
Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
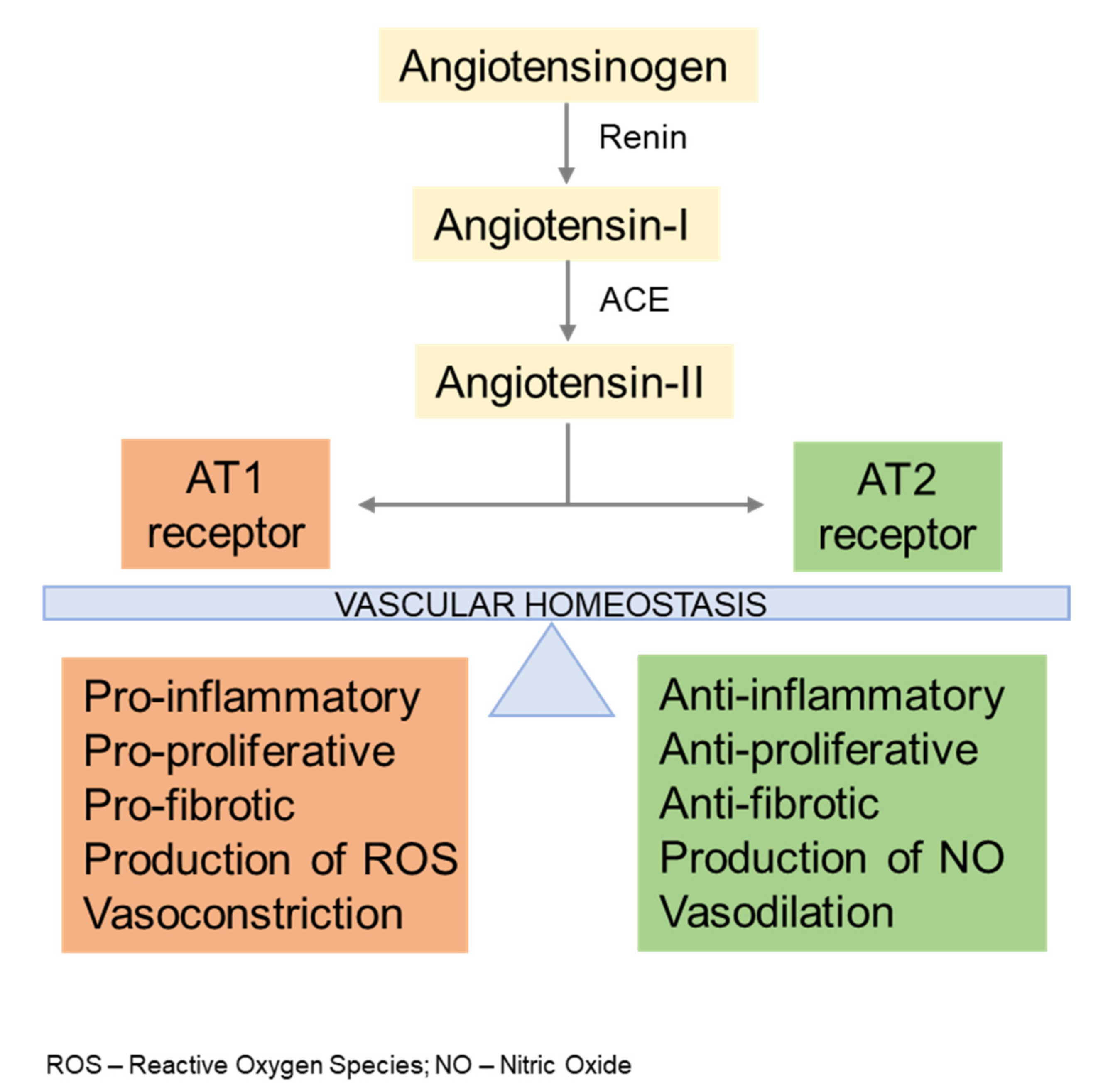
2. AngII Signaling
3. TGFβ Signaling
4. Endothelial Dysfunction
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Milewicz, D.M.; Ramirez, F. Therapies for thoracic aortic aneurysms and acute aortic dissections. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, F.; Sakai, L.Y. Biogenesis and function of fibrillin assemblies. Cell Tissue Res. 2010, 339, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef] [Green Version]
- Carta, L.; Smaldone, S.; Zilberberg, L.; Loch, D.; Dietz, H.C.; Rifkin, D.B.; Ramirez, F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J. Biol. Chem. 2009, 284, 5630–5636. [Google Scholar] [CrossRef] [Green Version]
- Habashi, J.P.; Judge, D.P.; Holm, T.M.; Cohn, R.D.; Loeys, B.L.; Cooper, T.K.; Myers, L.; Klein, E.C.; Liu, G.; Calvi, C.; et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006, 312, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Meisinger, T.; Knispel, R.; Worth, J.M.; Baxter, B.T. MMP-2 regulates Erk1/2 phosphorylation and aortic dilation in Marfan syndrome. Circ. Res. 2012, 110, e92–e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.R.; Clayton, N.P.; Carta, L.; Galatioto, J.; Chiu, E.; Smaldone, S.; Nelson, C.A.; Cheng, S.H.; Wentworth, B.M.; Ramirez, F. Dimorphic effects of TGFβ signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Tehrani, A.Y.; Ciufolini, M.A.; Bernatchez, P. Nitric oxide in the Marfan vasculature: Friend or foe? Nitric. Oxide. 2021, 116, 27–34. [Google Scholar] [CrossRef]
- Toral, M.; de la Fuente-Alonso, A.; Campanero, M.R.; Redondo, J.M. The NO signaling pathway in aortic aneurysm and dissection. Br. J. Pharmacol. 2021, 1–17. [Google Scholar] [CrossRef]
- Lu, H.; Rateri, D.L.; Bruemmer, F.; Cassis, L.A.; Daugherty, A. Involvement of the renin-angiotensin system in abdominal and thoracic aortic aneurysms. Clin. Sci. 2012, 123, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Jeremy, R.W. Angiotensin, transforming growth factorβ and aortic dilatation in Marfan syndrome: Of mice and humans. Int. J. Cardiol. Heart. Vasc. 2018, 18, 71–80. [Google Scholar]
- Yetman, A.T.; Bornermeier, R.A.; McCrindle, B.W. Usefulness of enalapril versus propranolol or atenolol for prevention of aortic dilation in patients with the Marfan syndrome. Am. J. Cardiol. 2005, 95, 1125–1127. [Google Scholar] [CrossRef]
- Brooke, B.S.; Habashi, J.P.; Judge, D.P.; Patel, N.; Loeys, B.; Dietz, H.C., III. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N. Engl. J. Med. 2008, 358, 2787–2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habashi, J.P.; Doyle, J.J.; Holm, T.M.; Aziz, H.; Schoenhoff, F.; Bedja, D.; Chen, Y.-C.; Modiri, A.N.; Judge, D.P.; Dietz, H.C. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011, 332, 361–365. [Google Scholar] [CrossRef]
