
Adipose Triglyceride Lipase in Hepatic Physiology and Pathophysiology


Abstract
:1. Introduction
2. The Discovery of ATGL and ATGL-Mediated Lipolysis
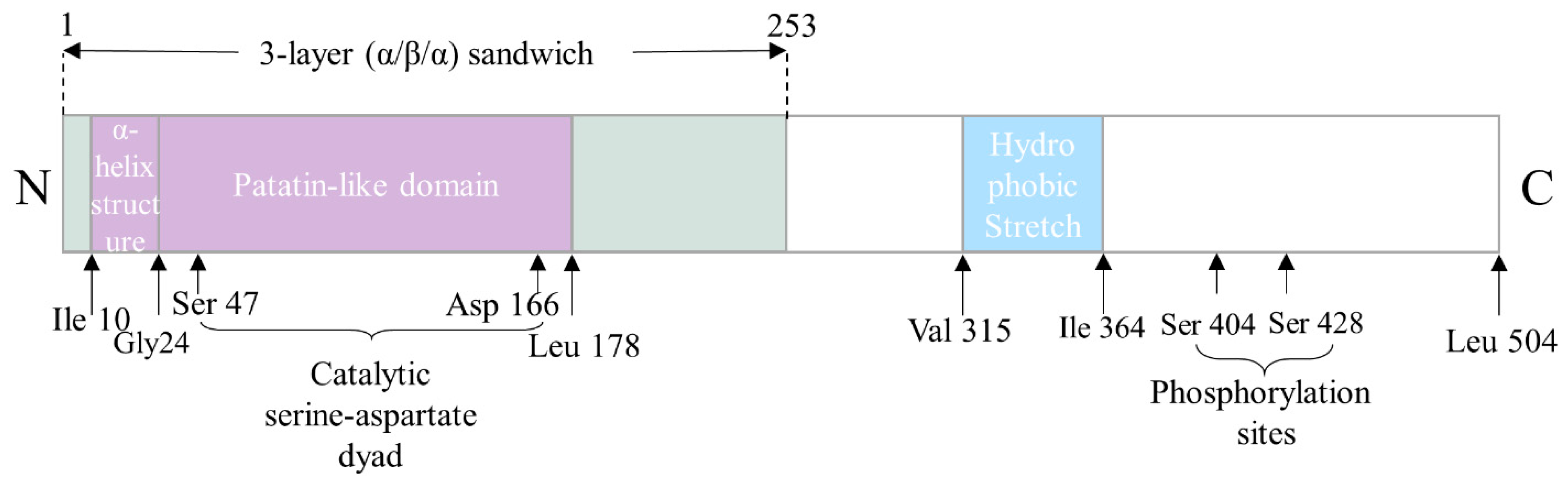
3. The Structure–Function Relationship of ATGL
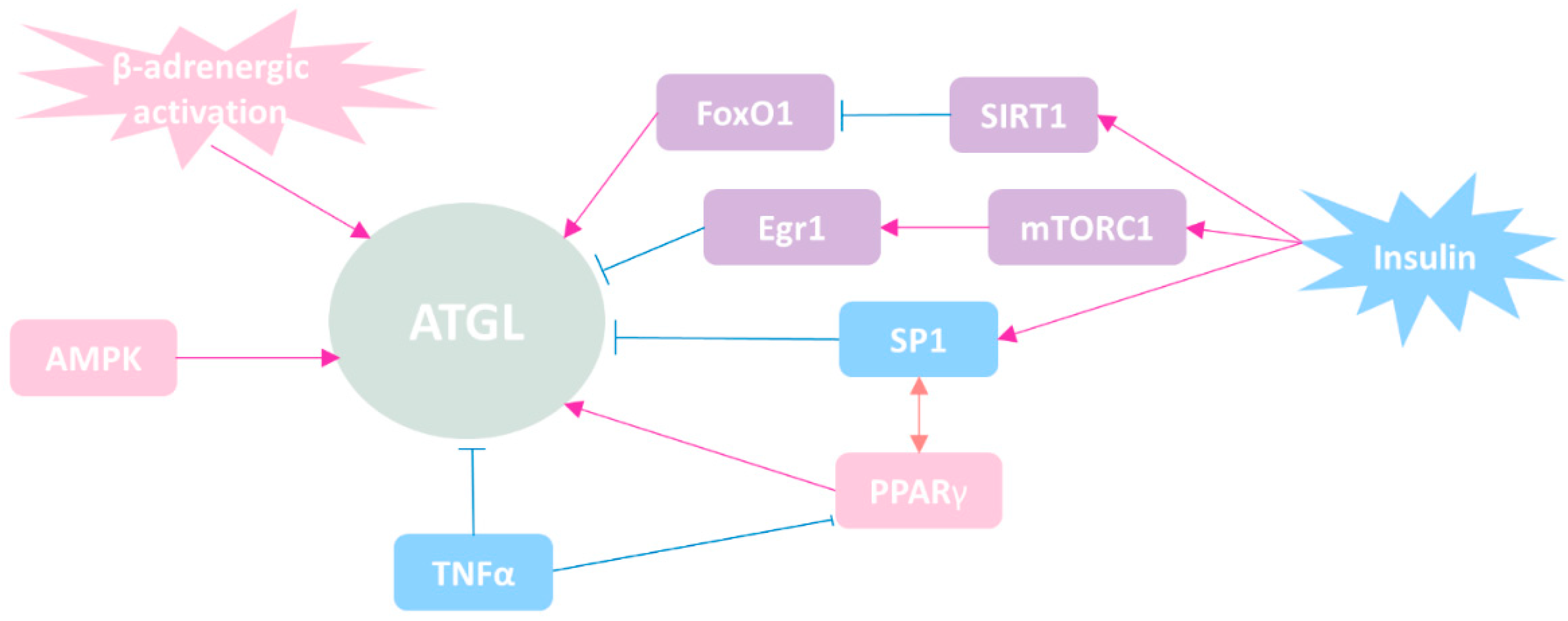
4. The Regulatory Mechanisms of ATGL
4.1. Regulation of ATGL Expression at Transcriptional Level
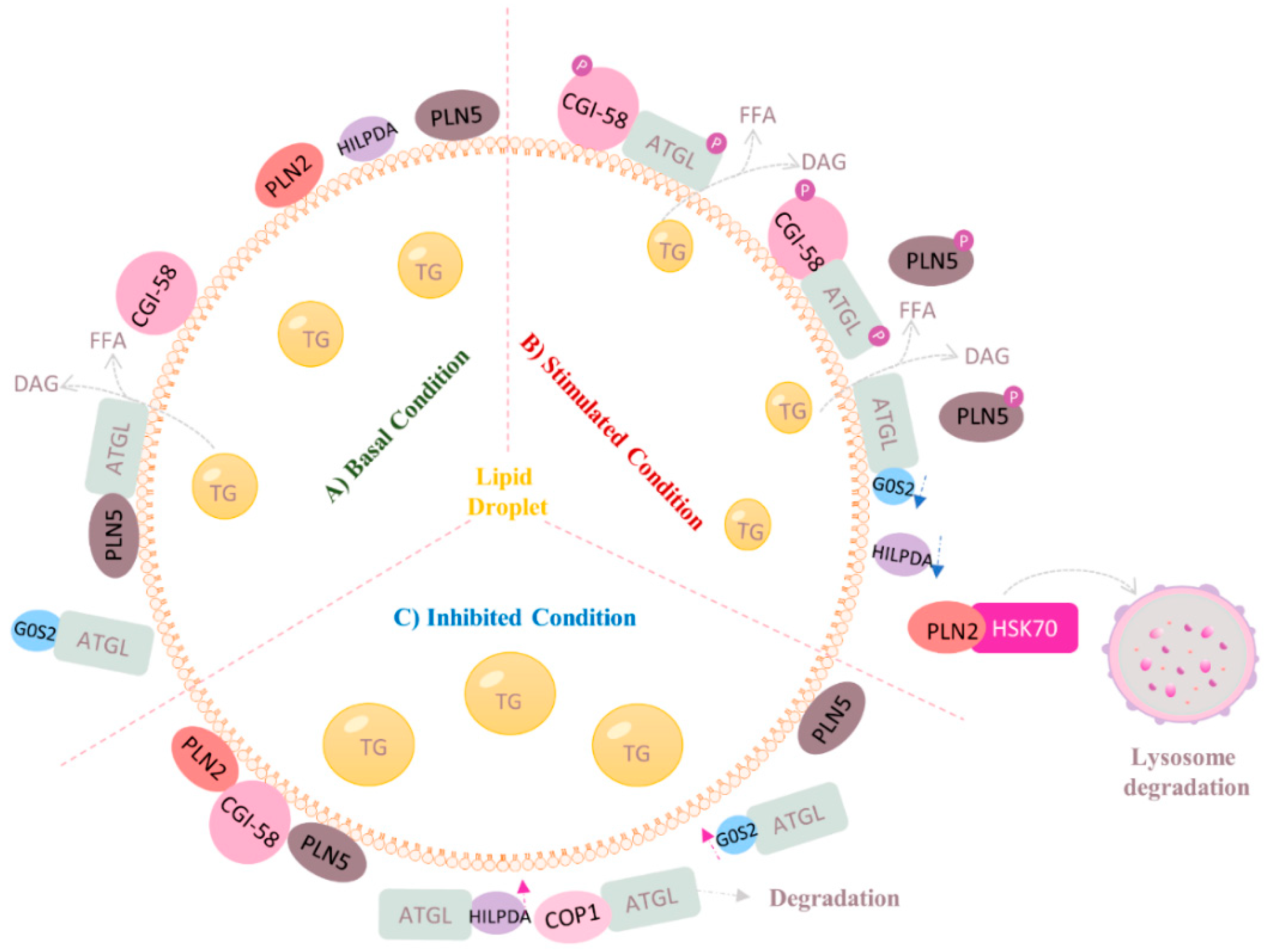
4.2. Regulation of ATGL at Post-Transcriptional Level (Protein–Protein Interaction)
4.3. Regulation of ATGL by Small Molecules
4.4. Regulation of ATGL by Lipid Intermediates
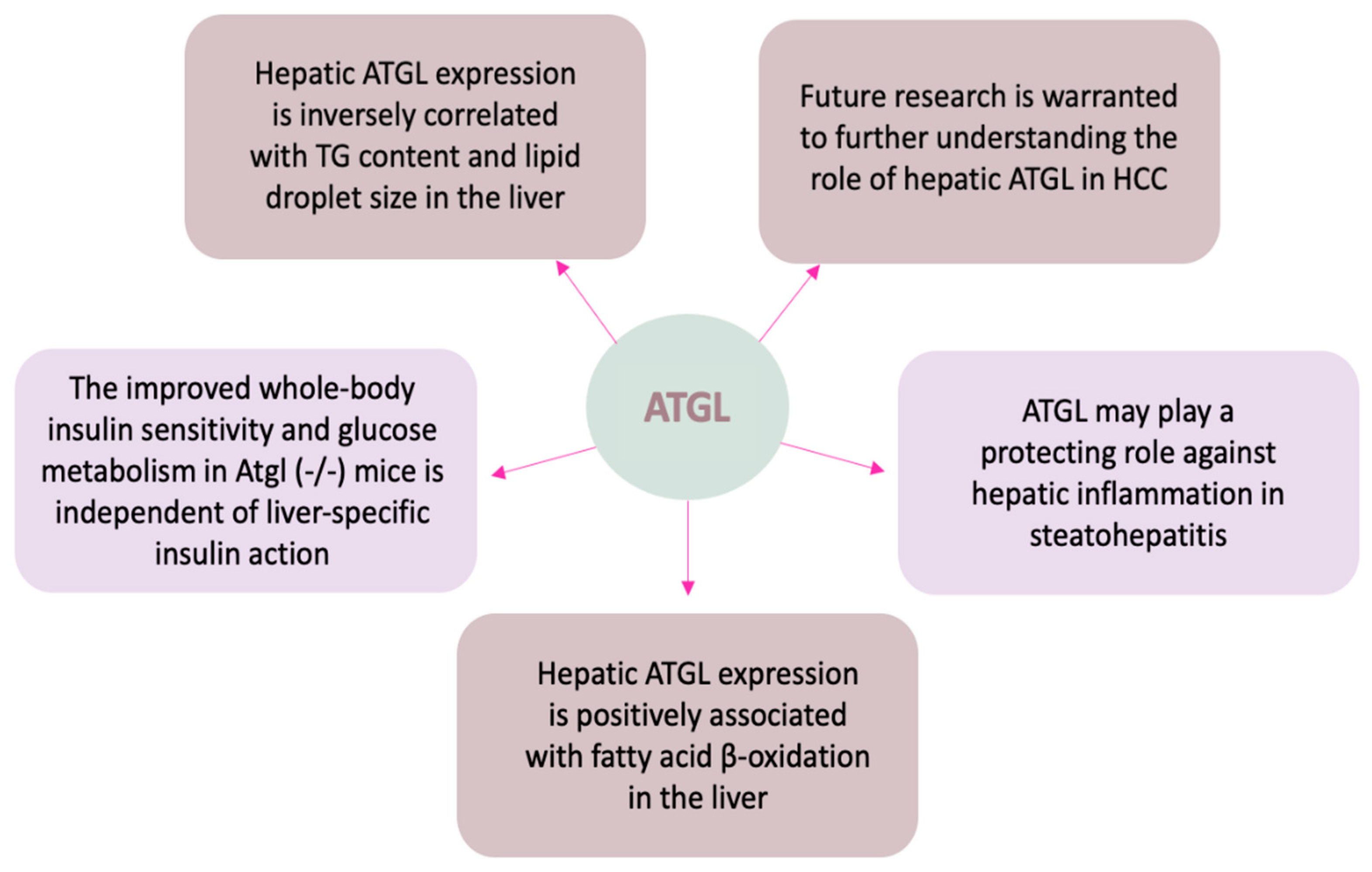
5. The Roles of Hepatic ATGL in Health and Disease
5.1. Lessons Learned from Humans with ATGL-Related Mutations
5.2. ATGL in Hepatic TG Accumulation
5.3. ATGL in Hepatic FA Oxidation
5.4. ATGL in Hepatic Inflammation
5.5. ATGL in Hepatic Glucose Metabolism
5.6. ATGL in Hepatocellular Carcinoma (HCC)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABDH5 | alpha-betahydrolasedomain-containing5 | iNOS | induciblenitricoxidesynthase |
| ACBP | acyl-CoA-binding protein | ||
| AFLD | alcohol-relatedfattyliverdisease | LPS | lipopolysaccharide |
| LC-CoA | long-chain acyl-coenzyme A | ||
| ALT | alanineaminotransferase | LD | Lipid droplet |
| L-FABP | liver fatty acid binding protein | ||
| Arf1 | ADP-ribosylationfactor1 | MAG | monoacylglycerol |
| AMPK | AMP-activatedproteinkinase | MAL | monoacylglycerollipase |
| AST | aspartateaminotransferase | MCD | methionine-choline-deficient |
| ATGL | adiposetriglyceridelipase | MCP-1 | monocytechemotacticprotein-1 |
| CGI-58 | comparativegeneidentification-58 | mTORC1 | mechanistictargetofrapamycincomplex1 |
| CIDEC | celldeathinducingDEFAlikeeffectorC | NAFLD | non-alcoholicfattyliverdisease |
| COPI | coatproteincomplexI | NLSD-I | neutrallipidstoragelipidstoragediseasewithichthyosis |
| CPT-1α | carnitinepalmitoyltransferase-1alpha | NLSD-M | neutrallipidstoragelipidstoragediseasewithmyopathy |
| DAG | diacylglycerol | PEDF | pigmentepithelium-derivedfactor |
| DIO | diet-inducedobesity | PKA | proteinkinaseA |
| Egr1 | earlygrowthresponse1 | iPLA2zeta | calcium-independentphospholipaseA2-zeta |
| ER | endoplasmicreticulum | Plin1 | perilipin1 |
| FA | fattyacid | PNPLA | patatin-likephospholipasedomain-containingfamily |
| FoxO1 | forkheadboxproteinO1 | PPARγ | peroxisomeproliferator-activatedreceptorgamma |
| FSP27 | fat-specificprotein27 | SERPINF1 | seepingfamilyFmember1 |
| G0S2 | G0/G1switchgene2 | SIRT1 | sirtuin1 |
| GBF1 | Golgi-specificbrefeldinAresistancefactor1 | Sp1 | specificityprotein1 |
| HCC | hepatocellular carcinoma | ||
| HILPDA | hypoxia-induciblelipiddroplet-associatedprotein | TG | triglyceride |
| HSC | hepaticstellatecell | TNF-α | tumornecrosisfactor-alpha |
| HSL | hormone-sensitivelipase | VLDL | verylow-densitylipoprotein |
| IL-1β | interleukin-1beta | WT | wildtype |
References
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef]
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osuga, J.; Ishibashi, S.; Oka, T.; Yagyu, H.; Tozawa, R.; Fujimoto, A.; Shionoiri, F.; Yahagi, N.; Kraemer, F.B.; Tsutsumi, O.; et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Zimmermann, R.; Hayn, M.; Theussl, C.; Waeg, G.; Wagner, E.; Sattler, W.; Magin, T.M.; Wagner, E.F.; Zechner, R. Hormone-sensitive lipase deficiency in mice causes diglyceride accumulation in adipose tissue, muscle, and testis. J. Biol. Chem. 2002, 277, 4806–4815. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [Green Version]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.H.; Sul, H.S. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef] [Green Version]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. [Google Scholar] [CrossRef]
