
Shortened Hinge Design of Fab x sdAb-Fc Bispecific Antibodies Enhances Redirected T-Cell Killing of Tumor Cells
 ,
, 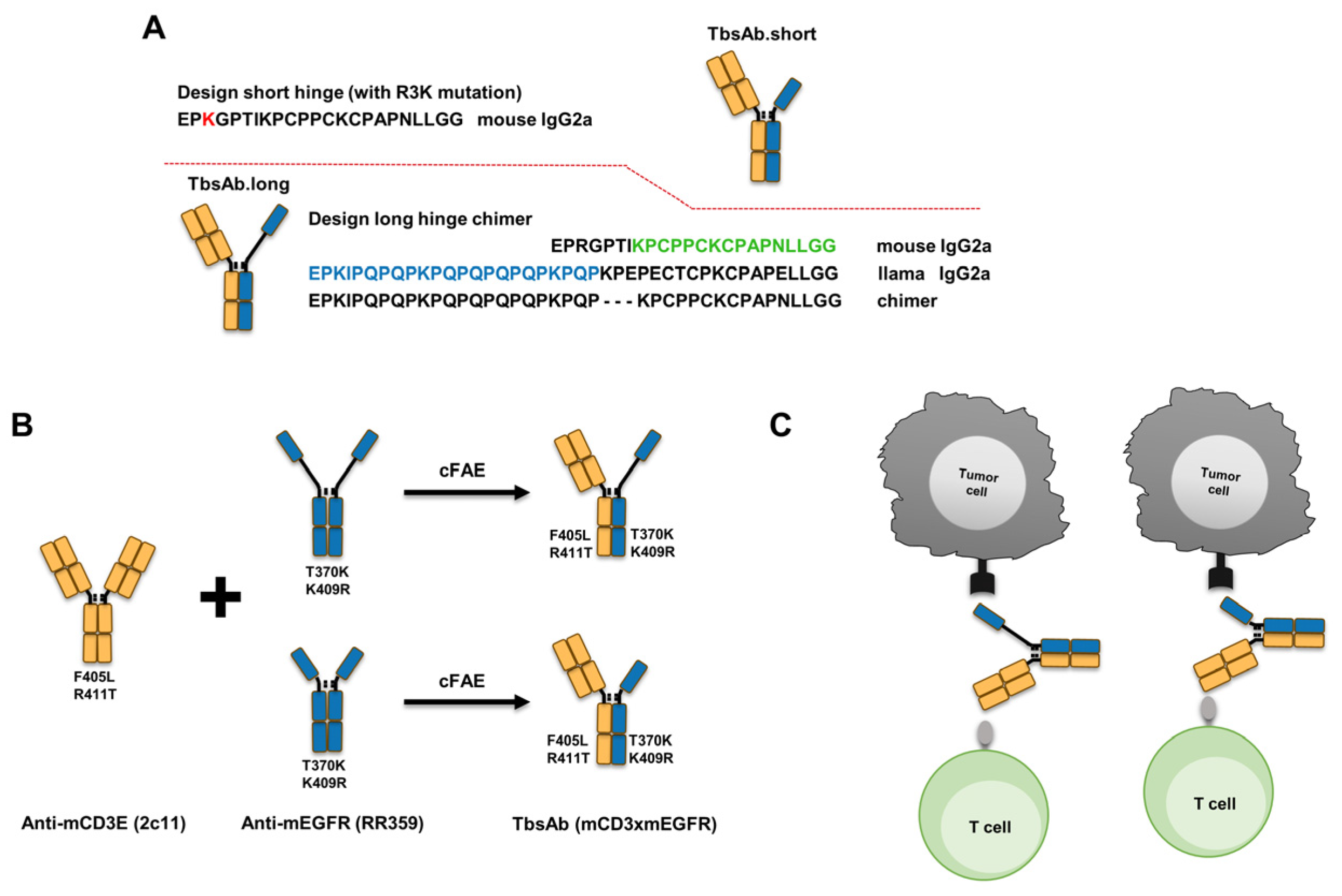{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Designing and Preparation of mCD3E x mEGFR TbsAbs with Different Hinges
2.2. Generation of TbsAb Negative Control Antibody
2.3. mCD3E x mEGFR TbsAb.short Molecule Mediated Enhanced T Cell Redirected Killing In Vitro
2.4. mCD3E × mEGFR TbsAb.short Mediated Enhanced Cell–Cell Association In Vitro
2.5. mCD3E × mEGFR TbsAb.short Mediated Enhanced T Cell Activation In Vitro
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Designation and Construction of Expression Vectors for Bispecific Antibodies
4.3. Generation of Bispecific Antibodies
4.4. Generation of Control TbsAb by Mutagenesis
4.5. Size-Exclusion Chromatography (HP-SEC)
4.6. In Vitro Cytotoxicity Assays
4.7. Microscopy
4.8. Cell-Cell Association Assays
4.9. T Cell Activation Assays
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blanco, B.; Compte, M.; Lykkemark, S.; Sanz, L.; Alvarez-Vallina, L. T Cell-Redirecting Strategies to ‘STAb’ Tumors: Beyond CARs and Bispecific Antibodies. Trends Immunol. 2019, 40, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and Approval of the Trifunctional Antibody Catumaxomab (Anti-EpCAM× anti-CD3) as a Targeted Cancer Immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.-W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.-J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; van Duijnhoven, S.M.J.; Sijts, A.J.A.M.; van Elsas, A. Bispecific Antibodies Targeting Dual Tumor-Associated Antigens in Cancer Therapy. J. Cancer Res. Clin. Oncol. 2020, 146, 3111–3122. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R.; Naso, M. Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells. Antibodies 2019, 8, 41. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Park, J.; Liu, X.; Hu, Y.; Wang, T.; McFarland, K.; Betenbaugh, M.J. Design and Production of Bispecific Antibodies. Antibodies 2019, 8, 43. [Google Scholar] [CrossRef]
- Schaefer, W.; Regula, J.T.; Bähner, M.; Schanzer, J.; Croasdale, R.; Dürr, H.; Gassner, C.; Georges, G.; Kettenberger, H.; Imhof-Jung, S.; et al. Immunoglobulin Domain Crossover as a Generic Approach for the Production of Bispecific IgG Antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 11187–11192. [Google Scholar] [CrossRef]
- Huang, S.; Segués, A.; Hulsik, D.L.; Zaiss, D.M.; Sijts, A.J.A.M.; van Duijnhoven, S.M.J.; van Elsas, A. A Novel Efficient Bispecific Antibody Format, Combining a Conventional Antigen-Binding Fragment with a Single Domain Antibody, Avoids Potential Heavy-Light Chain Mis-Pairing. J. Immunol. Methods 2020, 483, 112811. [Google Scholar] [CrossRef]