- Sellers, S.L.; Milad, N.; Chan, R.; Mielnik, M.; Jermilova, U.; Huang, P.L.; de Crom, R.; Hirota, J.A.; Hogg, J.C.; Sandor, G.G.; et al. Inhibitiion of Marfan syndrome aortic root dilation by Losartan: Role of angiotensin II receptor type 1—Independent activation of endothelial function. Am. J. Pathol. 2018, 183, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Z.; Sawada, H.; Ye, D.; Katsumata, Y.; Kukida, M.; Ohno-Urabe, S.; Moorleghen, J.J.; Franklin, M.K.; Howatt, D.A.; Sheppard, M.B.; et al. Deletion of AT1a (Angiotensin II Type 1a) receptor or inhibition of angiotensinogen synthesis attenuates thoracic aortopathies in fibrillin 1C1041G/+ mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Galatioto, J.; Caescu, C.I.; Hansen, J.; Cook, J.; Miramontes, I.; Iyengar, R.; Ramirez, F. Cell type-specific contributions of the Ang II type 1a receptor to aorta homeostasis and aneurysmal disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 588–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbrugghe, P.; Verhoeven, J.; Clijsters, M.; Vervoort, D.; Schepens, J.; Meuris, B.; Herijgers, P. The effect of a nonpeptide angiotensin II Type 2 receptor agonist, Compound 21, on aortic aneurysm growth in a mouse model of Marfan syndrome. J. Cardiovasc. Pharmacol. 2018, 71, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Peters, A.M.; Wang, S.; Janda, A.; Chen, J.; Zhou, P.; Arthur, E.; Kwartler, C.S.; Milewicz, D.M. Reversal of aortic enlargement induced by increased biomechanical forces requires AT1R inhibition in conjunction with AT2R activation. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 459–466. [Google Scholar] [CrossRef] [Green Version]
- Gomez, D.; Al Haj Zen, A.; Borges, L.F.; Phillippe, M.; Sampaio Gutierrez, P.; Jondeau, G.; Michel, J.-B.; Vranckx, R. Syndromic and non-syndromic aneurysms of the human ascending aorta share activation of the Smad2 pathway. J. Pathol. 2009, 218, 131–142. [Google Scholar] [CrossRef]
- Gomez, D.; Coyet, A.; Olivier, V.; Jeunemaitre, X.; Jondeau, G.; Michel, J.-B.; Vranckx, R. Epigenetic control of vascular smooth muscle cells in Marfan and non Marfan thoracic aortic aneurysms. Cardiovasc. Res. 2011, 89, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, M.E.; Dietz, H.C. The genetic basis of aortic aneurysm. Cold Spring Har. Perspect. Med. 2014, 4, a015909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Gerisson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Invest. 2014, 124, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Hong Hu, J.; Angelov, S.N.; Fox, K.; Yan, J.; Enstrom, R.; Smith, A.; Dichek, D.A. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered transforming growth factor β signaling. J. Amer. Heart Assoc. 2017, 24, e004968. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massaguè, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell. Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, L.; Phoon, C.K.L.; Robertson, I.; Dabovic, B.; Ramirez, F.; Rifkin, D.B. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 14012–14017. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Angelov, S.; Yildirim, I.A.; Wei, H.; Hu, J.H.; Majesky, M.W.; Brozovich, F.V.; Kim, F.; Dichek, D.A. Loss of transforming growth factor beta signaling in aortic smooth muscle cells causes endothelial dysfunction and aortic hypercontractility. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1956–1971. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T. Oxidative stress, endothelial dysfunction, and its impact on smooth muscle signaling. In Muscle; Hill, J.A., Olson, E.N., Eds.; Academic Press: London, UK, 2012; Volume 2, pp. 1219–1229. [Google Scholar]