- Cornaciu, I.; Boeszoermenyi, A.; Lindermuth, H.; Nagy, H.M.; Cerk, I.K.; Ebner, C.; Salzburger, B.; Gruber, A.; Schweiger, M.; Zechner, R.; et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS ONE 2011, 6, e26349. [Google Scholar] [CrossRef] [Green Version]
- Duncan, R.E.; Wang, Y.; Ahmadian, M.; Lu, J.; Sarkadi-Nagy, E.; Sul, H.S. Characterization of desnutrin functional domains: Critical residues for triacylglycerol hydrolysis in cultured cells. J. Lipid Res. 2010, 51, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Lake, A.C.; Sun, Y.; Li, J.L.; Kim, J.E.; Johnson, J.W.; Li, D.; Revett, T.; Shih, H.H.; Liu, W.; Paulsen, J.E.; et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J. Lipid Res. 2005, 46, 2477–2487. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Thiam, A.R.; Olarte, M.J.; Wang, J.; Beck, R.; Gould, T.J.; Allgeyer, E.S.; Pincet, F.; Bewersdorf, J.; Farese, R.V., Jr.; et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. eLife 2014, 3, e01607. [Google Scholar] [CrossRef]
- Smirnova, E.; Goldberg, E.B.; Makarova, K.S.; Lin, L.; Brown, W.J.; Jackson, C.L. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 2006, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Lefevre, C.; Morava, E.; Mussini, J.M.; Laforet, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Inoguchi, T.; Maeda, Y.; Nakashima, N.; Kuwano, A.; Eto, E.; Ueno, N.; Sasaki, S.; Sawada, F.; Fujii, M.; et al. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J. Clin. Endocrinol. Metab. 2008, 93, 2877–2884. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, M.; Schoiswohl, G.; Lass, A.; Radner, F.P.; Haemmerle, G.; Malli, R.; Graier, W.; Cornaciu, I.; Oberer, M.; Salvayre, R.; et al. The C-terminal region of human adipose triglyceride lipase affects enzyme activity and lipid droplet binding. J. Biol. Chem. 2008, 283, 17211–17220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartz, R.; Zehmer, J.K.; Zhu, M.; Chen, Y.; Serrero, G.; Zhao, Y.; Liu, P. Dynamic activity of lipid droplets: Protein phosphorylation and GTP-mediated protein translocation. J. Proteome Res. 2007, 6, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Vegliante, R.; Di Leo, L.; Ciccarone, F.; Ciriolo, M.R. Hints on ATGL implications in cancer: Beyond bioenergetic clues. Cell Death Dis. 2018, 9, 316. [Google Scholar] [CrossRef]
- Schott, M.B.; Rasineni, K.; Weller, S.G.; Schulze, R.J.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. beta-Adrenergic induction of lipolysis in hepatocytes is inhibited by ethanol exposure. J. Biol. Chem. 2017, 292, 11815–11828. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Tillison, K.; Lee, J.H.; Rearick, D.A.; Smas, C.M. The adipose tissue triglyceride lipase ATGL/PNPLA2 is downregulated by insulin and TNF-alpha in 3T3-L1 adipocytes and is a target for transactivation by PPARgamma. Am. J. Physiology. Endocrinol. Metab. 2006, 291, E115–E127. