem. 2001, 276, 24449–24452. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, A.; Li, Y.; Tang, B.; Hanze, J.; Eul, B.; Bohle, R.M.; Wilhelm, J.; Morty, R.E.; Brau, M.E.; Weir, E.K.; et al. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ. Res. 2006, 98, 1072–1080. [Google Scholar] [CrossRef]
- Olschewski, A.; Veale, E.L.; Nagy, B.M.; Nagaraj, C.; Kwapiszewska, G.; Antigny, F.; Lambert, M.; Humbert, M.; Czirjak, G.; Enyedi, P.; et al. TASK-1 (KCNK3) channels in the lung: From cell biology to clinical implications. Eur. Respir. J. 2017, 50, 1700754. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; Kempsill, F.E. Potassium channels underlying the resting potential of pulmonary artery smooth muscle cells. Clin. Exp. Pharmacol. Physiol. 2002, 29, 330–333. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef]
- Gardener, M.J.; Johnson, I.T.; Burnham, M.P.; Edwards, G.; Heagerty, A.M.; Weston, A.H. Functional evidence of a role for two-pore domain potassium channels in rat mesenteric and pulmonary arteries. Br. J. Pharmacol. 2004, 142, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Bohnen, M.S.; Roman-Campos, D.; Terrenoire, C.; Jnani, J.; Sampson, K.J.; Chung, W.K.; Kass, R.S. The Impact of Heterozygous KCNK3 Mutations Associated With Pulmonary Arterial Hypertension on Channel Function and Pharmacological Recovery. J. Am. Heart Assoc. 2017, 6, e006465. [Google Scholar] [CrossRef] [PubMed]
- Best, D.H.; Sumner, K.L.; Smith, B.P.; Damjanovich-Colmenares, K.; Nakayama, I.; Brown, L.M.; Ha, Y.; Paul, E.; Morris, A.; Jama, M.A.; et al. EIF2AK4 Mutations in Patients Diagnosed With Pulmonary Arterial Hypertension. Chest 2017, 151, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Higasa, K.; Ogawa, A.; Terao, C.; Shimizu, M.; Kosugi, S.; Yamada, R.; Date, H.; Matsubara, H.; Matsuda, F. A burden of rare variants in BMPR2 and KCNK3 contributes to a risk of familial pulmonary arterial hypertension. BMC Pulm. Med. 2017, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Navas, P.; Tenorio, J.; Quezada, C.A.; Barrios, E.; Gordo, G.; Arias, P.; Lopez Meseguer, M.; Santos-Lozano, A.; Palomino Doza, J.; Lapunzina, P.; et al. Molecular Analysis of BMPR2, TBX4, and KCNK3 and Genotype-Phenotype Correlations in Spanish Patients and Families With Idiopathic and Hereditary Pulmonary Arterial Hypertension. Rev. Esp. Cardiol. 2016, 69, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Antigny, F.; Hautefort, A.; Meloche, J.; Belacel-Ouari, M.; Manoury, B.; Rucker-Martin, C.; Pechoux, C.; Potus, F.; Nadeau, V.; Tremblay, E.; et al. Potassium Channel Subfamily K Member 3 (KCNK3) Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2016, 133, 1371–1385. [Google Scholar] [CrossRef]
- Lambert, M.; Capuano, V.; Boet, A.; Tesson, L.; Bertero, T.; Nakhleh, M.K.; Remy, S.; Anegon, I.; Pechoux, C.; Hautefort, A.; et al. Characterization of Kcnk3-Mutated Rat, a Novel Model of Pulmonary Hypertension. Circ. Res. 2019, 125, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Lamalle, C.; Oliveira, R.; Reid, J.; Gurney, A.M. Contractile and electrophysiological properties of pulmonary artery smooth muscle are not altered in TASK-1 knockout mice. J. Physiol. 2011, 589, 3231–3246. [Google Scholar] [CrossRef]
- Pandit, L.M.; Lloyd, E.E.; Reynolds, J.O.; Lawrence, W.S.; Reynolds, C.; Wehrens, X.H.; Bryan, R.M. TWIK-2 channel deficiency leads to pulmonary hypertension through a rho-kinase-mediated process. Hypertension 2014, 64, 1260–1265. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef]