- Ellerman, D. Bispecific T-Cell Engagers: Towards Understanding Variables Influencing the in Vitro Potency and Tumor Selectivity and Their Modulation to Enhance Their Efficacy and Safety. Methods 2019, 154, 102–117. [Google Scholar] [CrossRef]
- Dickopf, S.; Georges, G.J.; Brinkmann, U. Format and Geometries Matter: Structure-Based Design Defines the Functionality of Bispecific Antibodies. Comput. Struct. Biotechnol. J. 2020, 18, 1221–1227. [Google Scholar] [CrossRef]
- Klein, D.; Jacobs, S.; Sheri, M.; Anderson, M.; Attar, R.; Barnakov, A.; Brosnan, K.; Bushey, B.; Chevalier, K.; Chin, D.; et al. Abstract LB-312: Bispecific Centyrin Simultaneously Targeting EGFR and c-Met Demonstrates Improved Activity Compared to the Mixture of Single Agents. Cancer Res. 2013, 73, LB-312. [Google Scholar] [CrossRef]
- Wuellner, U.; Klupsch, K.; Buller, F.; Attinger-Toller, I.; Santimaria, R.; Zbinden, I.; Henne, P.; Grabulovski, D.; Bertschinger, J.; Brack, S. Bispecific CD3/HER2 Targeting FynomAb Induces Redirected T Cell-Mediated Cytolysis with High Potency and Enhanced Tumor Selectivity. Antibodies 2015, 4, 426–440. [Google Scholar] [CrossRef]
- Santich, B.H.; Park, J.A.; Tran, H.; Guo, H.-F.; Huse, M.; Cheung, N.-K.V. Interdomain Spacing and Spatial Configuration Drive the Potency of IgG-[L]-ScFv T Cell Bispecific Antibodies. Sci. Transl. Med. 2020, 12, eaax1315. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.A.; Zhang, W.; Rainey, G.J.; Burke, S.; Li, H.; Huang, L.; Gorlatov, S.; Veri, M.C.; Aggarwal, S.; Yang, Y.; et al. Application of Dual Affinity Retargeting Molecules to Achieve Optimal Redirected T-Cell Killing of B-Cell Lymphoma. Blood 2011, 117, 4542–4551. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef]
- Bluemel, C.; Hausmann, S.; Fluhr, P.; Sriskandarajah, M.; Stallcup, W.B.; Baeuerle, P.A.; Kufer, P. Epitope Distance to the Target Cell Membrane and Antigen Size Determine the Potency of T Cell-Mediated Lysis by BiTE Antibodies Specific for a Large Melanoma Surface Antigen. Cancer Immunol. Immunother. 2010, 59, 1197–1209. [Google Scholar] [CrossRef]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Antigen Sensitivity of CD22-Specific Chimeric TCR Is Modulated by Target Epitope Distance from the Cell Membrane. J. Immunol. 2008, 180, 7028–7038. [Google Scholar] [CrossRef]
- Root, A.R.; Cao, W.; Li, B.; LaPan, P.; Meade, C.; Sanford, J.; Jin, M.; O’Sullivan, C.; Cummins, E.; Lambert, M.; et al. Development of PF-06671008, a Highly Potent Anti-P-Cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies 2016, 5, 6. [Google Scholar] [CrossRef]
- Qi, J.; Li, X.; Peng, H.; Cook, E.M.; Dadashian, E.L.; Wiestner, A.; Park, H.; Rader, C. Potent and Selective Antitumor Activity of a T Cell-Engaging Bispecific Antibody Targeting a Membrane-Proximal Epitope of ROR1. Proc. Natl. Acad. Sci. USA 2018, 115, E5467–E5476. [Google Scholar] [CrossRef]