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Hamm, J.K.; Verhagen, L.A.; Peroni, O.; Katic, M.; Flier, J.S. Adipose triglyceride lipase: Function, regulation by insulin, and comparison with adiponutrin. Diabetes 2006, 55, 148–157. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; English, T.; Karki, S.; Qiang, L.; Tao, R.; Kim, J.; Luo, Z.; Farmer, S.R.; Kandror, K.V. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 2011, 52, 1693–1701. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, P.; English, T.; Shi, J.; Smas, C.M.; Kandror, K.V. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes 2010, 59, 775–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.; Farabaugh, K.T.; Wu, J.; Charrier, A.; Smas, C.; Hatzoglou, M.; Thirumurugan, K.; Buchner, D.A. Coordinated transcriptional control of adipocyte triglyceride lipase (Atgl) by transcription factors Sp1 and peroxisome proliferator-activated receptor gamma (PPARgamma) during adipocyte differentiation. J. Biol. Chem. 2017, 292, 14827–14835. [Google Scholar] [CrossRef] [Green Version]
- Kralisch, S.; Klein, J.; Lossner, U.; Bluher, M.; Paschke, R.; Stumvoll, M.; Fasshauer, M. Isoproterenol, TNFalpha, and insulin downregulate adipose triglyceride lipase in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2005, 240, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Heckmann, B.L.; Lu, X.; Liu, J. Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-alpha (TNF-alpha)-induced lipolysis in adipocytes. J. Biol. Chem. 2011, 286, 40477–40485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, A.; Cornaciu, I.; Lass, A.; Schweiger, M.; Poeschl, M.; Eder, C.; Kumari, M.; Schoiswohl, G.; Wolinski, H.; Kohlwein, S.D.; et al. The N-terminal region of comparative gene identification-58 (CGI-58) is important for lipid droplet binding and activation of adipose triglyceride lipase. J. Biol. Chem. 2010, 285, 12289–12298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, M.A.; Zhang, H.; Mladenovic, L.; Tseng, Y.Y.; Granneman, J.G. Molecular Basis of ABHD5 Lipolysis Activation. Sci. Rep. 2017, 7, 42589. [Google Scholar] [CrossRef] [Green Version]
- Boeszoermenyi, A.; Nagy, H.M.; Arthanari, H.; Pillip, C.J.; Lindermuth, H.; Luna, R.E.; Wagner, G.; Zechner, R.; Zangger, K.; Oberer, M. Structure of a CGI-58 motif provides the molecular basis of lipid droplet anchoring. J. Biol. Chem. 2015, 290, 26361–26372. [Google Scholar] [CrossRef] [Green Version]
- Kulminskaya, N.; Oberer, M. Protein-protein interactions regulate the activity of Adipose Triglyceride Lipase in intracellular lipolysis. Biochimie 2020, 169, 62–68. [Google Scholar] [CrossRef]
- Russell, L.; Forsdyke, D.R. A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA Cell Biol. 1991, 10, 581–591. [Google Scholar] [CrossRef]
- Siderovski, D.P.; Blum, S.; Forsdyke, R.E.; Forsdyke, D.R. A set of human putative lymphocyte G0/G1 switch genes includes genes homologous to rodent cytokine and zinc finger protein-encoding genes. DNA Cell Biol. 1990, 9, 579–587. [Google Scholar] [CrossRef]