- Hopper, R.K.; Moonen, J.R.; Diebold, I.; Cao, A.; Rhodes, C.J.; Tojais, N.F.; Hennigs, J.K.; Gu, M.; Wang, L.; Rabinovitch, M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation 2016, 133, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Le Ribeuz, H.; Courboulin, A.; Ghigna, M.R.; Lambert, M.; Hautefort, A.; Humbert, M.; Montani, D.; Cohen-Kaminsky, S.; Perros, F.; Antigny, F. In vivo miR-138-5p inhibition alleviates monocrotaline-induced pulmonary hypertension and normalizes pulmonary KCNK3 and SLC45A3 expression. Respir. Res. 2020, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Li, H.; Zhang, P.; Yu, X.; Jiang, J.; Chen, S. Down-regulation of lncRNA Gas5 promotes hypoxia-induced pulmonary arterial smooth muscle cell proliferation by regulating KCNK3 expression. Eur. J. Pharmacol. 2020, 889, 173618. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Song, N.; Hu, X.; Zhu, A.; Wei, X.; Liu, J.; Yuan, S.; Mao, W.; Chen, X. Inhibition of RELM-beta prevents hypoxia-induced overproliferation of human pulmonary artery smooth muscle cells by reversing PLC-mediated KCNK3 decline. Life Sci. 2020, 246, 117419. [Google Scholar] [CrossRef]
- Le Ribeuz, H.; Dumont, F.; Ruellou, G.; Lambert, M.; Balliau, T.; Quatredeniers, M.; Girerd, B.; Cohen-Kaminsky, S.; Mercier, O.; Yen-Nicolay, S.; et al. Proteomic Analysis of KCNK3 Loss of Expression Identified Dysregulated Pathways in Pulmonary Vascular Cells. Int. J. Mol. Sci. 2020, 21, 7400. [Google Scholar] [CrossRef] [PubMed]
- West, J.D.; Austin, E.D.; Rizzi, E.M.; Yan, L.; Tanjore, H.; Crabtree, A.L.; Moore, C.S.; Muthian, G.; Carrier, E.J.; Jacobson, D.A.; et al. KCNK3 Mutation Causes Altered Immune Function in Pulmonary Arterial Hypertension Patients and Mouse Models. Int. J. Mol. Sci. 2021, 22, 5014. [Google Scholar] [CrossRef] [PubMed]
- Putzke, C.; Wemhoner, K.; Sachse, F.B.; Rinne, S.; Schlichthorl, G.; Li, X.T.; Jae, L.; Eckhardt, I.; Wischmeyer, E.; Wulf, H.; et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc. Res. 2007, 75, 59–68. [Google Scholar] [CrossRef]
- Wiedmann, F.; Kiper, A.K.; Bedoya, M.; Ratte, A.; Rinne, S.; Kraft, M.; Waibel, M.; Anad, P.; Wenzel, W.; Gonzalez, W.; et al. Identification of the A293 (AVE1231) Binding Site in the Cardiac Two-Pore-Domain Potassium Channel TASK-1: A Common Low Affinity Antiarrhythmic Drug Binding Site. Cell. Physiol. Biochem. 2019, 52, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.K.; Netter, M.F.; Kempf, F.; Walecki, M.; Rinne, S.; Bollepalli, M.K.; Preisig-Muller, R.; Renigunta, V.; Daut, J.; Baukrowitz, T.; et al. A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J. Biol. Chem. 2011, 286, 13977–13984. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, D.P.; Simpson, D.S.; Miller, M.; Maki, B.E.; Zou, B.; Shi, J.; Wu, M.; McManus, O.B.; Aube, J.; Li, M.; et al. Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold. Bioorg. Med. Chem. Lett. 2014, 24, 3968–3973. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.P.; Holden, R.G.; Escribano-Subias, P.M.; Cogolludo, A.; Veale, E.L.; Mathie, A. Characterization and regulation of wild-type and mutant TASK-1 two pore domain potassium channels indicated in pulmonary arterial hypertension. J. Physiol. Lond. 2019, 597, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Wischmeyer, E.; Liu, G.X.; Muller, R.P.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel—An extracellular histidine as pH sensor. J. Biol. Chem. 2000, 275, 16650–16657. [Google Scholar] [CrossRef]
- Kim, Y.; Bang, H.; Kim, D. TASK-3, a new member of the tandem pore K+ channel family. J. Biol. Chem. 2000, 275, 9340–9347. [Google Scholar] [CrossRef]
- Czirjak, G.; Enyedi, P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem. 2002, 277, 5426–5432. [Google Scholar] [CrossRef]