- Nakamura, M.; Itoh, S.; Makita, A.; Ohira, N.; Hiramori, K. Peripheral resistance vessel dysfunction in Marfan syndrome. Am. Heart. J. 2000, 139, 661–666. [Google Scholar] [CrossRef]
- Takata, M.; Amiya, E.; Watanabe, M.; Omori, K.; Imai, Y.; Fujita, D.; Nishimura, H.; Kato, M.; Morota, T.; Nawata, K.; et al. Impairment of flow-mediated dilation correlates with aortic dilation in patients with Marfan syndrome. Heart Vessels. 2014, 29, 478–485. [Google Scholar] [CrossRef]
- Yan, C.; Kim, D.; Aizawa, T.; Berk, B.C. Functional interplay between angiotensin II and Nitric Oxide: Cyclic GMP as a key mediator. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Walshe, T.E.; Dela Paz, N.G.; D’Amore, P.A. The role of shear-induced transforming growth factor-β signaling in the endothelium. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2608–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emrich, F.; Penov, K.; Arakawa, M.; Dhablania, N.; Burdon, G.; Pedroza, A.J.; Koyano, T.K.; Kim, Y.M.; Raaz, U.; Connolly, A.J.; et al. Anatomically specific reactive oxygen species production participates in Marfan syndrome aneurysm formation. J. Cell Mol. Med. 2019, 23, 7000–7009. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.G.; Bellamy, M.F.; Ramsey, M.W.; Goodfellow, J.; Brownlee, M.; Davies, S.; Wilson, J.F.; Lewis, M.J.; Stuart, A.G. Endothelial function in Marfan syndrome: Selective impairment of flow-mediated vasodilation. Circulation. 1999, 99, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Pluijm, I.; Burger, J.; van Heijningen, P.M.; Jpma, A.I.; van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.-J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oller, J.; Gabande-Rodriguez, E.; Ruiz-Rodriguez, M.J.; Desdin-Mico, G.; Aranda, J.F.; Rodriguez-Diez, R.; Ballesteros-Martinez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuna, P.; et al. Extracellular tuning of mitrochondrial respiration leads to aortic aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef]
- Chung, A.W.Y.; Yeung, K.A.; Cortes, S.F.; Sandor, G.G.S.; Judge, D.P.; Dietz, H.C.; van Breemen, C. Endothelial dysfunction and compromise eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br. J. Pharmacol. 2007, 150, 1075–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.H.C.; van Breemen, C.; Chung, A.W.Y. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vasc. Pharmacol. 2010, 52, 37–45. [Google Scholar] [CrossRef]
- Jimenez-Altayo, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; Gorbenko del Blanco, D.; Lopez-Luque, J.; Mas-Stachurska, A.; Siegert, A.-M.; Bonorino, F.; Barbera, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58. [Google Scholar] [CrossRef]
- Huang, K.; Wang, Y.; Siu, K.L.; Zhang, Y.; Cai, H. Targeting feed-forward signaling of TGFβ/NOX4/DHFR/eNOS uncoupling/TGFβ axis with anti-TGFβ and folic acid attenuates formation of aortic aneurysms: Novel mechanisms and therapeutics. Redox Biol. 2021, 38, 101757. [Google Scholar] [CrossRef] [PubMed]
- Oller, J.; Mendez-Barbero, N.; Ruiz, E.J.; Villahoz, S.; Renard, M.; Canelas, L.I.; Briones, A.M.; Alberca, R.; Lozano-Vidal, N.; Hurle, M.A.; et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat. Med. 2017, 23, 200–212. [Google Scholar] [CrossRef]