- Zhang, X.; Heckmann, B.L.; Campbell, L.E.; Liu, J. G0S2: A small giant controller of lipolysis and adipose-liver fatty acid flux. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Paar, M.; Eder, C.; Brandis, J.; Moser, E.; Gorkiewicz, G.; Grond, S.; Radner, F.P.; Cerk, I.; Cornaciu, I.; et al. G0/G1 switch gene-2 regulates human adipocyte lipolysis by affecting activity and localization of adipose triglyceride lipase. J. Lipid Res. 2012, 53, 2307–2317. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Yang, X.; Liu, J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle 2010, 9, 2719–2725. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xie, X.; Heckmann, B.L.; Saarinen, A.M.; Czyzyk, T.A.; Liu, J. Targeted disruption of G0/G1 switch gene 2 enhances adipose lipolysis, alters hepatic energy balance, and alleviates high-fat diet-induced liver steatosis. Diabetes 2014, 63, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Lopez-Aguiar, A.G.; Li, A.; Lu, Y.; Sekula, D.; Nattie, E.E.; Freemantle, S.; Dmitrovsky, E. Mice lacking G0S2 are lean and cold-tolerant. Cancer Biol. Ther. 2014, 15, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Choi, Y.M.; Han, J.Y.; Lee, K. Inhibition of lipolysis in the novel transgenic quail model overexpressing G0/G1 switch gene 2 in the adipose tissue during feed restriction. PLoS ONE 2014, 9, e100905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasaemle, D.L. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, A.R.; Sztalryd, C. The Perilipins: Major Cytosolic Lipid Droplet-Associated Proteins and Their Roles in Cellular Lipid Storage, Mobilization, and Systemic Homeostasis. Annu. Rev. Nutr. 2016, 36, 471–509. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.; Zechner, R. Breaking the Barrier--Chaperone-Mediated Autophagy of Perilipins Regulates the Lipolytic Degradation of Fat. Cell Metab. 2015, 22, 60–61. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Yang, W.; Kozusko, K.; Saudek, V.; Savage, D.B. Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 9163–9168. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Sathyanarayan, A.; Mashek, M.T.; Mashek, D.G. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Chang, B.; Wu, X.; Li, L.; Sleeman, M.; Chan, L. Inactivation of Plin4 downregulates Plin5 and reduces cardiac lipid accumulation in mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E770–E779. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Omatsu, N.; Matsushita, S.; Osumi, T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J. Biol. Chem. 2004, 279, 30490–30497. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bell, M.; Sreenivasan, U.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S.; et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Togashi, A.; Katagiri, T.; Ashida, S.; Fujioka, T.; Maruyama, O.; Wakumoto, Y.; Sakamoto, Y.; Fujime, M.; Kawachi, Y.; Shuin, T.; et al. Hypoxia-inducible protein 2 (HIG2), a novel diagnostic marker for renal cell carcinoma and potential target for molecular therapy. Cancer Res. 2005, 65, 4817–4826. [Google Scholar] [CrossRef] [Green Version]
- DiStefano, M.T.; Danai, L.V.; Roth Flach, R.J.; Chawla, A.; Pedersen, D.J.; Guilherme, A.; Czech, M.P. The Lipid Droplet Protein Hypoxia-inducible Gene 2 Promotes Hepatic Triglyceride Deposition by Inhibiting Lipolysis. J. Biol. Chem. 2015, 290, 15175–15184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattijssen, F.; Georgiadi, A.; Andasarie, T.; Szalowska, E.; Zota, A.; Krones-Herzig, A.; Heier, C.; Ratman, D.; De Bosscher, K.; Qi, L.; et al. Hypoxia-inducible lipid droplet-associated (HILPDA) is a novel peroxisome proliferator-activated receptor (PPAR) target involved in hepatic triglyceride secretion. J. Biol. Chem. 2014, 289, 19279–19293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabha Das, K.M.; Wechselberger, L.; Liziczai, M.; De la Rosa Rodriguez, M.; Grabner, G.F.; Heier, C.; Viertlmayr, R.; Radler, C.; Lichtenegger, J.; Zimmermann, R.; et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J. Lipid Res. 2018, 59, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, M.; Niyogi, S.; Bhattacharyya, M.; Adak, M.; Nayak, D.K.; Chakrabarti, S.; Chakrabarti, P. Ubiquitin Ligase COP1 Controls Hepatic Fat Metabolism by Targeting ATGL for Degradation. Diabetes 2016, 65, 3561–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahn, T.H.M.; Kaur, R.; Yin, J.; Schweiger, M.; Sharma, V.M.; Lee, M.J.; Ido, Y.; Smas, C.M.; Zechner, R.; Lass, A.; et al. Fat-specific protein 27 (FSP27) interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J. Biol. Chem. 2014, 289, 12029–12039. [Google Scholar] [CrossRef] [Green Version]
- Niyogi, S.; Ghosh, M.; Adak, M.; Chakrabarti, P. PEDF promotes nuclear degradation of ATGL through COP1. Biochem. Biophys. Res. Commun. 2019, 512, 806–811. [Google Scholar] [CrossRef]
- Borg, M.L.; Andrews, Z.B.; Duh, E.J.; Zechner, R.; Meikle, P.J.; Watt, M.J. Pigment epithelium-derived factor regulates lipid metabolism via adipose triglyceride lipase. Diabetes 2011, 60, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, N.; Schweiger, M.; Romauch, M.; Grabner, G.F.; Eichmann, T.O.; Fuchs, E.; Ivkovic, J.; Heier, C.; Mrak, I.; Lass, A.; et al. Development of small-molecule inhibitors targeting adipose triglyceride lipase. Nat. Chem. Biol. 2013, 9, 785–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerk, I.K.; Salzburger, B.; Boeszoermenyi, A.; Heier, C.; Pillip, C.; Romauch, M.; Schweiger, M.; Cornaciu, I.; Lass, A.; Zimmermann, R.; et al. A peptide derived from G0/G1 switch gene 2 acts as noncompetitive inhibitor of adipose triglyceride lipase. J. Biol. Chem. 2014, 289, 32559–32570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.B.; Chen, F. Isolation and purification of baicalein, wogonin and oroxylin A from the medicinal plant Scutellaria baicalensis by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1074, 107–110. [Google Scholar] [CrossRef]
- Jin, H.; Lian, N.; Bian, M.; Zhang, C.; Chen, X.; Shao, J.; Wu, L.; Chen, A.; Guo, Q.; Zhang, F.; et al. Oroxylin A prevents alcohol-induced hepatic steatosis through inhibition of hypoxia inducible factor 1alpha. Chem. Biol. Interact. 2018, 285, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Z.; Yao, Z.; Wang, L.; Zhang, F.; Shao, J.; Chen, A.; Zheng, S. Activation of autophagy is required for Oroxylin A to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. Int. Immunopharmacol. 2018, 56, 148–155. [Google Scholar] [CrossRef]
- Zhu, R.; Zeng, G.; Chen, Y.; Zhang, Q.; Liu, B.; Liu, J.; Chen, H.; Li, M. Oroxylin A accelerates liver regeneration in CCl(4)-induced acute liver injury mice. PLoS ONE 2013, 8, e71612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Guo, M.; Shen, M.; Li, Y.; Tan, S.; Shao, J.; Zhang, F.; Chen, A.; Wang, S.; Zheng, S. Oroxylin A regulates the turnover of lipid droplet via downregulating adipose triglyceride lipase (ATGL) in hepatic stellate cells. Life Sci. 2019, 238, 116934. [Google Scholar] [CrossRef]