- Berg, A.P.; Talley, E.M.; Manger, J.P.; Bayliss, D.A. Motoneurons express heteromeric TWIK-related acid-sensitive K+ (TASK) channels containing TASK-1 (KCNK3) and TASK-3 (KCNK9) subunits. J. Neurosci. 2004, 24, 6693–6702. [Google Scholar] [CrossRef] [PubMed]
- Rinne, S.; Kiper, A.K.; Schlichthorl, G.; Dittmann, S.; Netter, M.F.; Limberg, S.H.; Silbernagel, N.; Zuzarte, M.; Moosdorf, R.; Wulf, H.; et al. TASK-1 and TASK-3 may form heterodimers in human atrial cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 81, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cavanaugh, E.J.; Kim, I.; Carroll, J.L. Heteromeric TASK-1/TASK-3 is the major oxygen-sensitive background K+ channel in rat carotid body glomus cells. J. Physiol.-London 2009, 587, 2963–2975. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Foster, M.N.; Coetzee, W.A. KATP Channels in the Cardiovascular System. Physiol. Rev. 2016, 96, 177–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aittoniemi, J.; Fotinou, C.; Craig, T.J.; de Wet, H.; Proks, P.; Ashcroft, F.M. Review. SUR1: A unique ATP-binding cassette protein that functions as an ion channel regulator. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, N.; Gonoi, T.; Clement, J.P.t.; Namba, N.; Inazawa, J.; Gonzalez, G.; Aguilar-Bryan, L.; Seino, S.; Bryan, J. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science 1995, 270, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Ashfield, R.; Ammala, C.; Ashcroft, F.M. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J. Physiol. 1997, 498 Pt 1, 87–98. [Google Scholar] [CrossRef]
- Lee, K.P.K.; Chen, J.; MacKinnon, R. Molecular structure of human KATP in complex with ATP and ADP. eLife 2017, 6, e32481. [Google Scholar] [CrossRef] [PubMed]
- McClenaghan, C.; Woo, K.V.; Nichols, C.G. Pulmonary Hypertension and ATP-Sensitive Potassium Channels. Hypertension 2019, 74, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Brundel, B.J.; Van Gelder, I.C.; Henning, R.H.; Tuinenburg, A.E.; Wietses, M.; Grandjean, J.G.; Wilde, A.A.; Van Gilst, W.H.; Crijns, H.J. Alterations in potassium channel gene expression in atria of patients with persistent and paroxysmal atrial fibrillation: Differential regulation of protein and mRNA levels for K+ channels. J. Am. Coll. Cardiol. 2001, 37, 926–932. [Google Scholar] [CrossRef]
- Flagg, T.P.; Kurata, H.T.; Masia, R.; Caputa, G.; Magnuson, M.A.; Lefer, D.J.; Coetzee, W.A.; Nichols, C.G. Differential structure of atrial and ventricular KATP: Atrial KATP channels require SUR1. Circ. Res. 2008, 103, 1458–1465. [Google Scholar] [CrossRef]
- Babenko, A.P.; Gonzalez, G.; Aguilar-Bryan, L.; Bryan, J. Reconstituted human cardiac KATP channels: Functional identity with the native channels from the sarcolemma of human ventricular cells. Circ. Res. 1998, 83, 1132–1143. [Google Scholar] [CrossRef]
- Bao, L.; Kefaloyianni, E.; Lader, J.; Hong, M.; Morley, G.; Fishman, G.I.; Sobie, E.A.; Coetzee, W.A. Unique properties of the ATP-sensitive K(+) channel in the mouse ventricular cardiac conduction system. Circ. Arrhythm. Electrophysiol. 2011, 4, 926–935. [Google Scholar] [CrossRef]
- Yoshida, H.; Feig, J.E.; Morrissey, A.; Ghiu, I.A.; Artman, M.; Coetzee, W.A. K ATP channels of primary human coronary artery endothelial cells consist of a heteromultimeric complex of Kir6.1, Kir6.2, and SUR2B subunits. J. Mol. Cell. Cardiol. 2004, 37, 857–869. [Google Scholar] [CrossRef]
- Lago-Docampo, M.; Tenorio, J.; Hernandez-Gonzalez, I.; Perez-Olivares, C.; Escribano-Subias, P.; Pousada, G.; Baloira, A.; Arenas, M.; Lapunzina, P.; Valverde, D. Characterization of rare ABCC8 variants identified in Spanish pulmonary arterial hypertension patients. Sci. Rep. 2020, 10, 15135. [Google Scholar] [CrossRef] [PubMed]