- Chu, T.H.; Patz, E.F.; Ackerman, M.E. Coming Together at the Hinges: Therapeutic Prospects of IgG3. mAbs 2021, 13, 1882028. [Google Scholar] [CrossRef]
- Wang, F.; Tsai, J.C.; Davis, J.H.; Chau, B.; Dong, J.; West, S.M.; Hogan, J.M.; Wheeler, M.L.; Bee, C.; Morishige, W.; et al. Design and Characterization of Mouse IgG1 and IgG2a Bispecific Antibodies for Use in Syngeneic Models. mAbs 2019, 12, 1685350. [Google Scholar] [CrossRef] [PubMed]
- Griffin, L.M.; Snowden, J.R.; Lawson, A.D.G.; Wernery, U.; Kinne, J.; Baker, T.S. Analysis of Heavy and Light Chain Sequences of Conventional Camelid Antibodies from Camelus Dromedarius and Camelus Bactrianus Species. J. Immunol. Methods 2014, 405, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.J.; Duehr, J.; Dulin, H.; Broecker, F.; Brown, J.A.; Arumemi, F.O.; Bermúdez González, M.C.; Leyva-Grado, V.H.; Evans, M.J.; Simon, V.; et al. Human Antibodies Targeting Zika Virus NS1 Provide Protection against Disease in a Mouse Model. Nat. Commun. 2018, 9, 4560. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Gedeon, P.C.; Kuan, C.-T.; Sanchez-Perez, L.; Archer, G.E.; Bigner, D.D.; Sampson, J.H. Rational Design and Generation of Recombinant Control Reagents for Bispecific Antibodies through CDR Mutagenesis. J. Immunol. Methods 2013, 395, 14–20. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Diem, M.; Reeves, D.; Wang, W.; Schulingkamp, R.; Sproesser, K.; Mattson, B.; Heidrich, B.; Mendonça, M.; Joseph, J.; et al. A Novel C2 Domain Binding CD33×CD3 Bispecific Antibody with Potent T-Cell Redirection Activity against Acute Myeloid Leukemia. Blood Adv. 2020, 4, 906–919. [Google Scholar] [CrossRef]
- Chen, W.; Yang, F.; Wang, C.; Narula, J.; Pascua, E.; Ni, I.; Ding, S.; Deng, X.; Chu, M.L.-H.; Pham, A.; et al. One Size Does Not Fit All: Navigating the Multi-Dimensional Space to Optimize T-Cell Engaging Protein Therapeutics. mAbs 2021, 13, 1871171. [Google Scholar] [CrossRef]
- Kapelski, S.; Cleiren, E.; Attar, R.M.; Philippar, U.; Häsler, J.; Chiu, M.L. Influence of the Bispecific Antibody IgG Subclass on T Cell Redirection. mAbs 2019, 11, 1012–1024. [Google Scholar] [CrossRef]
- Aleksic, M.; Dushek, O.; Zhang, H.; Shenderov, E.; Chen, J.-L.; Cerundolo, V.; Coombs, D.; van der Merwe, P.A. Dependence of T Cell Antigen Recognition on T Cell Receptor-Peptide MHC Confinement Time. Immunity 2010, 32, 163–174. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Knackmuss, S.; Weichel, M.; Gantke, T.; Molkenthin, V.; Zhukovsky, E.A.; Tesar, M.; Treder, M. Highly Specific and Effective Targeting of EGFRvIII-Positive Tumors with TandAb Antibodies. Front. Oncol. 2017, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Kipriyanov, S.M.; Moldenhauer, G.; Schuhmacher, J.; Cochlovius, B.; Von der Lieth, C.