- Hu, L.; Deeney, J.T.; Nolan, C.J.; Peyot, M.L.; Ao, A.; Richard, A.M.; Luc, E.; Faergeman, N.J.; Knudsen, J.; Guo, W.; et al. Regulation of lipolytic activity by long-chain acyl-coenzyme A in islets and adipocytes. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E1085–E1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, H.M.; Paar, M.; Heier, C.; Moustafa, T.; Hofer, P.; Haemmerle, G.; Lass, A.; Zechner, R.; Oberer, M.; Zimmermann, R. Adipose triglyceride lipase activity is inhibited by long-chain acyl-coenzyme A. Biochim. Biophys. Acta 2014, 1841, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Eichmann, T.O.; Grumet, L.; Taschler, U.; Hartler, J.; Heier, C.; Woblistin, A.; Pajed, L.; Kollroser, M.; Rechberger, G.; Thallinger, G.G.; et al. ATGL and CGI-58 are lipid droplet proteins of the hepatic stellate cell line HSC-T6. J. Lipid Res. 2015, 56, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mello, T.; Nakatsuka, A.; Fears, S.; Davis, W.; Tsukamoto, H.; Bosron, W.F.; Sanghani, S.P. Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun. 2008, 374, 460–464. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.; Radner, F.P.; Moustafa, T.; Schreiber, R.; Grond, S.; Eichmann, T.O.; Schweiger, M.; Schmidt, A.; Cerk, I.K.; Oberer, M.; et al. G0/G1 Switch Gene 2 Regulates Cardiac Lipolysis. J. Biol. Chem. 2015, 290, 26141–26150. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Higuchi, N.; Enjoji, M. Reduced hepatic expression of adipose tissue triglyceride lipase and CGI-58 may contribute to the development of non-alcoholic fatty liver disease in patients with insulin resistance. Scand. J. Gastroenterol. 2008, 43, 1018–1019. [Google Scholar] [CrossRef]
- Missaglia, S.; Coleman, R.A.; Mordente, A.; Tavian, D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells 2019, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Xu, M.J.; Gao, B. Hepatocytes: A key cell type for innate immunity. Cell. Mol. Immunol. 2016, 13, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, K.; Kuroda, H.; Izumi, R.; Tateyama, M.; Kato, M.; Sugimura, K.; Sakata, Y.; Ikeda, Y.; Hirano, K.; Aoki, M. A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: A case report and literature review. Neuromuscul. Disord. NMD 2014, 24, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E.M.; Arca, M.; Bertini, E.; Bruno, C.; Cassandrini, D.; D’Amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Neutral Lipid Storage Diseases: Clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet J. Rare Dis. 2017, 12, 90. [Google Scholar] [CrossRef] [Green Version]
- Yavuz, A.; Unverengil, G.; Yildirim, A.N.T.; Marasli, H.S.; Tuncer, I. Late-Onset Lipid Storage Myopathy with Fatal Hepatosteatosis. Eur. J. Case Rep. Intern. Med. 2020, 7, 001980. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.T.; Mashek, M.T.; Davidson, N.O.; Mashek, D.G. Hepatic ATGL mediates PPAR-α signaling and fatty acid channeling through an L-FABP independent mechanism. J. Lipid Res. 2014, 55, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, V.; Missaglia, S.; Marozzo, R.; Tavian, D.; Angelini, C. MiRNAs as biomarkers of phenotype in neutral lipid storage disease with myopathy. Muscle Nerve 2020, 61, 253–257. [Google Scholar] [CrossRef]
- Turpin, S.M.; Hoy, A.J.; Brown, R.D.; Rudaz, C.G.; Honeyman, J.; Matzaris, M.; Watt, M.J. Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 2011, 54, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Reid, B.N.; Ables, G.P.; Otlivanchik, O.A.; Schoiswohl, G.; Zechner, R.; Blaner, W.S.; Goldberg, I.J.; Schwabe, R.F.; Chua, S.C., Jr.; Huang, L.S. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008, 283, 13087–13099. [Google Scholar] [CrossRef] [Green Version]