- Yildizdas, D.; Erdem, S.; Kucukosmanoglu, O.; Yilmaz, M.; Yuksel, B. Pulmonary hypertension, heart failure and neutropenia due to diazoxide therapy. Adv. Ther. 2008, 25, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Klinke, W.P.; Gilbert, J.A. Diazoxide in primary pulmonary hypertension. N. Engl. J. Med. 1980, 302, 91–92. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.S.; McLay, J.; Kenmure, A.C. Reversibility of primary pulmonary hypertension during six years of treatment with oral diazoxide. Br. Heart J. 1987, 57, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Tran, S.; Tinker, A.; Clapp, L.H. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. Am. J. Respir. Cell. Mol. Biol. 2002, 26, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Le Ribeuz, H.; Masson, B.; Capuano, V.; Dutheil, M.; Gooroochurn, H.; Boet, A.; Ghigna, M.R.; De Montpreville, V.; Girerd, B.; Lambert, M.; et al. SUR1 As a New Therapeutic Target for Pulmonary Arterial Hypertension. Am. J. Respir. Cell. Mol. Biol. 2022, 66, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Earley, S. TRPM4 channels in smooth muscle function. Pflugers Arch. 2013, 465, 1223–1231. [Google Scholar] [CrossRef]
- Yang, X.R.; Lin, M.J.; McIntosh, L.S.; Sham, J.S. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L1267–L1276. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Wang, S.; Nichols, C.G. On potential interactions between non-selective cation channel TRPM4 and sulfonylurea receptor SUR1. J. Biol. Chem. 2012, 287, 8746–8756. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, S.; Hiller, S.; Osswald, H.; Losle, M.; Grenz, A.; Hambrock, A. 17beta-Estradiol modulates apoptosis in pancreatic beta-cells by specific involvement of the sulfonylurea receptor (SUR) isoform SUR1. J. Biol. Chem. 2009, 284, 4905–4913. [Google Scholar] [CrossRef] [PubMed]
- Hambrock, A.; de Oliveira Franz, C.B.; Hiller, S.; Grenz, A.; Ackermann, S.; Schulze, D.U.; Drews, G.; Osswald, H. Resveratrol binds to the sulfonylurea receptor (SUR) and induces apoptosis in a SUR subtype-specific manner. J. Biol. Chem. 2007, 282, 3347–3356. [Google Scholar] [CrossRef] [PubMed]
- Kakei, M.; Noma, A.; Shibasaki, T. Properties of adenosine-triphosphate-regulated potassium channels in guinea-pig ventricular cells. J. Physiol. 1985, 363, 441–462. [Google Scholar] [CrossRef]
- Dunne, M.J.; Petersen, O.H. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett. 1986, 208, 59–62. [Google Scholar] [CrossRef]
- Dunne, M.J.; Illot, M.C.; Peterson, O.H. Interaction of diazoxide, tolbutamide and ATP4- on nucleotide-dependent K+ channels in an insulin-secreting cell line. J. Membr. Biol. 1987, 99, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bryan, L.; Nichols, C.G.; Wechsler, S.W.; Clement, J.P.t.; Boyd, A.E., III; Gonzalez, G.; Herrera-Sosa, H.; Nguy, K.; Bryan, J.; Nelson, D.A. Cloning of the beta cell high-affinity sulfonylurea receptor: A regulator of insulin secretion. Science 1995, 268, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Dorschner, H.; Brekardin, E.; Uhde, I.; Schwanstecher, C.; Schwanstecher, M. Stoichiometry of sulfonylurea-induced ATP-sensitive potassium channel closure. Mol. Pharmacol. 1999, 55, 1060–1066. [Google Scholar] [CrossRef]
- Coetzee, W.A. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 2013, 140, 167–175. [Google Scholar] [CrossRef]