-W.; Matys, E.R.; Little, M. Bispecific Tandem Diabody for Tumor Therapy with Improved Antigen Binding and Pharmacokinetics11Edited by J. Karn. J. Mol. Biol. 1999, 293, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, S.S.; Guo, H.; Wu, Z.; Hatano, M.N.; Cheung, N.-K.V. A Potent Tetravalent T-Cell–Engaging Bispecific Antibody against CD3 in Acute Myeloid Leukemia. Blood Adv. 2018, 2, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Harwood, S.L.; Alvarez-Cienfuegos, A.; Nuñez-Prado, N.; Compte, M.; Hernández-Pérez, S.; Merino, N.; Bonet, J.; Navarro, R.; Van Bergen en Henegouwen, P.M.P.; Lykkemark, S.; et al. ATTACK, a Novel Bispecific T Cell-Recruiting Antibody with Trivalent EGFR Binding and Monovalent CD3 Binding for Cancer Immunotherapy. OncoImmunology 2018, 7, e1377874. [Google Scholar] [CrossRef] [PubMed]
- Kipriyanov, S.M.; Moldenhauer, G.; Strauss, G.; Little, M. Bispecific CD3 × CD19 Diabody for T Cell-Mediated Lysis of Malignant Human B Cells. Int. J. Cancer 1998, 77, 763–772. [Google Scholar] [CrossRef]
- Feldmann, A.; Stamova, S.; Bippes, C.C.; Bartsch, H.; Wehner, R.; Schmitz, M.; Temme, A.; Cartellieri, M.; Bachmann, M. Retargeting of T Cells to Prostate Stem Cell Antigen Expressing Tumor Cells: Comparison of Different Antibody Formats. Prostate 2011, 71, 998–1011. [Google Scholar] [CrossRef]
- Durben, M.; Schmiedel, D.; Hofmann, M.; Vogt, F.; Nübling, T.; Pyz, E.; Bühring, H.-J.; Rammensee, H.-G.; Salih, H.R.; Große-Hovest, L.; et al. Characterization of a Bispecific FLT3 X CD3 Antibody in an Improved, Recombinant Format for the Treatment of Leukemia. Mol. Ther. 2015, 23, 648–655. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; van Loosdregt, J.; Gorlani, A.; Bekker, C.P.J.; Gröne, A.; Sibilia, M.; van Bergen en Henegouwen, P.M.P.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin Enhances Regulatory T Cell-Suppressive Function via the Epidermal Growth Factor Receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Segués, A.; Waterfall, M.; Wright, D.; Vayssiere, C.; van Duijnhoven, S.M.J.; van Elsas, A.; Sijts, A.J.A.M.; Zaiss, D.M. Shortened Hinge Design of Fab x sdAb-Fc Bispecific Antibodies Enhances Redirected T-Cell Killing of Tumor Cells. Biomolecules 2022, 12, 1331. https://doi.org/10.3390/biom12101331
Huang S, Segués A, Waterfall M, Wright D, Vayssiere C, van Duijnhoven SMJ, van Elsas A, Sijts AJAM, Zaiss DM. Shortened Hinge Design of Fab x sdAb-Fc Bispecific Antibodies Enhances Redirected T-Cell Killing of Tumor Cells. Biomolecules. 2022; 12(10):1331. https://doi.org/10.3390/biom12101331
Chicago/Turabian StyleHuang, Shuyu, Aina Segués, Martin Waterfall, David Wright, Charlotte Vayssiere, Sander M. J. van Duijnhoven, Andrea van Elsas, Alice J. A. M. Sijts, and Dietmar M. Zaiss. 2022. "Shortened Hinge Design of Fab x sdAb-Fc Bispecific Antibodies Enhances Redirected T-Cell Killing of Tumor Cells" Biomolecules 12, no. 10: 1331. https://doi.org/10.3390/biom12101331