- Wohlers, L.M.; Spangenburg, E.E. 17beta-estradiol supplementation attenuates ovariectomy-induced increases in ATGL signaling and reduced perilipin expression in visceral adipose tissue. J. Cell. Biochem. 2010, 110, 420–427. [Google Scholar] [CrossRef]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Greenberg, A.S.; Mashek, D.G. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology 2011, 53, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Tavian, D.; Missaglia, S.; Redaelli, C.; Pennisi, E.M.; Invernici, G.; Wessalowski, R.; Maiwald, R.; Arca, M.; Coleman, R.A. Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: A strikingly low amount of lipase activity may preserve cardiac function. Hum. Mol. Genet. 2012, 21, 5318–5328. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, M.V.; Lodhi, I.J.; Yin, L.; Malapaka, R.R.; Xu, H.E.; Turk, J.; Semenkovich, C.F. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 2009, 138, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, P.; Claudel, T.; Baghdasaryan, A.; Mueller, M.; Halilbasic, E.; Das, S.K.; Lass, A.; Zimmermann, R.; Zechner, R.; Hoefler, G.; et al. Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology 2014, 59, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.P.; Patel, J.; Byrne, C.D. Causes of Mortality in Non-Alcoholic Fatty Liver Disease (NAFLD) and Alcohol Related Fatty Liver Disease (AFLD). Curr. Pharm. Des. 2020, 26, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.G.; Thosani, N.C.; Pan, J.J. Noninvasive biomarkers for the diagnosis of steatohepatitis and advanced fibrosis in NAFLD. Biomark. Res. 2013, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, C.D.; Claudel, T.; Kumari, P.; Haemmerle, G.; Pollheimer, M.J.; Stojakovic, T.; Scharnagl, H.; Halilbasic, E.; Gumhold, J.; Silbert, D.; et al. Absence of adipose triglyceride lipase protects from hepatic endoplasmic reticulum stress in mice. Hepatology 2012, 56, 270–280. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Lee, D.; Pulinilkunnil, T.; Brenner, D.S.; Cai, L.; Magnes, C.; Koefeler, H.C.; Streith, I.E.; Rechberger, G.N.; Haemmerle, G.; et al. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J. Biol. Chem. 2009, 284, 30218–30229. [Google Scholar] [CrossRef] [Green Version]
- Ong, K.T.; Mashek, M.T.; Bu, S.Y.; Mashek, D.G. Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Al-Zoughbi, W.; Pichler, M.; Gorkiewicz, G.; Guertl-Lackner, B.; Haybaeck, J.; Jahn, S.W.; Lackner, C.; Liegl-Atzwanger, B.; Popper, H.; Schauer, S.; et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget 2016, 7, 33832–33840. [Google Scholar] [CrossRef] [Green Version]
- Tomin, T.; Fritz, K.; Gindlhuber, J.; Waldherr, L.; Pucher, B.; Thallinger, G.G.; Nomura, D.K.; Schittmayer, M.; Birner-Gruenberger, R. Deletion of Adipose Triglyceride Lipase Links Triacylglycerol Accumulation to a More-Aggressive Phenotype in A549 Lung Carcinoma Cells. J. Proteome Res. 2018, 17, 1415–1425. [Google Scholar] [CrossRef]