- Sikimic, J.; Hoffmeister, T.; Gresch, A.; Kaiser, J.; Barthlen, W.; Wolke, C.; Wieland, I.; Lendeckel, U.; Krippeit-Drews, P.; Dufer, M.; et al. Possible New Strategies for the Treatment of Congenital Hyperinsulinism. Front. Endocrinol. 2020, 11, 545638. [Google Scholar] [CrossRef]
- Barros, F.; Pardo, L.A.; Domínguez, P.; Sierra, L.M.; de la Peña, P. New Structures and Gating of Voltage-Dependent Potassium (Kv) Channels and Their Relatives: A Multi-Domain and Dynamic Question. Int. J. Mol. Sci. 2019, 20, 248. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.J. Voltage-gated K+ currents regulate resting membrane potential and [Ca2+]i in pulmonary arterial myocytes. Circ. Res. 1995, 77, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef]
- Archer, S.L.; Wu, X.C.; Thébaud, B.; Nsair, A.; Bonnet, S.; Tyrrell, B.; McMurtry, M.S.; Hashimoto, K.; Harry, G.; Michelakis, E.D. Preferential expression and function of voltage-gated, O2-sensitive K+ channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: Ionic diversity in smooth muscle cells. Circ. Res. 2004, 95, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Platoshyn, O.; Sweeney, M.; Kim, H.; Yuan, J.X. Activation of K+ channels induces apoptosis in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2001, 280, C970–C979. [Google Scholar] [CrossRef]
- Brevnova, E.E.; Platoshyn, O.; Zhang, S.; Yuan, J.X.-J. Overexpression of human KCNA5 increases IK(V) and enhances apoptosis. Am. J. Physiol. Cell Physiol. 2004, 287, C715–C722. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Platoshyn, O.; McDaniel, S.S.; Rubin, L.J.; Yuan, J.X. Augmented K(+) currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L887–L894. [Google Scholar] [CrossRef]
- Ekhterae, D.; Platoshyn, O.; Krick, S.; Yu, Y.; McDaniel, S.S.; Yuan, J.X. Bcl-2 decreases voltage-gated K+ channel activity and enhances survival in vascular smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2001, 281, C157–C165. [Google Scholar] [CrossRef]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Souil, E.; Dinh-Xuan, A.T.; Schremmer, B.; Mercier, J.C.; El Yaagoubi, A.; Nguyen-Huu, L.; Reeve, H.L.; Hampl, V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J. Clin. Investig. 1998, 101, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.L.; Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Powell, F.; Haddad, G.H.; Yuan, J.X. Hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Ann. N. Y. Acad. Sci. 2009, 1177, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Lodi, F.; Frazziano, G.; Cobeño, L.; Tamargo, J.; Perez-Vizcaino, F. Serotonin inhibits voltage-gated K+ currents in pulmonary artery smooth muscle cells: Role of 5-HT2A receptors, caveolin-1, and KV1.5 channel internalization. Circ. Res. 2006, 98, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Holmes, T.C.; Fadool, D.A.; Ren, R.; Levitan, I.B. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science 1996, 274, 2089–2091. [Google Scholar] [CrossRef] [PubMed]
- Nesti, E.; Everill, B.; Morielli, A.D. Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol. Biol. Cell 2004, 15, 4073–4088. [Google Scholar] [CrossRef] [PubMed]
- Cogolludo, A.; Moreno, L.; Bosca, L.; Tamargo, J.; Perez-Vizcaino, F. Thromboxane A2-induced inhibition of voltage-gated K+ channels and pulmonary vasoconstriction: Role of protein kinase Czeta. Circ. Res. 2003, 93, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Young, K.A.; Ivester, C.; West, J.; Carr, M.; Rodman, D.M. BMP signaling controls PASMC KV channel expression in vitro and in vivo. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L841–L848. [Google Scholar] [CrossRef]