- De la Fuente-Alonso, A.; Toral, M.; Alfayate, A.; Ruiz-Rodriguez, M.J.; Bonzon-Kulichenko, E.; Teixdo-Tura, G.; Martinez-Martinez, S.; Mendez-Olivares, M.J.; Lopez-Maderuelo, D.; Gonzalez-Validez, I.; et al. Aortic disease in Marfan syndrome is caused by overactivation of sGC-PRKG signaling by NO. Nat. Commun. 2021, 12, 2628. [Google Scholar] [CrossRef] [PubMed]
- Alfranca, A.; Campanero, M.R.; Redondo, J.M. New methods for disease modeling using lentiviral vectors. Trends Mol. Med. 2018, 24, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Wacker, B.K.; Stamatikos, A.; Sethuraman, M.; Komandur, K.; Dichek, D.A. Jugular vein injection of high-titer lentiviral vectors does not transduce the aorta. Arterioscler. Thromb. Vasc. Bio. 2021, 41, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Judge, D.P.; Biery, N.J.; Keene, D.R.; Geubtner, J.; Myers, L.; Huso, D.L.; Sakai, L.Y.; Dietz, H.C. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Invest. 2004, 114, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Lee, S.Y.; Gayraud, B.; Andrikopoulos, K.; Shapiro, S.D.; Bunton, T.; Biery, N.J.; Dietz, H.C.; Sakai, L.Y.; Ramirez, F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc. Natl. Acad. Sci. USA 1999, 96, 3819–3823. [Google Scholar] [CrossRef] [Green Version]
- Ohno-Urabe, S.; Kukida, M.; Franklin, M.K.; Katsumata, Y.; Su, W.; Gong, M.C.; Lu, H.S.; Daughterty, A.; Sawada, H. Authentication of in situ measurements for thoracic aortic aneurysms in mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2117–2119. [Google Scholar] [CrossRef]
- Angelov, S.N.; Hu, J.H.; Wei, H.; Airhart, N.; Shi, M.; Dichek, D.A. TGF-β (Transforming Growth Factor-β) signaling protects the thoracic and abdominal aorta from angiotensin II-induced pathology by distinct mechanisms. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2102–2113. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Rateri, D.; Sheppard, M.; Daugherty, A. Sexual dimorphism in a Marfan syndrome mouse model. Circulation 2017, 136, A20315. [Google Scholar]
- Ferruzzi, J.; Bersi, M.R.; Humphrey, J.D. Biomechanical phenotyping of central arteries in health and disease: Advantages of and methods for murine models. Ann. Biomed. Eng. 2013, 41, 1311–1330. [Google Scholar] [CrossRef] [Green Version]
- Del Campo, L.; Ferrer, M. Wire myography to study vascular tone and vascular structure of isolated mouse arteries. Methods Mol. Biol. 2015, 1339, 255–276. [Google Scholar] [PubMed]
- Griendling, K.K.; Rouyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.-R.; Harrison, D.G.; Bhatnagar, A. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef] [PubMed]
- Tavassoloy, I.; Goldfarb, J.; Iyengar, R. Systems biology primer: The basic methods and approaches. Essays Biochem. 2018, 62, 487–500. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asano, K.; Cantalupo, A.; Sedes, L.; Ramirez, F. Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome. Biomolecules 2022, 12, 128. https://doi.org/10.3390/biom12010128
Asano K, Cantalupo A, Sedes L, Ramirez F. Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome. Biomolecules. 2022; 12(1):128. https://doi.org/10.3390/biom12010128
Chicago/Turabian StyleAsano, Keiichi, Anna Cantalupo, Lauriane Sedes, and Francesco Ramirez. 2022. "Pathophysiology and Therapeutics of Thoracic Aortic Aneurysm in Marfan Syndrome" Biomolecules 12, no. 1: 128. https://doi.org/10.3390/biom12010128