- Di Leo, L.; Vegliante, R.; Ciccarone, F.; Salvatori, I.; Scimeca, M.; Bonanno, E.; Sagnotta, A.; Grazi, G.L.; Aquilano, K.; Ciriolo, M.R. Forcing ATGL expression in hepatocarcinoma cells imposes glycolytic rewiring through PPAR-α/p300-mediated acetylation of p53. Oncogene 2019, 38, 1860–1875. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Attané, C.; Milhas, D.; Dirat, B.; Dauvillier, S.; Guerard, A.; Gilhodes, J.; Lazar, I.; Alet, N.; Laurent, V.; et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2017, 2, e87489. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniëls, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yu, X.; Lin, L.; Deng, J.; Wang, K.; Xia, Y.; Tang, X.; Hong, H. ATGL promotes the proliferation of hepatocellular carcinoma cells via the p-AKT signaling pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22391. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | Year | Animal Model | Animal Age | Key Findings |
|---|---|---|---|---|
| Haemmerle et al. [67] | 2006 | Global ATGL inactivation by targeted homologous recombination | 8 to 14 weeks | ATGL is the rate-limiting enzyme of TG catabolism. The inactivation of ATGL increased glucose tolerance and insulin sensitivity. |
| Reid et al. [83] | 2008 | Adenovirus-mediated global ATGL knockout in ob/ob mice | 3 to 5 months old | ATGL possesses TG hydrolase activity and is essential in maintaining hepatic lipid homeostasis by mobilizing and partitioning stored TG into FA oxidation pathways. |
| Kienesberger et al. [84] | 2009 | Global ATGL knockout in mice with mixed genetic background (50% C57BL/6 and 50% 129/Ola) | <14 weeks | Global ATGL deficiency decreased insulin signaling in the liver of the mice. |
| Turpin et al. [85] | 2011 | Adenovirus-mediated liver-specific ATGL overexpression in ob/ob mice | 10 weeks | Liver-specific ATGL overexpression reduces hepatic steatosis and mildly enhances liver insulin sensitivity. |
| Wu et al. [86] | 2011 | Albumin Cre-mediated liver specific ATGL knockout | 6, 8, and 12 months old | ATGLLKO induced hepatic steatosis and suppressed β-oxidation in the liver. |
| Ong et al. [87] | 2011 | Adenovirus-mediated liver-specific ATGL knockdown in C57/B16 mice | 8 to 10 weeks old | Liver-specific knockdown of ATGL reduced TG hydrolase activity, and increased TG content in the liver. It also altered fatty acid composition with a significant reduction in C16:0, C18:0, and C18:3 but an increase in C18:1 in hepatic TG content. |
| Fuchs et al. [88] | 2012 | Global ATGL inactivation by targeted homologous recombination | N/A | The increased non-esterified oleic acid (OA) in the liver protected ATGL KO mice from TM-induced hepatic ER stress through interfering with palmitate (PA)-induced phosphoinositide-3-kinase inhibitor 1 (Pik3ip1) expression. |
| Jha et al. [89] | 2014 | Global ATGL inactivation by targeted homologous recombination | 7 to 10 weeks | ATGL deficiency enhanced MCD- and LPS-induced hepatic inflammation. The anti-inflammatory effect of ATGL in the liver was partially achieved by PPARα signaling pathway. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Guo, W.; Zhou, Z. Adipose Triglyceride Lipase in Hepatic Physiology and Pathophysiology. Biomolecules 2022, 12, 57. https://doi.org/10.3390/biom12010057
Li T, Guo W, Zhou Z. Adipose Triglyceride Lipase in Hepatic Physiology and Pathophysiology. Biomolecules. 2022; 12(1):57. https://doi.org/10.3390/biom12010057
Chicago/Turabian StyleLi, Tianjiao, Wei Guo, and Zhanxiang Zhou. 2022. "Adipose Triglyceride Lipase in Hepatic Physiology and Pathophysiology" Biomolecules 12, no. 1: 57. https://doi.org/10.3390/biom12010057