- Fantozzi, I.; Platoshyn, O.; Wong, A.H.; Zhang, S.; Remillard, C.V.; Furtado, M.R.; Petrauskene, O.V.; Yuan, J.X. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L993–L1004. [Google Scholar] [CrossRef]
- Zhang, S.; Fantozzi, I.; Tigno, D.D.; Yi, E.S.; Platoshyn, O.; Thistlethwaite, P.A.; Kriett, J.M.; Yung, G.; Rubin, L.J.; Yuan, J.X. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L740–L754. [Google Scholar] [CrossRef]
- West, J.; Fagan, K.; Steudel, W.; Fouty, B.; Lane, K.; Harral, J.; Hoedt-Miller, M.; Tada, Y.; Ozimek, J.; Tuder, R.; et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ. Res. 2004, 94, 1109–1114. [Google Scholar] [CrossRef]
- Wipff, J.; Dieudé, P.; Guedj, M.; Ruiz, B.; Riemekasten, G.; Cracowski, J.L.; Matucci-Cerinic, M.; Melchers, I.; Humbert, M.; Hachulla, E.; et al. Association of a KCNA5 gene polymorphism with systemic sclerosis-associated pulmonary arterial hypertension in the European Caucasian population. Arthr. Rheum. 2010, 62, 3093–3100. [Google Scholar] [CrossRef]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of Kv1.5 channels and genetic variations of KCNA5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell. Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.R.; Wang, W.; Lei, P.C.; Jia, H.P.; Dong, J.; Gou, Y.Q.; Chen, C.L.; Cao, J.; Wang, Y.F.; Zhu, Y.K. 5-HTT, BMPR2, EDN1, ENG, KCNA5 gene polymorphisms and susceptibility to pulmonary arterial hypertension: A meta-analysis. Gene 2019, 680, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir. Med. 2019, 7, 227–238. [Google Scholar] [CrossRef]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef]
- Fedida, D.; Eldstrom, J.; Hesketh, J.C.; Lamorgese, M.; Castel, L.; Steele, D.F.; Van Wagoner, D.R. Kv1.5 is an important component of repolarizing K+ current in canine atrial myocytes. Circ. Res. 2003, 93, 744–751. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Loboda, A. A Model for 4-Aminopyridine Action on K Channels: Similarities to Tetraethylammonium Ion Action. Biophys. J. 2001, 81, 895–904. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Morecroft, I.; Murray, A.; Nilsen, M.; Gurney, A.M.; MacLean, M.R. Treatment with the Kv7 potassium channel activator flupirtine is beneficial in two independent mouse models of pulmonary hypertension. Br. J. Pharmacol. 2009, 157, 1241–1249. [Google Scholar] [CrossRef]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef]
- Mondéjar-Parreño, G.; Moral-Sanz, J.; Barreira, B.; De la Cruz, A.; Gonzalez, T.; Callejo, M.; Esquivel-Ruiz, S.; Morales-Cano, D.; Moreno, L.; Valenzuela, C.; et al. Activation of K(v) 7 channels as a novel mechanism for NO/cGMP-induced pulmonary vasodilation. Br. J. Pharmacol. 2019, 176, 2131–2145. [Google Scholar] [CrossRef]
- Ayon, R.; Pohl, N.; Yamamura, A.; Yamamura, H.; Makino, A.; Yuan, J. miRNA-29b Directly Downregulates K+ Channel Expression and Function in IPAH-PASMC. FASEB J. 2015, 29, 662.616. [Google Scholar] [CrossRef]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thébaud, B.; Wu, X.C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redel-Traub, G.; Sampson, K.J.; Kass, R.S.; Bohnen, M.S. Potassium Channels as Therapeutic Targets in Pulmonary Arterial Hypertension. Biomolecules 2022, 12, 1341. https://doi.org/10.3390/biom12101341
Redel-Traub G, Sampson KJ, Kass RS, Bohnen MS. Potassium Channels as Therapeutic Targets in Pulmonary Arterial Hypertension. Biomolecules. 2022; 12(10):1341. https://doi.org/10.3390/biom12101341
Chicago/Turabian StyleRedel-Traub, Gabriel, Kevin J. Sampson, Robert S. Kass, and Michael S. Bohnen. 2022. "Potassium Channels as Therapeutic Targets in Pulmonary Arterial Hypertension" Biomolecules 12, no. 10: 1341. https://doi.org/10.3390/biom12101341